Genome-Wide Analysis of Long Non-Coding RNAs in Potato and Their Potential Role in Tuber Sprouting Process


Abstract
1. Introduction
2. Results
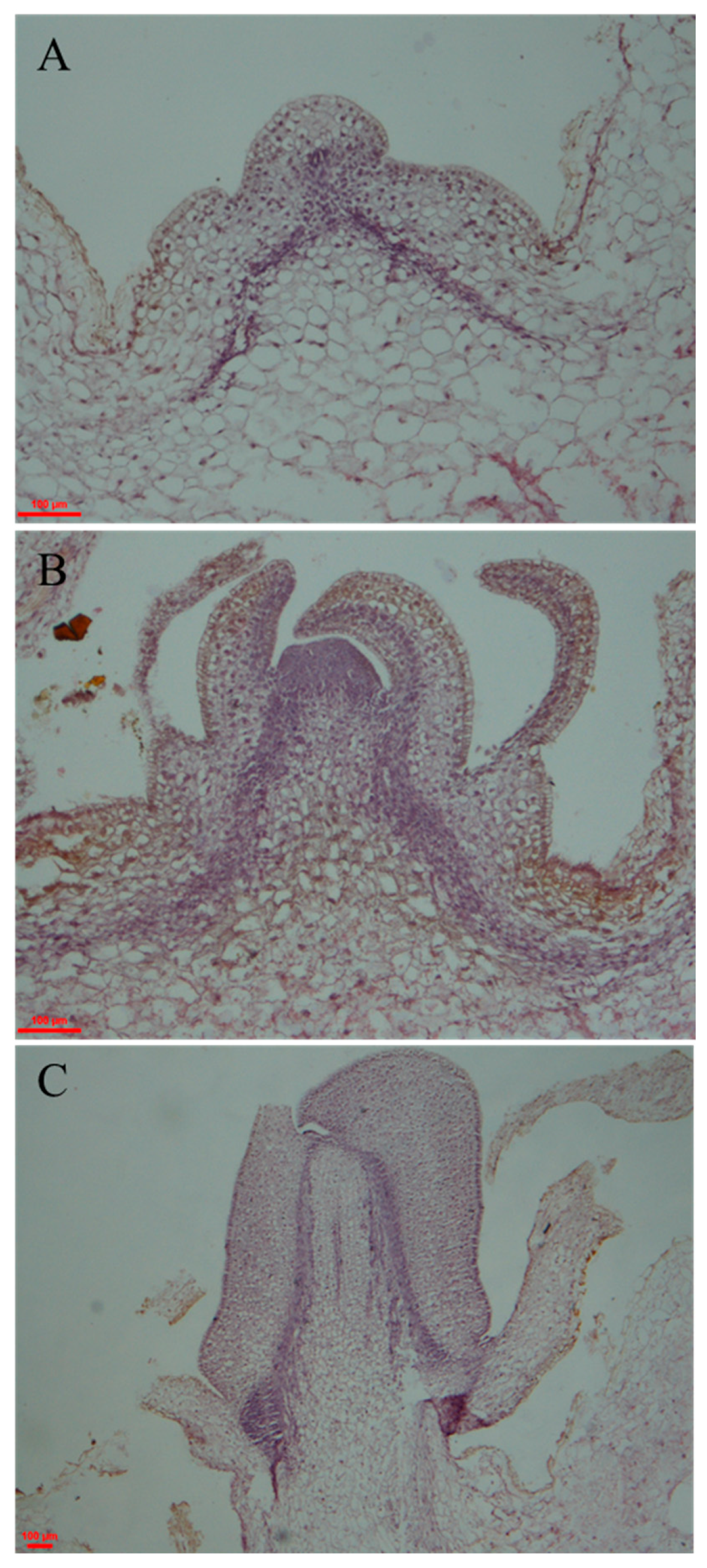
2.1. Observation and Identification of Potato Morphological Changes during Tuber Dormancy Release
2.2. Quality Assessment of Sequence Data
2.3. Sequence Alignment
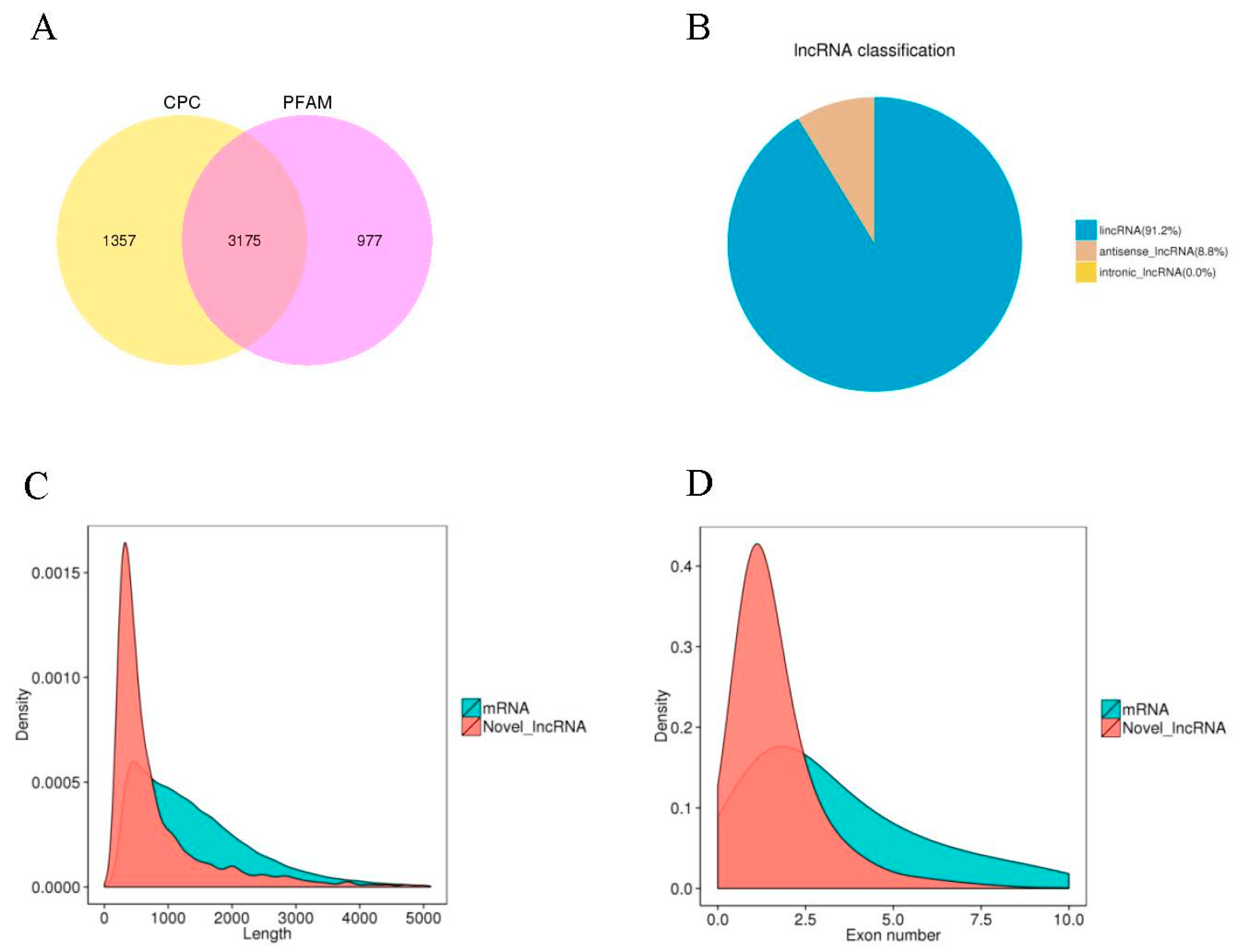
2.4. Characteristic Features of Potato LncRNAs
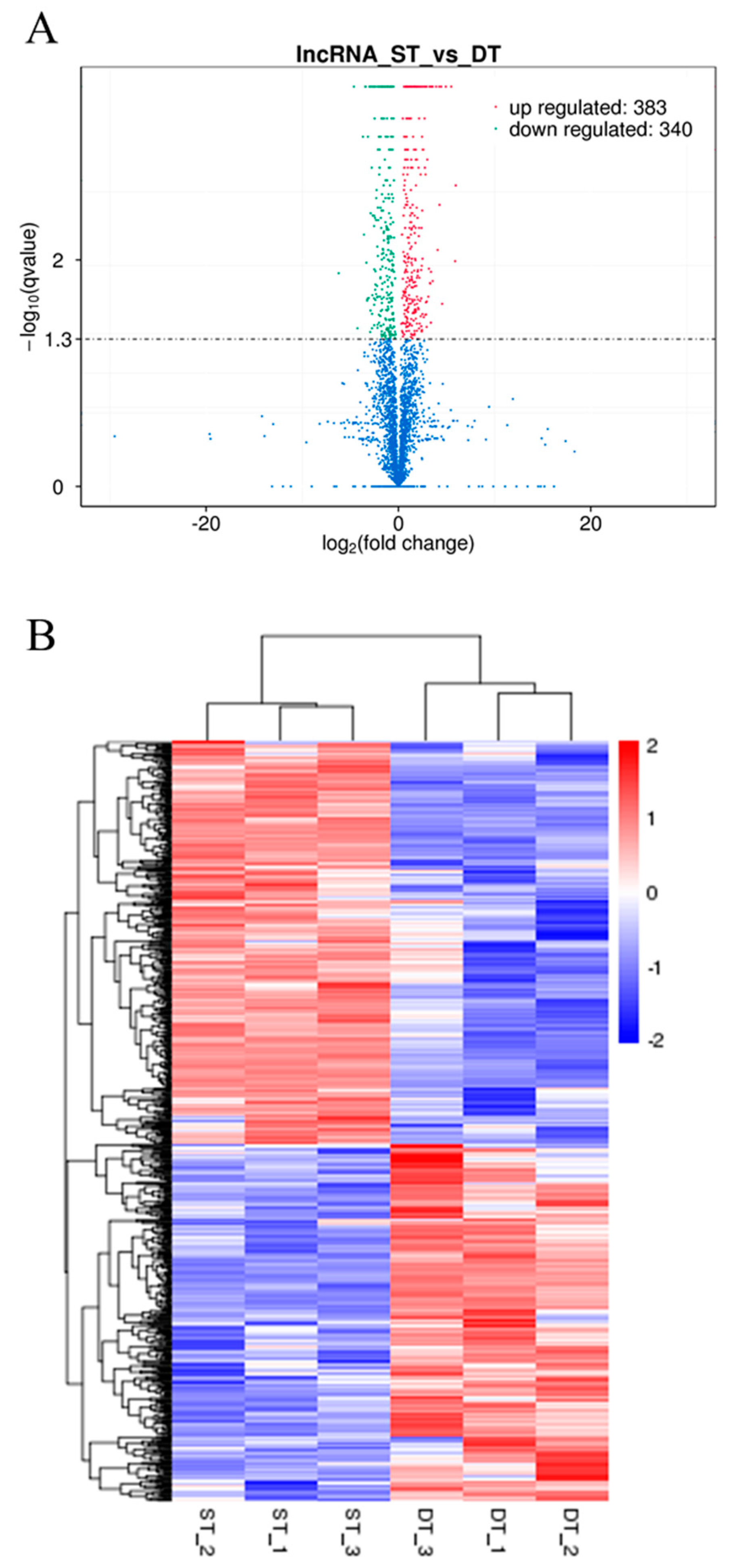
2.5. Analysis of Differentially Expressed LncRNAs
2.6. GO Enrichment of Differentially Expressed LncRNAs
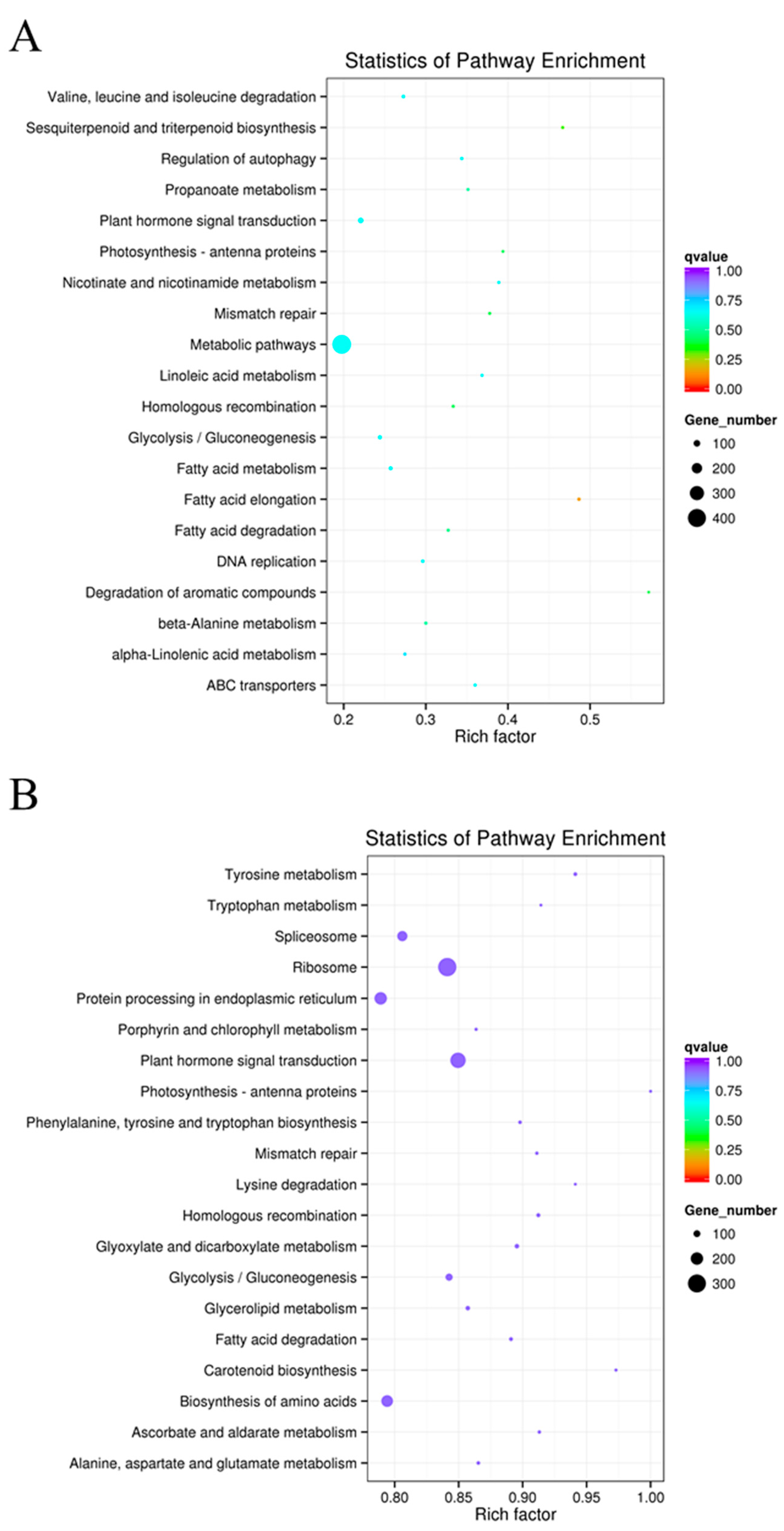
