
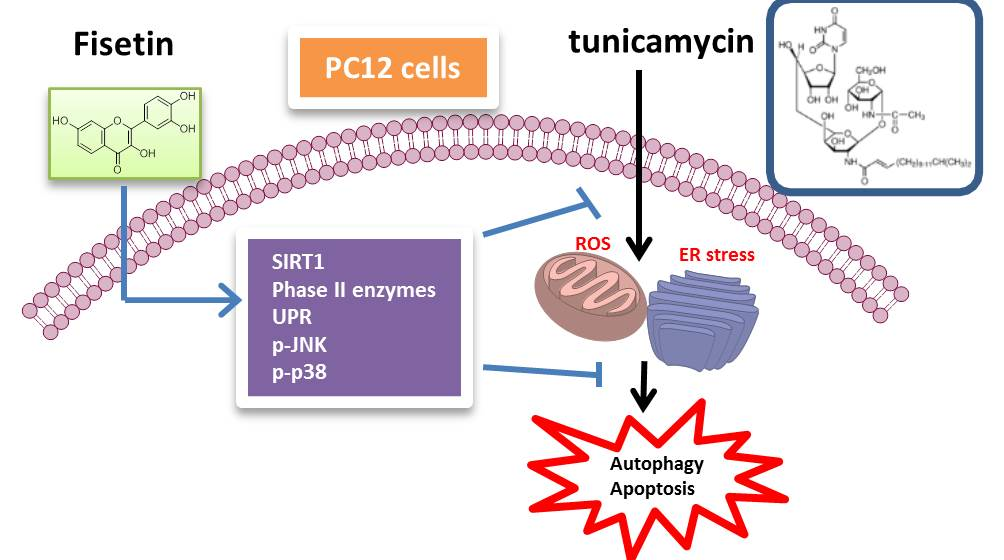
Fisetin Protects PC12 Cells from Tunicamycin-Mediated Cell Death via Reactive