Identification of Two Novel Fibrinogen Bβ Chain Mutations in Two Slovak Families with Quantitative Fibrinogen Disorders

 ,
, 
Abstract
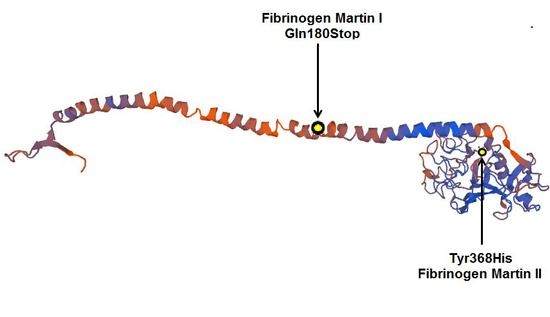
:
1. Introduction
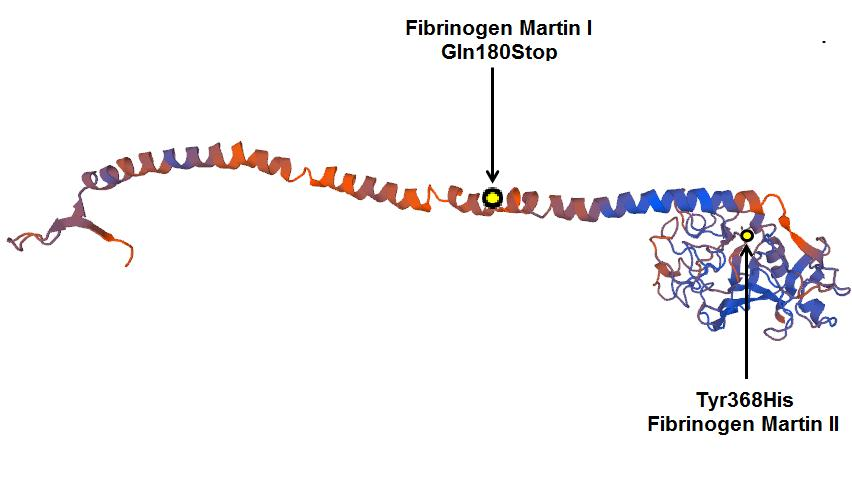
2. Results
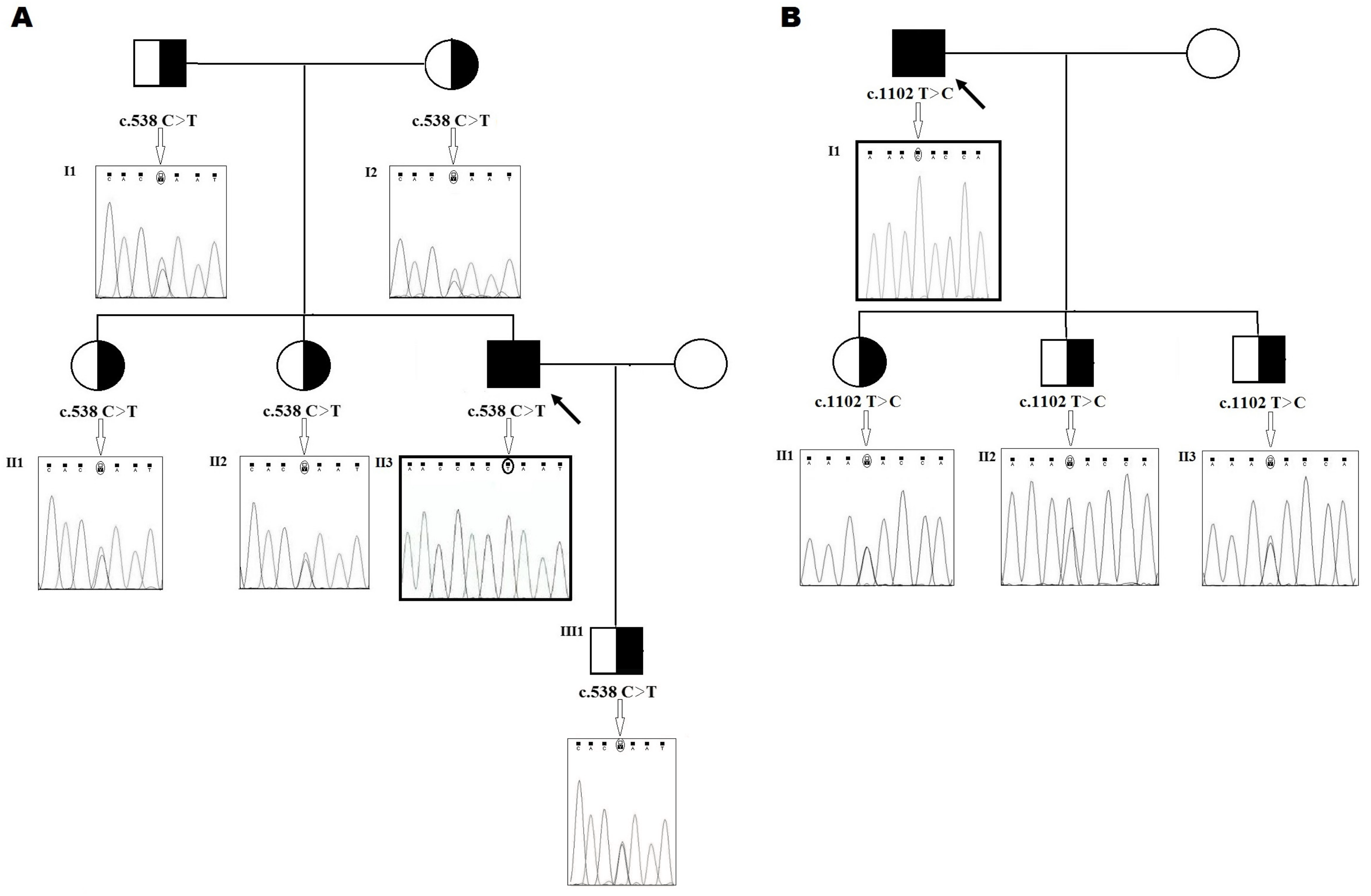
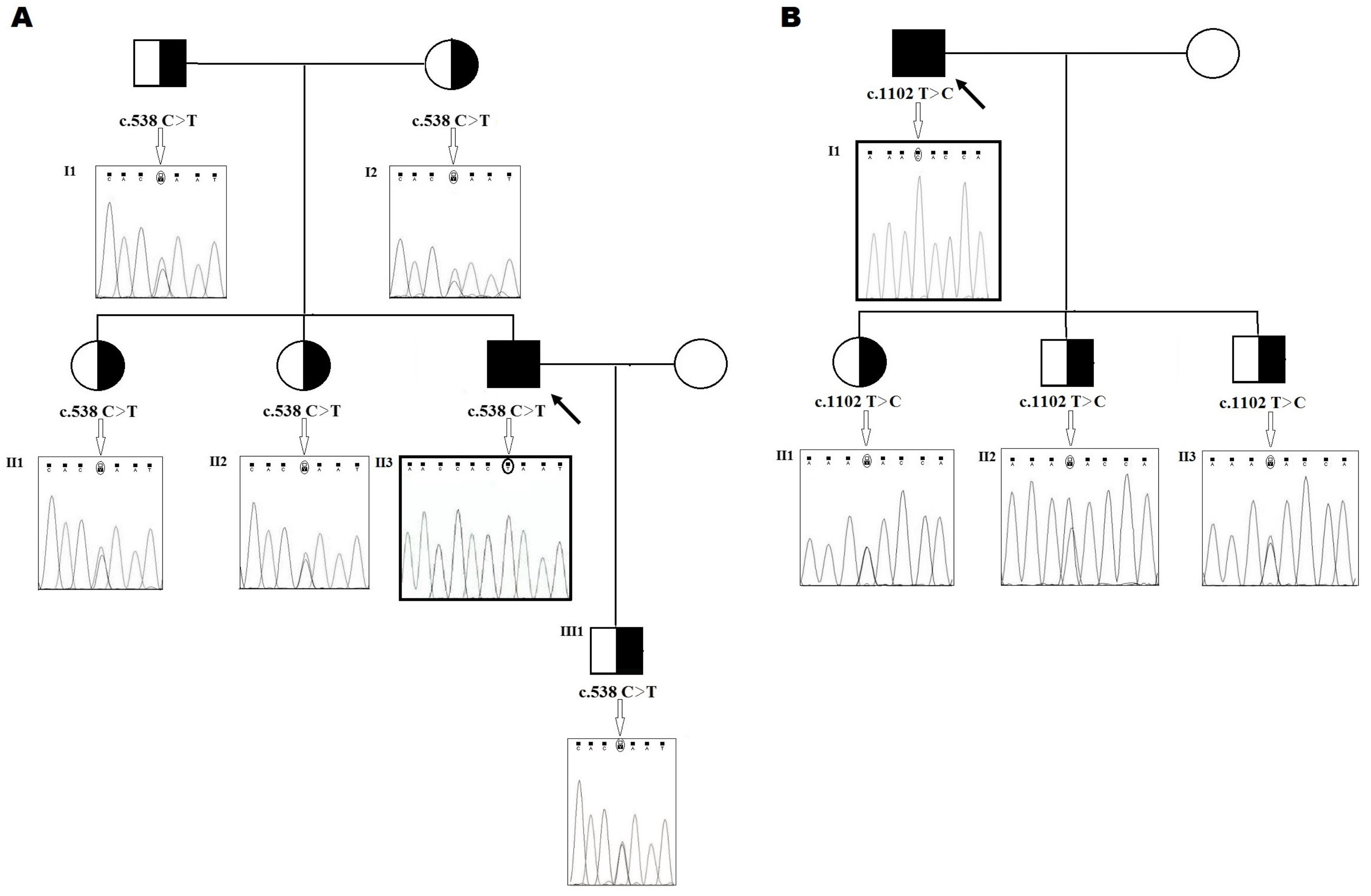
2.1. Family 1
2.2. Family 2
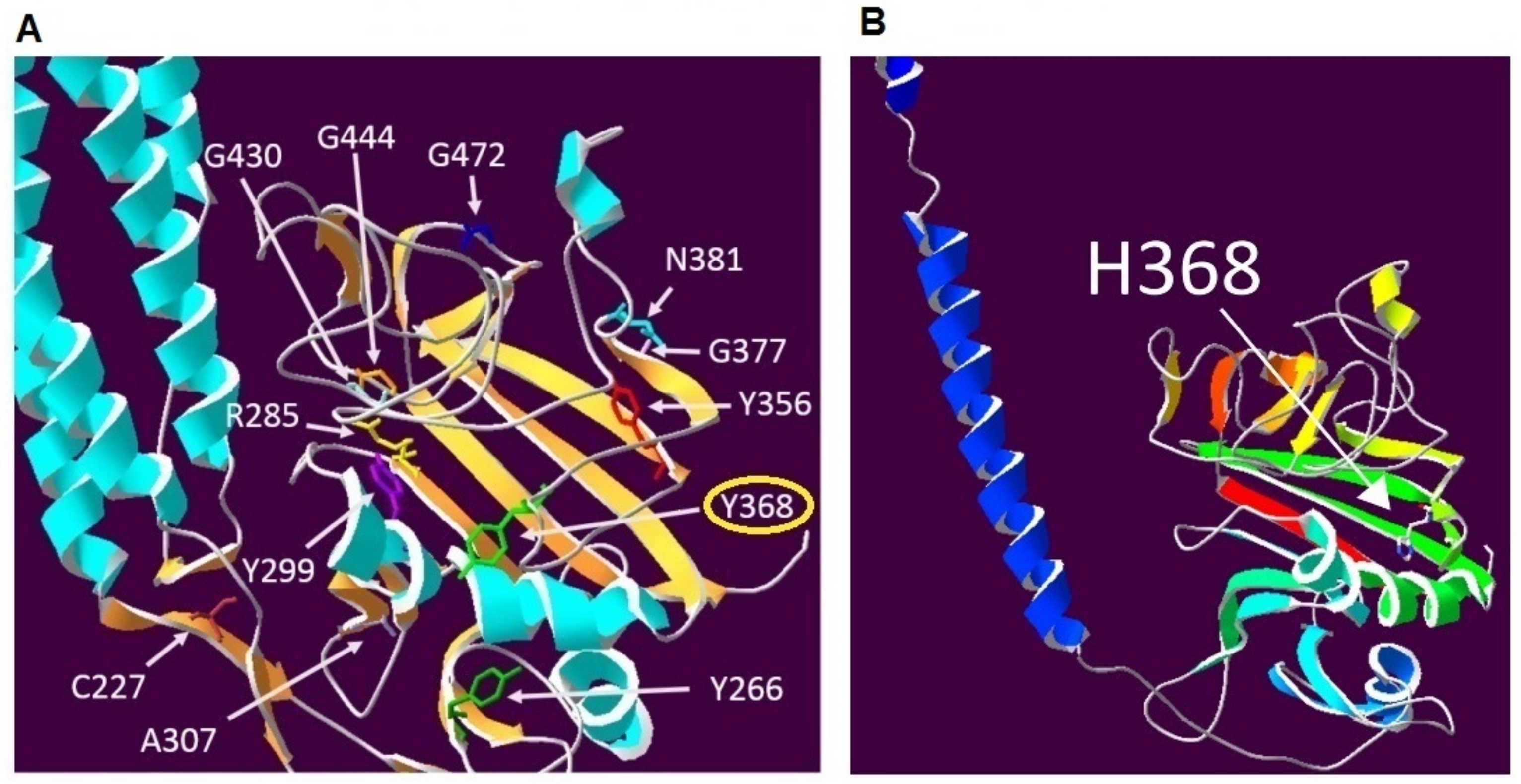
3. Discussion
4. Materials and Methods
4.1. Routine Coagulation Assays
4.2. Fibrinolytic System Assays
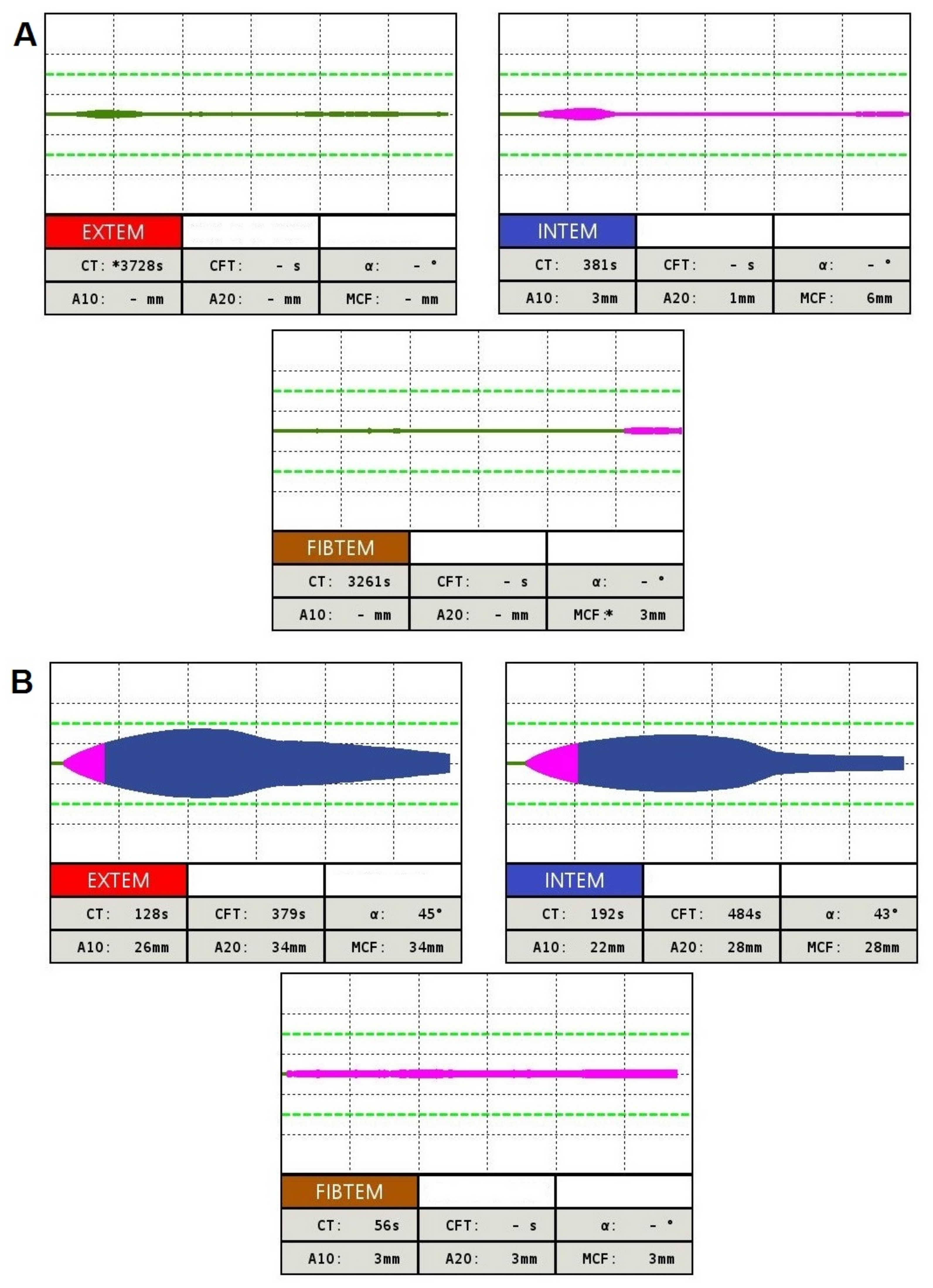
4.3. Global Hemostasis Testing