2.7. KEGG Functional Enrichment of Differentially Expressed LncRNAs
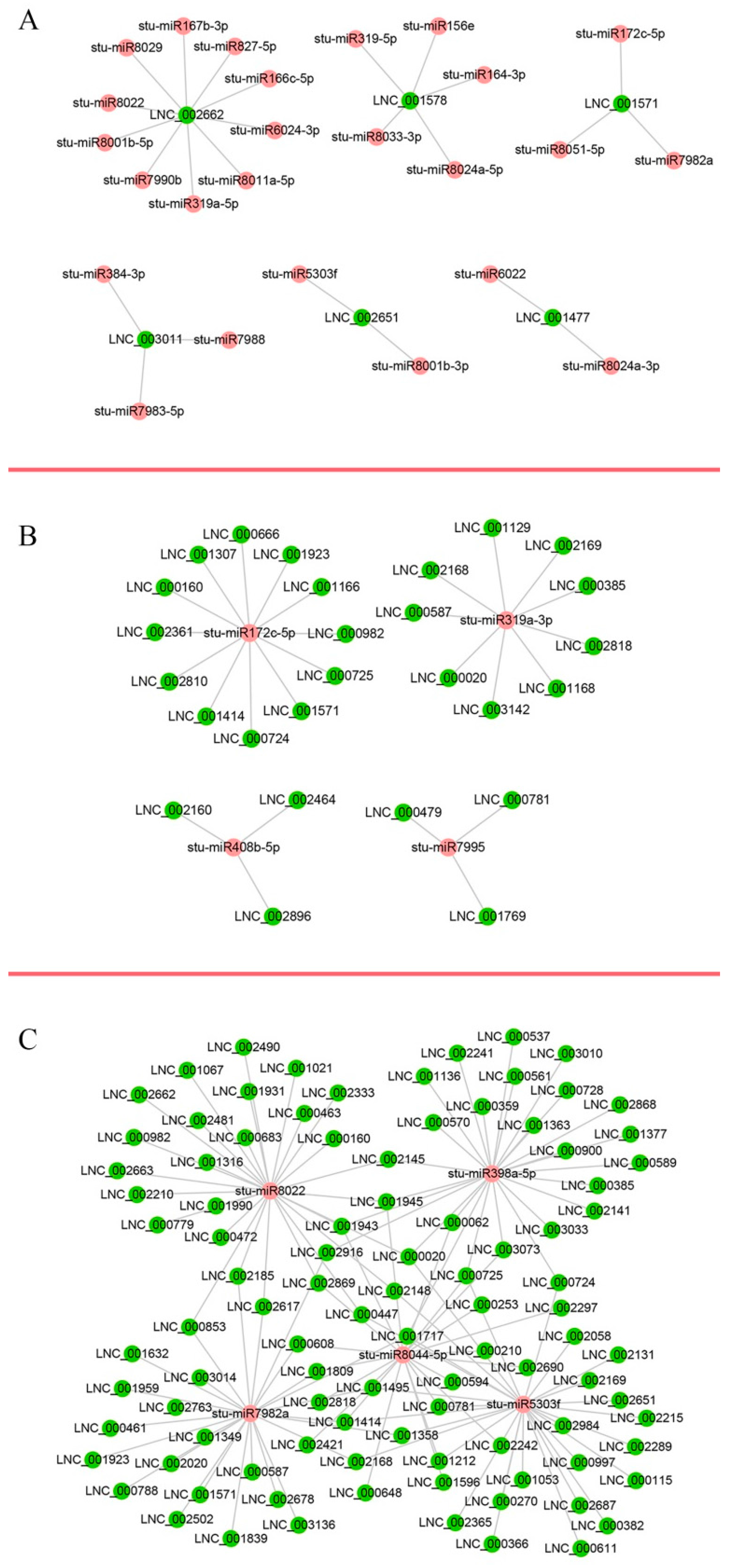
2.8. LncRNAs as Potential Regulators of miRNAs
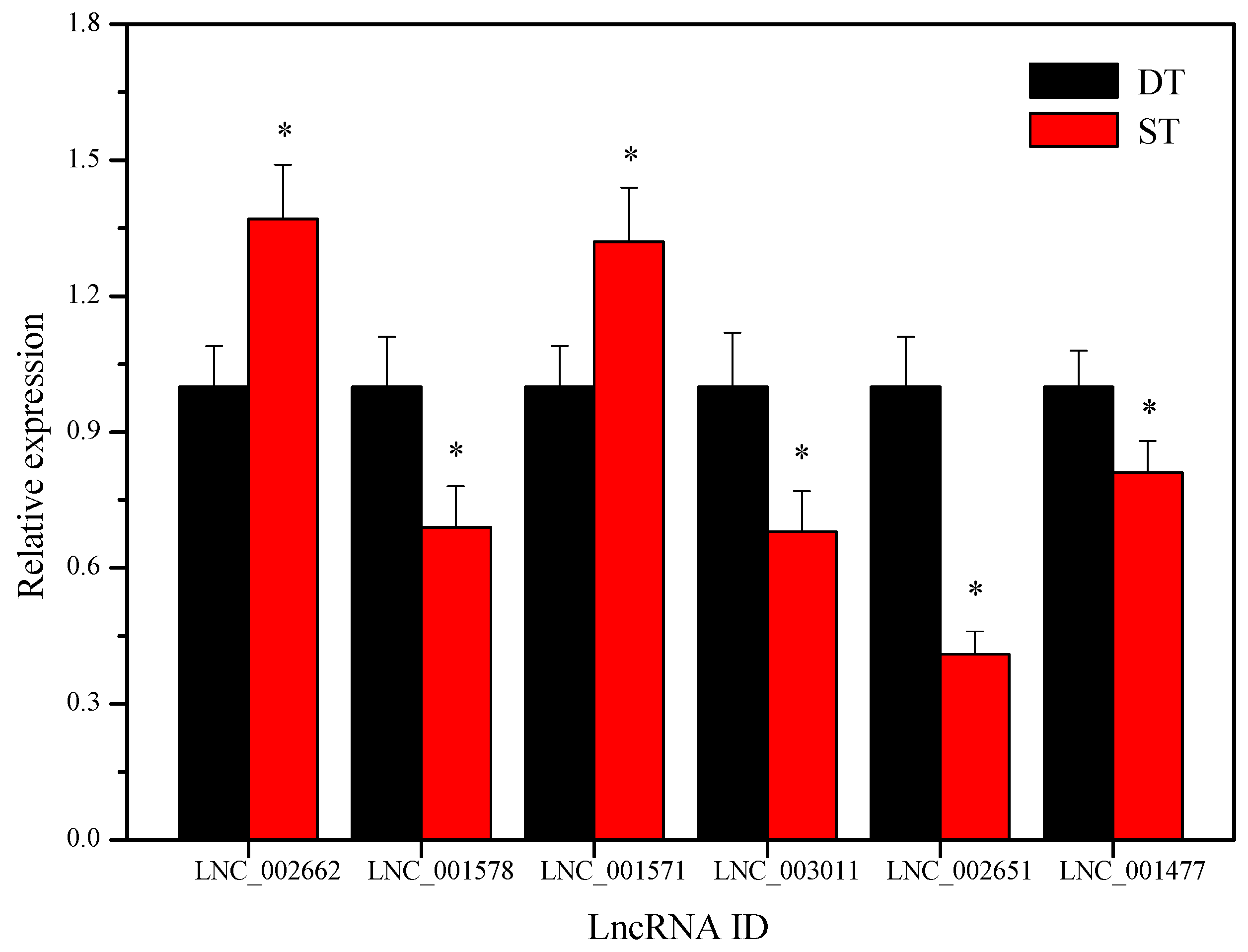
2.9. Validation of the Differentially Expressed LncRNAs by Using Quantitative Real-Time PCR
3. Discussion
3.1. Differentiation and Development of Potato Bud Primordia
3.2. Effects of Sprouting on LncRNA Expression in Potato Tubers
4. Materials and Methods
4.1. Plant Materials
4.2. Observations of Potato Morphological Changes during Tuber Dormancy Release
4.2.1. Dehydration, Transparency, Wax Dipping, and Embedding