Oxygen Species Scavenging and Modulation of Nrf2-Driven Gene Expression, SIRT1 and MAPK Signaling in PC12 Cells


Abstract
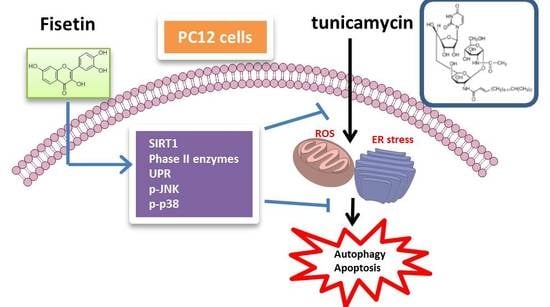
:
1. Introduction
2. Results
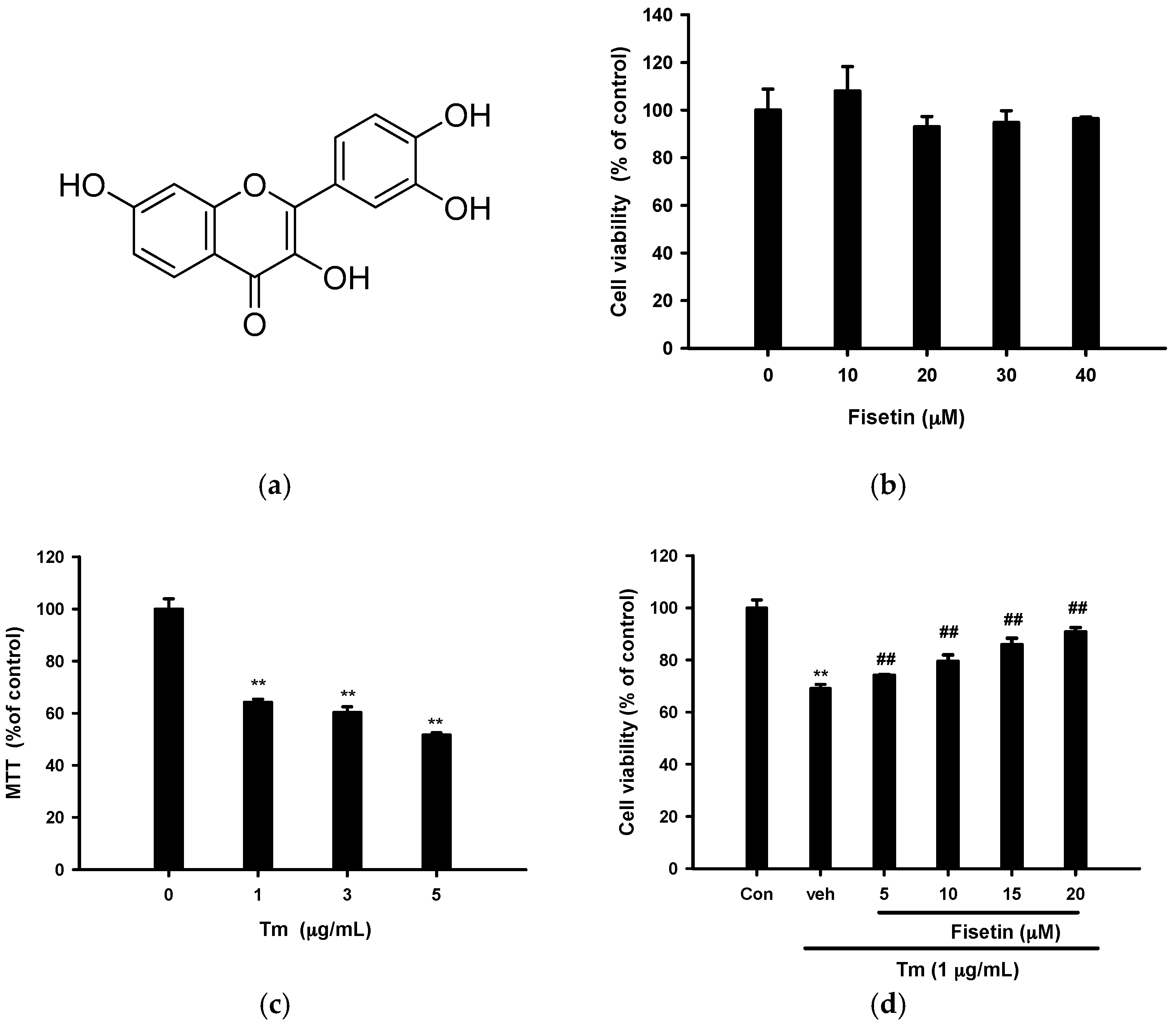