4.4. DNA Preparation and Genetic Analysis
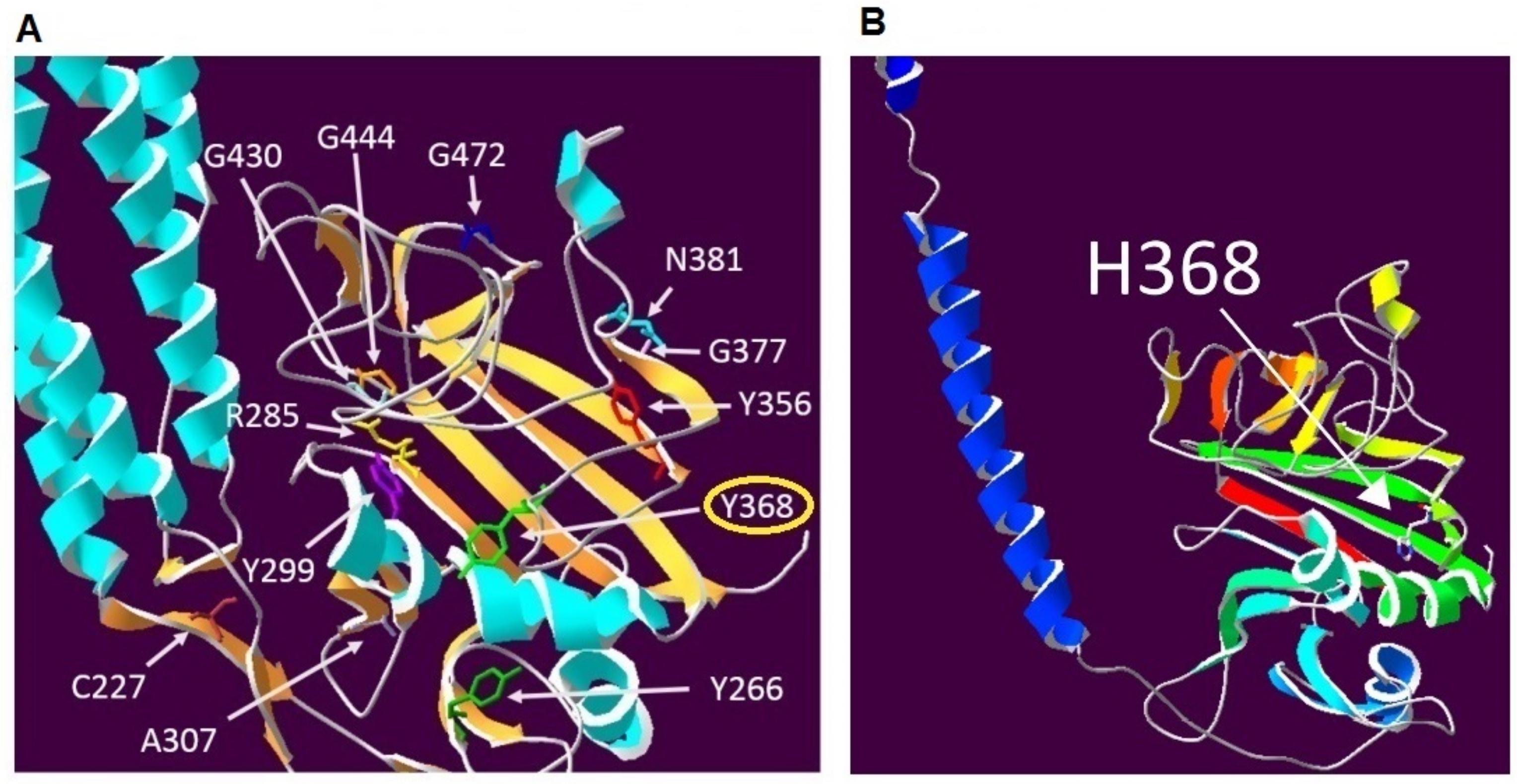
4.5. Protein Modelling
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mosesson, M.W.; Siebenlist, K.R.; Meh, D.A. The structure and biological features of fibrinogen and fibrin. Ann. N. Y. Acad. Sci. 2001, 936, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Asselta, R.; Platè, M.; Robusto, M.; Borhany, M.; Guella, I.; Soldà, G.; Afransiabi, A.; Menegatti, M.; Shamsi, T.; Peyvandi, F.; et al. Clinical and molecular characterisation of 21 patients affected by quantitative fibrinogen deficiency. Thromb. Haemost. 