- Dehydration: Potato tuber apical buds fixed with FAA fixative (5% formaldehyde, 5% acetic acid, 45% ethanol, and 45% distilled water) were sequentially immersed for 90 min in 50, 70, 85, 95 and 100% ethanol to dehydrate and harden the samples for optimal paraffin infiltration.
- Transparency: After a 30-min intermediate processing with anhydrous ethanol and xylene (volume ratio of 1:1), samples were processed with pure xylene for another 30 min to remove alcohol that infiltrated during the dehydration process and to facilitate wax dipping and embedding.
- Wax dipping: Xylene was removed from the apical buds of potato tubers, and the tubers were then placed in 100% paraffin for 90 min in an incubator at 56–60 °C.
- Embedding: The apical buds of potato tubers processed with 100% paraffin were embedded in cassettes by using a Leica embedding station (Leica EG 1160; Leica Biosystems, Nussloch, Germany) for sectioning.
4.2.2. Sectioning and Staining
- Sectioning: The wax blocks formed were trimmed with a blade and sectioned using a Leica RM 2145 microtome (Leica Biosystems) to a thickness of 8 μm. These sections were then placed on glass slides and heated for 60 min at 65 °C on a Leica HI 1220 slide warmer (Leica Biosystems).
- Dewaxing: Slides were immersed in xylene twice for 50 min each for dewaxing, and then processed in decreasing ethanol concentrations (100, 95, 80, 50 and 0%) for 5 min each.
- Staining: Slides were stained in haematoxylin for 8 min and rinsed with tap water for 5 min. They were subsequently immersed in and stained with eosin for another 2 min and rinsed with distilled water, following which they were subjected to rapid gradient dehydration in increasing ethanol concentrations (50, 80, 95 and 100%) for 2 min each. Samples were rendered transparent in xylene for 5 min.
- Paraffin section sealing and microscopic imaging: Stained paraffin sections were preserved in neutral gum and organised before observation and imaging by using a Nikon Eclipse 80i microscope (Nikon Corporation, Tokyo, Japan).