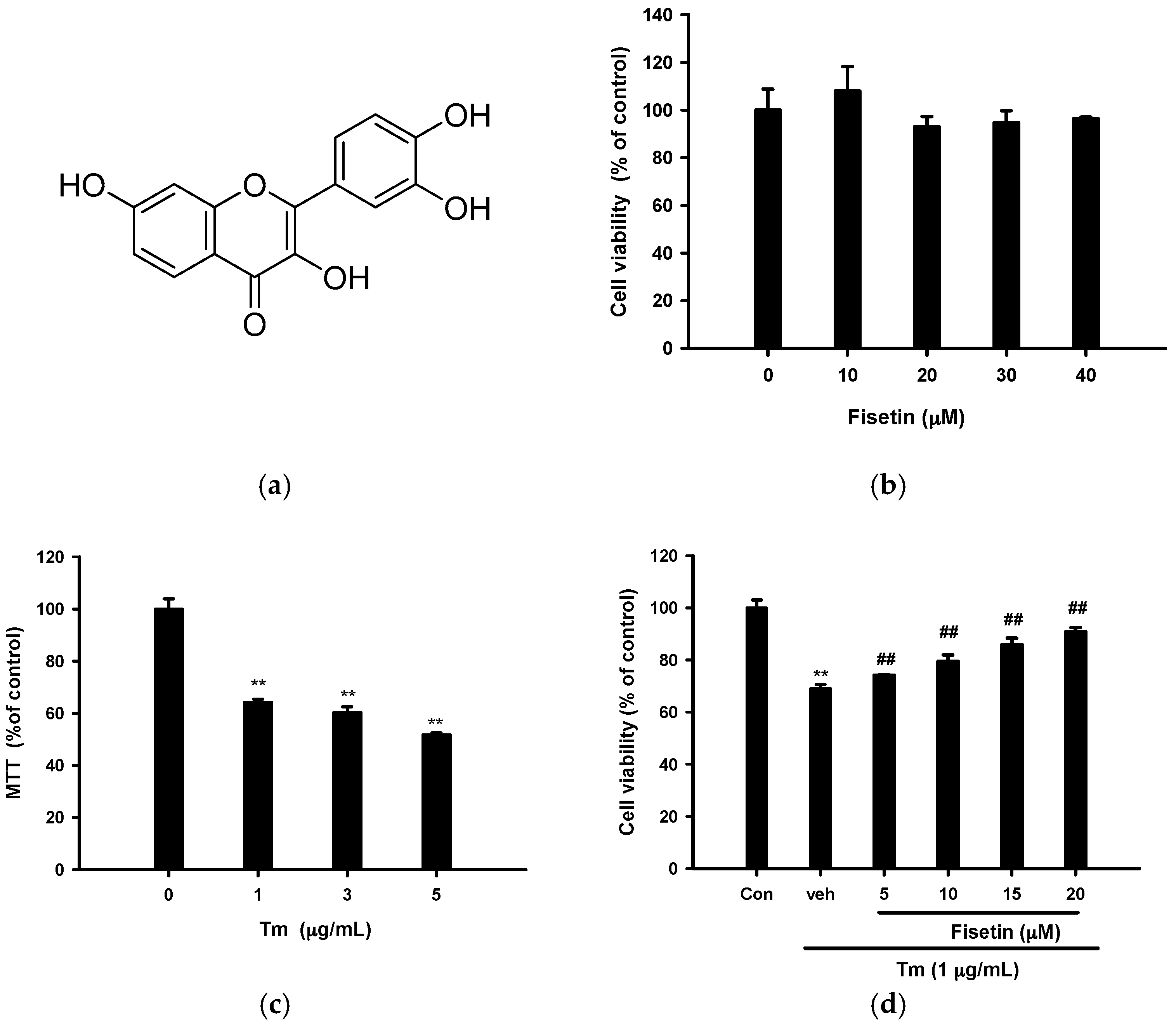
2.1. Fisetin Protects PC12 Cells from Tm-Mediated Cell Death