2015, 113, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Medved, L.; Weisel, J.W. Fibrinogen and Factor XIII Subcommittee of Scientific Standardization Committee of International Society on Thrombosis and Haemostasis. Recommendations for nomenclature on fibrinogen and fibrin. J. Thromb. Haemost. 2009, 7, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; de Moerloose, P.; Congenital Fibrinogen Disorders Group. Management of congenital quantitative fibrinogen disorders: A Delphi consensus. Haemophilia 2016, 22, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Mumford, A.D.; Ackroyd, S.; Alikhan, R.; Bowles, L.; Chowdary, P.; Grainger, J.; Mainwaring, J.; Marthias, M.; O’Connell, N. Guideline for the diagnosis and management of rare coagulation disorders: A United Kingdom Haemophilia Centre Doctors’ Organization guideline on behalf of the British Committee for Standards in Haematology. Br. J. Haematol. 2014, 167, 304–326. [Google Scholar] [CrossRef] [PubMed]
- De Moerloose, P.; Casini, A.; Neerman-Arbez, M. Congenital fibrinogen disorders: An update. Semin. Thromb. Hemost. 2013, 39, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Castaman, G.; Linari, S. Diagnosis and Treatment of von Willebrand Disease and Rare Bleeding Disorders. J. Clin. Med. 2017, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Palla, R.; Peyvandi, F.; Shapiro, A.D. Rare bleeding disorders: Diagnosis and treatment. Blood 2015, 125, 2052–2061. [Google Scholar] [CrossRef] [PubMed]
- Report on the WFH Annual Global Survey 2015. Available online: http://www1.wfh.org/publication/files/pdf-1669.pdf (accessed on 30 October 2017).