4.3. RNA Isolation, Library Preparation, and Sequencing
4.3.1. RNA Quantification and Qualification
4.3.2. Library Preparation for RNA Sequencing
4.3.3. RNA Sequencing
4.4. Quality Control, Mapping to the Reference Genome, and Transcriptome Assembly
4.4.1. Quality Control
4.4.2. Mapping to the Reference Genome
4.4.3. Transcriptome Assembly
4.5. Coding Potential Analysis, Conservative Analysis, and Target Gene Prediction
4.5.1. Coding Potential Analysis
4.5.2. Conservative Analysis
4.5.3. Target Gene Prediction
4.6. Quantification of Gene Expression Levels and Differential Expression Analysis
4.6.1. Quantification of Gene Expression Levels
4.6.2. Differential Expression Analysis
4.7. Enrichment Analysis of Differentially Expressed LncRNAs
4.7.1. GO Enrichment Analysis
4.7.2. KEGG Enrichment Analysis
4.8. Identification of Differentially Expressed LncRNAs as Targets of miRNAs
4.9. Validation of the Differentially Expressed LncRNAs by Using Quantitative Real-Time PCR
4.10. Statistical Analysis
4.11. GenBank Accession Code
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yan, N.; Liu, Y.; Gong, D.; Du, Y.; Zhang, H.; Zhang, Z. Solanesol: A review of its resources, derivatives, bioactivities, medicinal applications, and biosynthesis. Phytochem. Rev. 2015, 14, 403–417. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, N.; Wen, Y.; Jin, X.; Yang, J.; Si, H.; Wang, D. Transcriptomic changes during tuber dormancy release process revealed by RNA sequencing in potato. J. Biotechnol. 2015, 198, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, N.; Zhao, S.; Chang, J.; Wang, Z.; Zhang, G.; Si, H.; Wang, D. Proteomic changes during tuber dormancy release process revealed by iTRAQ quantitative proteomics in potato. Plant Physiol. Biochem. 2015, 86, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Zaheer, K.; Akhtar, M.H. Potato production, usage, and nutrition—A review. Crit. Rev. Food Sci. Nutr. 2016, 56, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Sonnewald, S.; Sonnewald, U. Regulation of potato tuber sprouting. Planta 2014, 239, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Koffi, G.Y.; Remaud-Simeon, M.; Due, A.E.; Combes, D. Isolation and chemoenzymatic treatment of glycoalkaloids from green, sprouting and rotting Solanum tuberosum potatoes for solanidine recovery. Food Chem. 2017, 220, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Uri, C.; Juhász, Z.; Polgár, Z.; Bánfalvi, Z. A GC-MS-based metabolomics study on the tubers of commercial potato cultivars upon storage. Food Chem. 2014, 159, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Knuthsena, P.; Jensenb, U.; Schmidta, B.; Larsenb, I.K. Glycoalkaloids in potatoes: Content of glycoalkaloids in potatoes for consumption. J. Food Compos. Anal. 2009, 22, 577–581. [Google Scholar] [CrossRef]
- Mensinga, T.T.; Sips, A.J.; Rompelberg, C.J.; van Twillert, K.; Meulenbelt, J.; van den Top, H.J.; van Egmond, H.P. Potato glycoalkaloids and adverse effects in humans: An ascending dose study. Regul. Toxicol. Pharmacol. 2005, 41, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhao, S.; Tan, F.; Zhao, H.; Wang, D.; Si, H.; Chen, Q. Changes in ROS production and antioxidant capacity during tuber sprouting in potato. Food Chem. 2017, 237, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Bachem, C.; van der Hoeven, R.; Lucker, J.; Oomen, R.; Casarini, E.; Jacobsen, E.; Visser, R. Functional genomic analysis of potato tuber life-cycle. Potato Res. 2000, 43, 297–312. [Google Scholar] [CrossRef]
- Kloosterman, B.; De Koeyer, D.; Griffiths, R.; Flinn, B.; Steuernagel, B.; Scholz, U.; Sonnewald, S.; Sonnewald, U.; Bryan, G.J.; Prat, S.; et al. Genes driving potato tuber initiation and growth: Identification based on transcriptional changes using the POCI array. Funct. Integr. Genomics 2008, 8, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, N.; Wen, Y.; Si, H.; Wang, D. Identification of differentially expressed genes in potato associated with tuber dormancy release. Mol. Biol. Rep. 2012, 39, 11277–11287. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.A.; Gleichsner, A.; Alsbury, R.; Horvath, D.; Suttle, J. The sprout inhibitors chlorpropham and 1,4-dimethylnaphthalene elicit different transcriptional profiles and do not suppress growth through a prolongation of the dormant state. Plant Mol. Biol. 2010, 73, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.A.; Gleichsner, A.; Hilldorfer, L.; Horvath, D.; Suttle, J. The sprout inhibitor 1,4-dimethylnaphthalene induces the expression of the cell cycle inhibitors KRP1 and KRP2 in potatoes. Funct. Integr. Genomics 2012, 12, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.; Suttle, J.; Douches, D.S.; Buell, C.R. Treatment of potato tubers with the synthetic cytokinin 1-(α-ethylbenzyl)-3-nitroguanidine results in rapid termination of endodormancy and induction of transcripts associated with cell proliferation and growth. Funct. Integr. Genomics 2014, 14, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.; Chang, H.Y. Long noncoding RNA in genome regulation: Prospects and mechanisms. RNA Biol. 2010, 7, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Chekanova, J.A. Long non-coding RNAs and their functions in plants. Curr. Opin. Plant Biol. 2015, 27, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.C.; Liao, J.Y.; Li, Z.Y.; Yu, Y.; Zhang, J.P.; Li, Q.F.; Qu, L.H.; Shu, W.S.; Chen, Y.Q. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014, 15, 512. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yuan, D.; Tu, L.; Gao, W.; He, Y.; Hu, H.; Wang, P.; Liu, N.; Lindsey, K.; Zhang, X. Long noncoding RNAs and their proposed functions in fibre development of cotton (Gossypium spp.). New Phytol. 2015, 207, 1181–1197. [Google Scholar] [CrossRef] [PubMed]
- Khemka, N.; Singh, V.K.; Garg, R.; Jain, M. Genome-wide analysis of long intergenic non-coding RNAs in chickpea and their potential role in flower development. Sci. Rep. 2016, 6, 33297. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hao, L.; Li, D.; Zhu, L.; Hu, S. Long non-coding RNAs and their biological roles in plants. Genom. Proteom. Bioinform. 2015, 13, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Schlesinger, F.; Davis, C.A.; Zhang, Y.; Li, R.; Salit, M.; Gingeras, T.R.; Oliver, B. Synthetic spike-in standards for RNA-seq experiments. Genome Res. 2011, 21, 1543–1551. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Du, Y.; Liu, X.; Zhang, H.; Liu, Y.; Shi, J.; Xue, S.J.; Zhang, Z. Analyses of effects of α-cembratrien-diol on cell morphology and transcriptome of Valsa mali var. mali. Food Chem. 2017, 214, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Liu, B.; Lu, W.; Zhang, N.; Si, H.; Wang, D. Observation and identification of potato morphological changes during tuber dormancy release. Chin. Potato J. 2013, 27, 14–18. [Google Scholar]
- Teper-Bamnolker, P.; Buskila, Y.; Lopesco, Y.; Ben-Dor, S.; Saad, I.; Holdengreber, V.; Belausov, E.; Zemach, H.; Ori, N.; Lers, A.; et al. Release of apical dominance in potato tuber is accompanied by programmed cell death in the apical bud meristem. Plant Physiol. 2012, 158, 2053–2067. [Google Scholar] [CrossRef] [PubMed]
- Eshel, D.; Teper-Bamnolker, P. Can loss of apical dominance in potato tuber serve as a marker of physiological age? Plant Signal. Behav. 2012, 7, 1158–1162. [Google Scholar] [CrossRef] [PubMed]
- Kwenda, S.; Birch, P.R.; Moleleki, L.N. Genome-wide identification of potato long intergenic noncoding RNAs responsive to Pectobacterium carotovorum subspecies brasiliense infection. BMC Genomics 2016, 17, 614. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Liu, Z. Global identification and analysis of long non-coding RNAs in diploid strawberry Fragaria vesca during flower and fruit development. BMC Genomics 2015, 16, 815. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.C.; Chen, Y.Q. Long noncoding RNAs: New regulators in plant development. Biochem. Biophys. Res. Commun. 2013, 436, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Helliwell, C.A.; Robertson, M.; Finnegan, E.J.; Buzas, D.M.; Dennis, E.S. Vernalization-repression of Arabidopsis FLC requires promoter sequences but not antisense transcripts. PLoS ONE 2011, 6, e21513. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.B.; Sung, S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science 2011, 331, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yan, B.; Qu, Y.; Qin, F.; Yang, Y.; Hao, X.; Yu, J.; Zhao, Q.; Zhu, D.; Ao, G. Zm401, a short-open reading-frame mRNA or noncoding RNA, is essential for tapetum and microspore development and can regulate the floret formation in maize. J. Cell. Biochem. 2008, 105, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Cao, J.S.; Yu, X.L.; Xiang, X. BcMF11, a putative pollen-specific non-coding RNA from Brassica campestris ssp. chinensis. J. Plant Physiol. 2007, 164, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Cao, J.S.; Wang, C.G. BcMF11, a novel non-coding RNA gene from Brassica campestris, is required for pollen development and male fertility. Plant Cell Rep. 2013, 32, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Wang, Z.M.; Wang, M.; Wang, X.J. Widespread long noncoding RNAs as endogenous target mimics for microRNAs in plants. Plant Physiol. 2013, 161, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Potato Genome Sequencing Consortium. Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189–195. [Google Scholar]