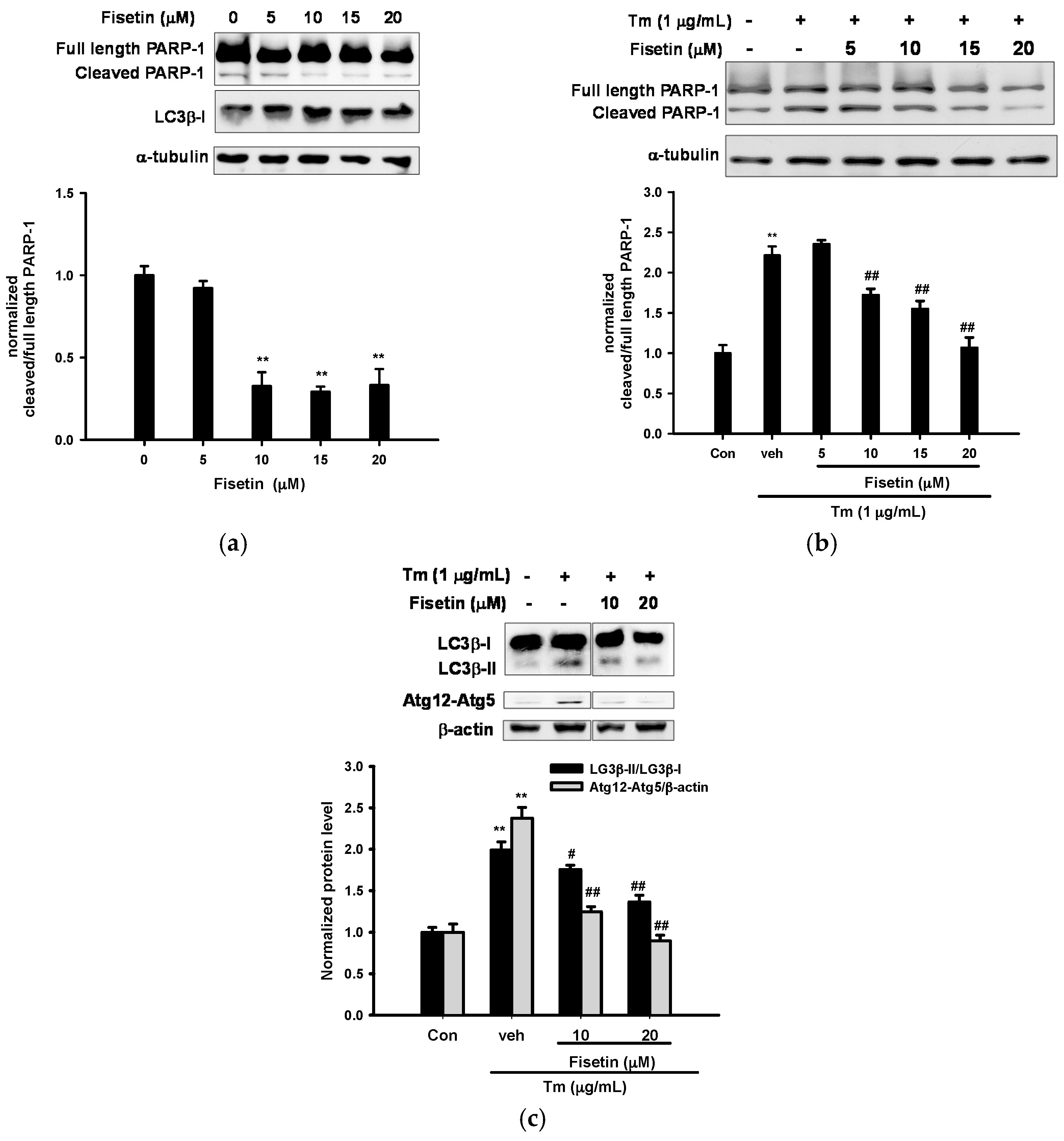
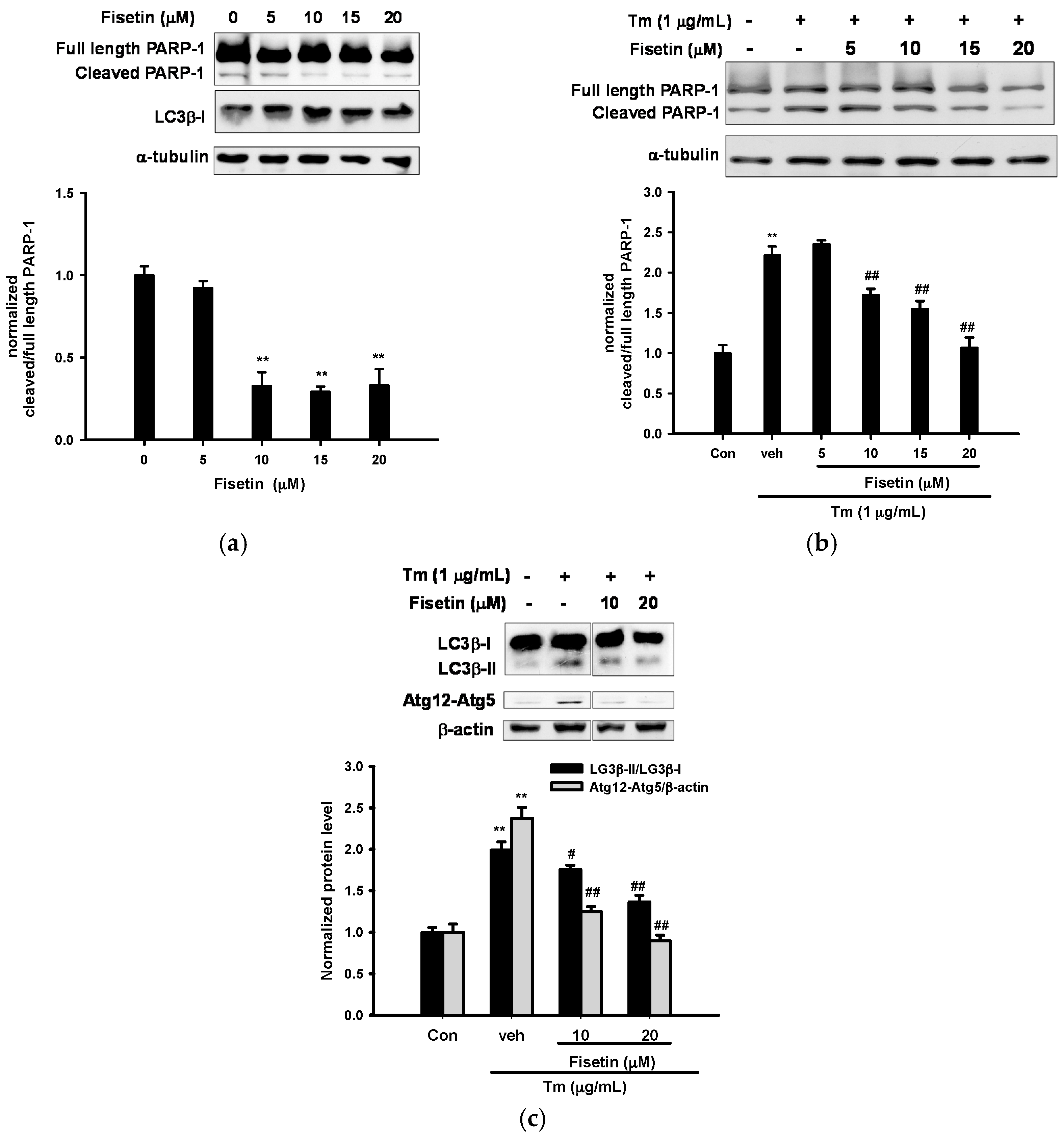
2.2. Fisetin Inhibits Tm-Mediated Apoptotic and