- Casini, A.; de Moerloose, P.; Neerman-Arbez, M. Clinical Features and Management of Congenital Fibrinogen Deficiencies. Semin. Thromb. Hemost. 2016, 42, 366–374. [Google Scholar] [PubMed]
- Castaman, G.; Rimoldi, V.; Giacomelli, S.H.; Duga, S. Congenital hypofibrinogenemia associated with novel homozygous fibrinogen Aα and heterozygous Bβ chain mutations. Thromb. Res. 2015, 136, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Simurda, T.; Snahnicanova, Z.; Loderer, D.; Sokol, J.; Stasko, J.; Lasabova, Z.; Kubisz, P. Fibrinogen Martin: A Novel Mutation in FGB (Gln180Stop) Causing Congenital Afibrinogenemia. Semin. Thromb. Hemost. 2016, 42, 455–458. [Google Scholar] [PubMed]
- Stikarová, J.; Blatný, J.; Kotlín, R.; Suttnar, J.; Zapletal, O.; Pimková, K.; Májek, P.; Hrachovinová, I.; Dyr, J.E. Novel homozygous fibrinogen Aα chain truncation causes severe afibrinogenemia with life threatening complications in a two-year-old boy. Thromb. Res. 2013, 132, 490–492. [Google Scholar] [CrossRef] [PubMed]
- Simurda, T.; Kubisz, P.; Dobrotova, M.; Necas, L.; Stasko, J. Perioperative coagulation management in a patient with congenital afibrinogenemia during revision total hip arthroplasty. Semin. Thromb. Hemost. 2016, 42, 689–692. [Google Scholar] [PubMed]
- Simurda, T.; Stanciakova, L.; Stasko, J.; Dobrotova, M.; Kubisz, P. Yes or no for secondary prophylaxis in afibrinogenemia? Blood Coagul. Fibrinolysis 2015, 26, 978–980. [Google Scholar] [CrossRef] [PubMed]
- Neerman-Arbez, M.; de Moerloose, P.; Casini, A. Laboratory and Genetic Investigation of Mutations Accounting for Congenital Fibrinogen Disorders. Semin. Thromb. Hemost. 2016, 42, 356–365. [Google Scholar] [PubMed]
- Platé, M.; Asselta, R.; Peyvandi, F.; Tenchini, M.L.; Duga, S. Molecular characterization of the first missense mutation in the fibrinogen Alpha-chain gene identified in a compound heterozygous afibrinogenemic patient. Biochim. Biophys. Acta 2007, 1772, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Naz, A.; Biswas, A.; Khan, T.N.; Goodeve, A.; Ahmed, N.; Saqlain, N.; Ahmed, S.; Ujjan, I.D.; Shamsi, T.S.; Oldenburg, J. Identification of novel mutations in congenital afibrinogenemia patients and molecular modelling of missense mutations in Pakistani population. Thromb. J. 2017, 15, 24. [Google Scholar] [CrossRef] [PubMed]
- Asselta, R.; Duga, S.; Tenchini, M.L. The molecular basis of quantitative fibrinogen disorders. J. Thromb. Haemost. 2006, 4, 2115–2129. [Google Scholar] [CrossRef] [PubMed]
- Platè, M.; Asselta, R.; Spena, S.; Spreafico, M.; Fagoonee, S.; Peyvandi, F.; Tenchini, M.L.; Duga, S. Congenital hypofibrinogenemia: Characterization of two missense mutations affecting fibrinogen assembly and secretion. Blood Cells Mol. Dis. 2008, 41, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Vilar, R.; Beauverd, Y.; Aslan, D.; Devreese, K.; Mondelaers, V.; Alberio, L.; Gubert, C.; de Moerloose, P.; Neerman-Arbez, M. Protein modelling to understand FGB mutations leading to congenital hypofibrinogenaemia. Haemophilia 2017, 23, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Lukowski, S.; Quintard, V.L.; Crutu, A.; Zak, M.; Regazzoni, S.; de Moerloose, P.; Neerman-Arbez, M. FGB mutations leading to congenital quantitative fibrinogen deficiencies: An update and report of four novel mutations. Thromb. Res. 2014, 133, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Fort, A.; Borel, C.; Migliavacca, E.; Antonarakis, S.E.; Fish, R.J.; Neerman-Arbez, M. Regulation of fibrinogen production by microRNAs. Blood 2010, 116, 2608–2615. [Google Scholar] [CrossRef] [PubMed]