- Guttman, M.; Garber, M.; Levin, J.Z.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.J.; Gnirke, A.; Nusbaum, C.; et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010, 28, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The PFAM protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef] [PubMed]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Marshall, D.; Bryan, G.J.; Hornyik, C. Identification and characterization of miRNA transcriptome in potato by high-throughput sequencing. PLoS ONE 2013, 8, e57233. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name a | Raw Reads | Clean Reads | Clean Bases (G) | Error Rate (%) | Q20 (%) b | Q30 (%) c | GC Content (%) |
|---|---|---|---|---|---|---|---|
| DT_1 | 88,474,758 | 81,270,030 | 12.19 | 0.03 | 95.28 | 88.53 | 44.91 |
| DT_2 | 101,834,712 | 94,506,478 | 14.18 | 0.02 | 97.04 | 92.28 | 46.02 |
| DT_3 | 98,802,096 | 93,941,306 | 14.09 | 0.02 | 96.85 | 91.91 | 44.12 |
| ST_1 | 93,598,076 | 87,083,084 | 13.06 | 0.02 | 97.04 | 92.49 | 45.34 |
| ST_2 | 95,140,804 | 88,600,856 | 13.29 | 0.02 | 97.19 | 92.80 | 45.39 |
| ST_3 | 109,587,878 | 101,669,792 | 15.25 | 0.02 | 95.98 | 90.32 | 45.13 |
| Sample Name | DT_1 | DT_2 | DT_3 | ST_1 | ST_2 | ST_3 |
|---|---|---|---|---|---|---|
| Total reads a | 81,270,030 | 94,506,478 | 93,941,306 | 87,083,084 | 88,600,856 | 101,669,792 |
| Total mapped b | 38,206,584 (47.01%) | 44,961,000 (47.57%) | 48,397,116 (51.52%) | 44,253,083 (50.82%) | 44,551,383 (50.28%) | 49,531,693 (48.72%) |
| Multiple mapped c | 1,803,952 (2.22%) | 2,019,548 (2.14%) | 2,434,047 (2.59%) | 2,227,833 (2.56%) | 2,004,069 (2.26%) | 1,967,805 (1.94%) |
| Uniquely mapped d | 36,402,632 (44.79%) | 42,941,452 (45.44%) | 45,963,069 (48.93%) | 42,025,250 (48.26%) | 42,547,314 (48.02%) | 47,563,888 (46.78%) |
| Read-1 e | 18,880,028 (23.23%) | 21,725,955 (22.99%) | 23,421,450 (24.93%) | 21,534,853 (24.73%) | 21,645,064 (24.43%) | 25,291,412 (24.88%) |
| Read-2 f | 17,522,604 (21.56%) | 21,215,497 (22.45%) | 22,541,619 (24%) | 20,490,397 (23.53%) | 20,902,250 (23.59%) | 22,272,476 (21.91%) |
| Reads map to ‘+’ g | 18,602,502 (22.89%) | 22,024,764 (23.31%) | 23,379,973 (24.89%) | 21,728,282 (24.95%) | 22,113,656 (24.96%) | 24,617,535 (24.21%) |
| Reads map to ‘−’ g | 17,800,130 (21.9%) | 20,916,688 (22.13%) | 22,583,096 (24.04%) | 20,296,968 (23.31%) | 20,433,658 (23.06%) | 22,946,353 (22.57%) |
| Non-splice reads h | 25,490,872 (31.37%) | 30,801,757 (32.59%) | 32,635,041 (34.74%) | 30,406,191 (34.92%) | 30,621,306 (34.56%) | 33,776,061 (33.22%) |
| Splice reads i | 10,911,760 (13.43%) | 12,139,695 (12.85%) | 13,328,028 (14.19%) | 11,619,059 (13.34%) | 11,926,008 (13.46%) | 13,787,827 (13.56%) |
| Reads mapped in proper pairs | 27,006,682 (33.23%) | 33,520,536 (35.47%) | 34,778,164 (37.02%) | 32,012,422 (36.76%) | 32,201,808 (36.34%) | 34,863,014 (34.29%) |
| GO ID | GO Terms | Type a | Corrected p-Value | Test b | Ref. c |
|---|---|---|---|---|---|
| GO:0005737 | Cytoplasm | P | 3.43 × 10−62 | 2551 | 3137 |
| GO:0044444 | Cytoplasmic part | P | 5.97 × 10−58 | 2299 | 2819 |
| GO:0005622 | Intracellular | P | 1.79 × 10−50 | 4071 | 5388 |
| GO:0005623 | Cell | P | 8.00 × 10−50 | 4400 | 5864 |
| GO:0044464 | Cell part | P | 8.00 × 10−50 | 4400 | 5864 |
| GO:0044424 | Intracellular part | P | 1.45 × 10−48 | 3954 | 5236 |
| GO:0005575 | Cellular component | P | 4.21 × 10−41 | 5755 | 7878 |
| GO:0043226 | Organelle | P | 2.10 × 10−39 | 3355 | 4460 |
| GO:0043229 | Intracellular organelle | P | 2.10 × 10−39 | 3353 | 4457 |
| GO:0044422 | Organelle part | P | 4.96 × 10−29 | 1362 | 1685 |
| GO:0044446 | Intracellular organelle Part | P | 4.96 × 10−29 | 1362 | 1685 |
| GO:0043227 | Membrane-bounded organelle | P | 7.31 × 10−26 | 2904 | 3905 |
| GO:0043231 | Intracellular membrane-bounded organelle | P | 8.76 × 10−26 | 2902 | 3903 |
| GO:0009987 | Cellular process | F | 2.49 × 10−25 | 6970 | 9831 |
| GO:0044237 | Cellular metabolic process | F | 2.37 × 10−23 | 5612 | 7863 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, X.; Du, Y.; Liu, X.; Zhang, H.; Liu, Y.; Yan, N.; Zhang, Z. Genome-Wide Analysis of Long Non-Coding RNAs in Potato and Their Potential Role in Tuber Sprouting Process. Int. J. Mol. Sci. 2018, 19, 101. https://doi.org/10.3390/ijms19010101
Hou X, Du Y, Liu X, Zhang H, Liu Y, Yan N, Zhang Z. Genome-Wide Analysis of Long Non-Coding RNAs in Potato and Their Potential Role in Tuber Sprouting Process. International Journal of Molecular Sciences. 2018; 19(1):101. https://doi.org/10.3390/ijms19010101
Chicago/Turabian StyleHou, Xiaodong, Yongmei Du, Xinmin Liu, Hongbo Zhang, Yanhua Liu, Ning Yan, and Zhongfeng Zhang. 2018. "Genome-Wide Analysis of Long Non-Coding RNAs in Potato and Their Potential Role in Tuber Sprouting Process" International Journal of Molecular Sciences 19, no. 1: 101. https://doi.org/10.3390/ijms19010101
APA StyleHou, X., Du, Y., Liu, X., Zhang, H., Liu, Y., Yan, N., & Zhang, Z. (2018). Genome-Wide Analysis of Long Non-Coding RNAs in Potato and Their Potential Role in Tuber Sprouting Process. International Journal of Molecular Sciences, 19(1), 101. https://doi.org/10.3390/ijms19010101