- Neerman-Arbez, M.; Germanos-Haddad, M.; Tzanidakis, K.; Vu, D.; Deutsch, S.; David, A.; Morris, M.A.; De Moerloose, P. Expression and analysis of a split premature termination codon in FGG responsible for congenital afibrinogenemia: Escape from RNA surveillance mechanisms in transfected cells. Blood 2004, 104, 3618–3623. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.E.; Parker, R. Nonsense-mediated mRNA decay: Terminating erroneous gene expression. Curr. Opin. Cell Biol. 2004, 16, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Asselta, R.; Spena, S.; Duga, S.; Peyvandi, F.; Malcovati, M.; Mannucci, P.M.; Tenchini, M.L. Analysis of Iranian patients allowed the identification of the first truncating mutation in the fibrinogen Bβ-chain gene causing afibrinogenemia. Haematologica 2002, 87, 855–859. [Google Scholar] [PubMed]
- Monaldini, L.; Asselta, R.; Duga, S.; Peyvandi, F.; Karimi, M.; Malcovati, M.; Tenchini, M.L. Mutational screening of six afibrinogenemic patients: Identification and characterization of four novel molecular defects. Thromb. Haemost. 2007, 97, 546–551. [Google Scholar] [PubMed]
- Marchi, R.; Brennan, S.; Meyer, M.; Rojas, H.; Kanzler, D.; De Agrela, M.; Ruiz-Saez, A. A novel mutation in the FGB: c.1105C>T turns the codon for amino acid Bβ Q339 into a stop codon causing hypofibrinogenemia. Blood. Cells Mol. Dis. 2013, 50, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Castaman, G.; Giacomelli, S.H.; Duga, S.; Rodeghiero, F. Congenital hypofibrinogenemia associated with novel heterozygous fibrinogen Bβ and gamma chain mutations. Haemophilia 2008, 14, 630–633. [Google Scholar] [CrossRef] [PubMed]
- Rottenstreich, A.; Lask, A.; Schliamser, L.; Zivelin, A.; Seligsohn, U.; Kalish, Y. Thromboembolic events in patients with severe inherited fibrinogen deficiency. J. Thromb. Thrombolysis 2016, 42, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Korte, Q.; Poon, M.C.; Iorio, M.; Makris, M. Thrombosis in Inherited Fibrinogen Disorders. Transfus. Med. Hemother. 2017, 44, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Neerman-Arbez, M.; Ariëns, R.A.; de Moerloose, P. Dysfibrinogenemia: From molecular anomalies to clinical manifestations and management. J. Thromb. Haemost. 2015, 13, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Horellou, M.H.; Chevreaud, C.; Mathieux, V.; Conard, J.; de Mazancourt, P. Fibrinogen Paris IX: A case of symptomatic hypofibrinogenemia with Bβ Y236C and Bbeta IVS7-1G→C mutations. J. Thromb. Haemost. 2006, 4, 1134–1136. [Google Scholar] [CrossRef] [PubMed]
- Grandone, E.; Tiscia, G.; Cappucci, F.; Favuzzi, G.; Santacroce, R.; Pisanelli, D.; Soli, F.; Legnani, C.; Rizzo, M.A.; Palareti, G.; Margaglione, M. Clinical histories and molecular characterization of two afibrinogenemic patients: Insights into clinical management. Haemophilia 2012, 18, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Mukaddam, A.; Kulkarni, B.; Jadli, A.; Ghosh, K.; Shetty, S. Spectrum of mutations in Indian patients with fibrinogen disorders and its application in genetic diagnosis of the affected families. Haemophilia 2015, 21, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Miosge, L.A.; Field, M.A.; Sontani, Y.; Cho, V.; Johnson, S.; Palkova, A.; Balakishnan, B.; Liang, R.; Zhang, Y.; Lyon, S.; et al. Comparison of predicted and actual consequences of missense mutations. Proc. Natl. Acad. Sci. USA 2015, 112, E5189–E5198. [Google Scholar] [CrossRef] [PubMed]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Lebreton, A.; Casini, A. Diagnosis of congenital fibrinogen disorders. Ann. Biol. Clin. 2016, 74, 405–412. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types of Congenital Fibrinogen Disorders | Bleeding Features | Thrombotic Features | Asymptomatic |
|---|---|---|---|
| afibrinogenemia | 1 | - | - |
| hypofibrinogenemia | 2 | 5 | 6 |
| hypodysfibrinogenemia | - | - | 1 |
| dysfibrinogenemia | 10 | 1 | 28 |
| Family Members | Clotting Time (s) | Fibrinogen (g/L) | |||||
|---|---|---|---|---|---|---|---|
| PT | APTT | TT | RT | Clauss | LIA | ||
| A. Family 1 | |||||||
| Proband (28) | II–3 | >300 | >300 | >300 | >150 | UD | 0.0 |
| Son (1) | III–1 | 11.7 | 23.6 | 19.5 | 20.1 | 1.1 | 1.2 |
| Elder sister (34) | II–2 | 11.7 | 35.6 | 17.6 | 19.9 | 1.6 | 1.4 |
| The eldest sister (38) | II–1 | 11.5 | 31.1 | 18.3 | 18.6 | 1.7 | 1.5 |
| Father (64) | I–1 | 11.8 | 28.0 | 16.0 | 18.1 | 1.3 | 1.3 |
| Mother (62) | I–2 | 11.1 | 23.8 | 13.9 | 19.7 | 1.5 | 1.5 |
| Normal range | - | 10.4–12.6 | 22–32 | 15–22 | 16–22 | 1.8–4.2 | 1.8–4.2 |
| B. Family 2 | |||||||
| Proband (62) | I–1 | 14.2 | >300 | 16.4 | 26.4 | 0.5 | 0.5 |
| Daughter (23) | II–1 | 14.2 | 26.3 | 10.6 | 15.8 | 1.2 | 1.1 |
| Elder son (34) | II–2 | 12.7 | 23.8 | 12.4 | 17.4 | 1.0 | 1.3 |
| The eldest son (37) | II–3 | 12.5 | 24.8 | 12.9 | 17.9 | 1.4 | 1.7 |
| Normal range | - | 10.4–12.6 | 22–32 | 15–22 | 16–22 | 1.8–4.2 | 1.8–4.2 |
| Exon | cDNA | Status | Type | Bleeding | Thrombosis | Reference |
|---|---|---|---|---|---|---|
| 2 | 3282C>T (17)Arg>stop | homozyg | afib | yes | no | [26] |
| 3 | c.352C>T (88)Glu>stop | compound heterozyg | afib | yes | no | [22] |
| 6 | c.887G>A (266)Trp>stop | homozyg | afib | yes | - | [27] |
| 7 | c.1105C>T (339)Gln>stop | heterozyg | hypofib | yes | no | [28] |
| 8 | nt.7893C>T (393)Gln>stop | heterozyg | hypofib | yes | no | [29] |
| Exon | cDNA | Status | Type | Bleeding | Thrombosis | Reference |
|---|---|---|---|---|---|---|
| 5 | 5909A>G (236)Tyr>Cys | compound heterozyg | hypofib | yes | yes | [33] |
| 6 | c.285T>C (269)Tyr>His | homozyg | afib | yes | yes | [22] |
| 6 | c.919G>T (277)Ala>Ser | homozyg | afib | no | yes | [34] |
| 8 | c.G1391A (434)Gly>Asp | homozyg | afib | yes | yes | [35] |
| 8 | c.1415G>T (442)Gly>Val | homozyg | hypofib | no | yes | [22] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simurda, T.; Zolkova, J.; Snahnicanova, Z.; Loderer, D.; Skornova, I.; Sokol, J.; Hudecek, J.; Stasko, J.; Lasabova, Z.; Kubisz, P. Identification of Two Novel Fibrinogen Bβ Chain Mutations in Two Slovak Families with Quantitative Fibrinogen Disorders. Int. J. Mol. Sci. 2018, 19, 100. https://doi.org/10.3390/ijms19010100
Simurda T, Zolkova J, Snahnicanova Z, Loderer D, Skornova I, Sokol J, Hudecek J, Stasko J, Lasabova Z, Kubisz P. Identification of Two Novel Fibrinogen Bβ Chain Mutations in Two Slovak Families with Quantitative Fibrinogen Disorders. International Journal of Molecular Sciences. 2018; 19(1):100. https://doi.org/10.3390/ijms19010100
Chicago/Turabian StyleSimurda, Tomas, Jana Zolkova, Zuzana Snahnicanova, Dusan Loderer, Ingrid Skornova, Juraj Sokol, Jan Hudecek, Jan Stasko, Zora Lasabova, and Peter Kubisz. 2018. "Identification of Two Novel Fibrinogen Bβ Chain Mutations in Two Slovak Families with Quantitative Fibrinogen Disorders" International Journal of Molecular Sciences 19, no. 1: 100. https://doi.org/10.3390/ijms19010100
APA StyleSimurda, T., Zolkova, J., Snahnicanova, Z., Loderer, D., Skornova, I., Sokol, J., Hudecek, J., Stasko, J., Lasabova, Z., & Kubisz, P. (2018). Identification of Two Novel Fibrinogen Bβ Chain Mutations in Two Slovak Families with Quantitative Fibrinogen Disorders. International Journal of Molecular Sciences, 19(1), 100. https://doi.org/10.3390/ijms19010100