Autophagic Marker Protein Expression
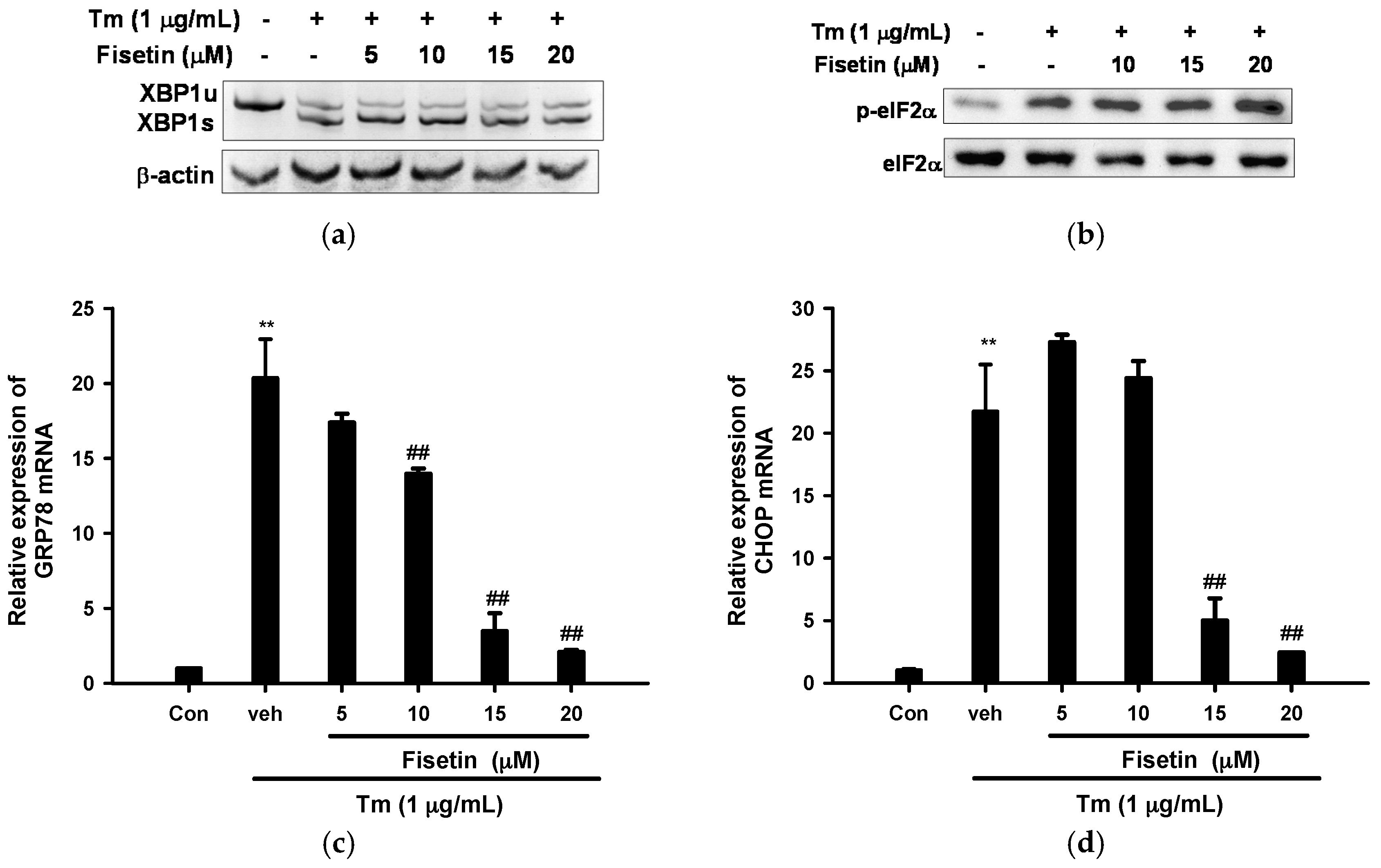
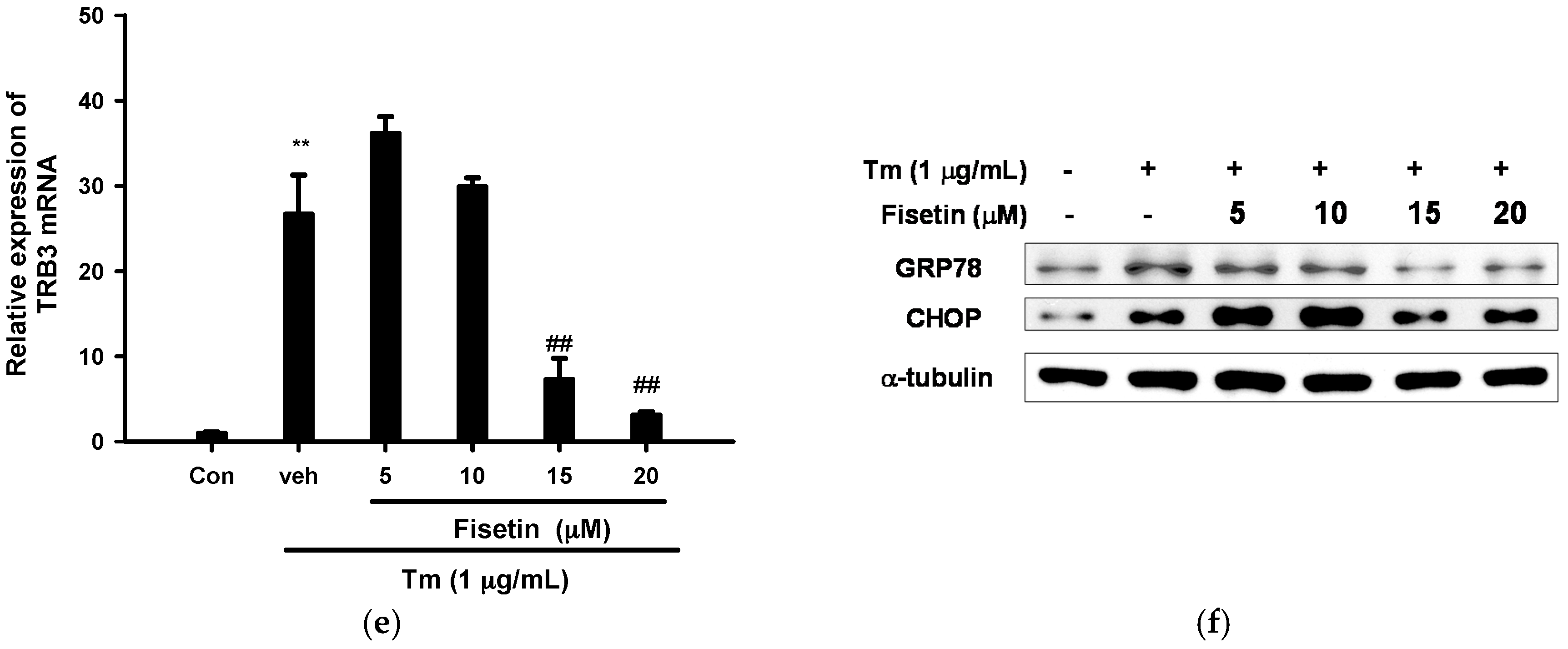
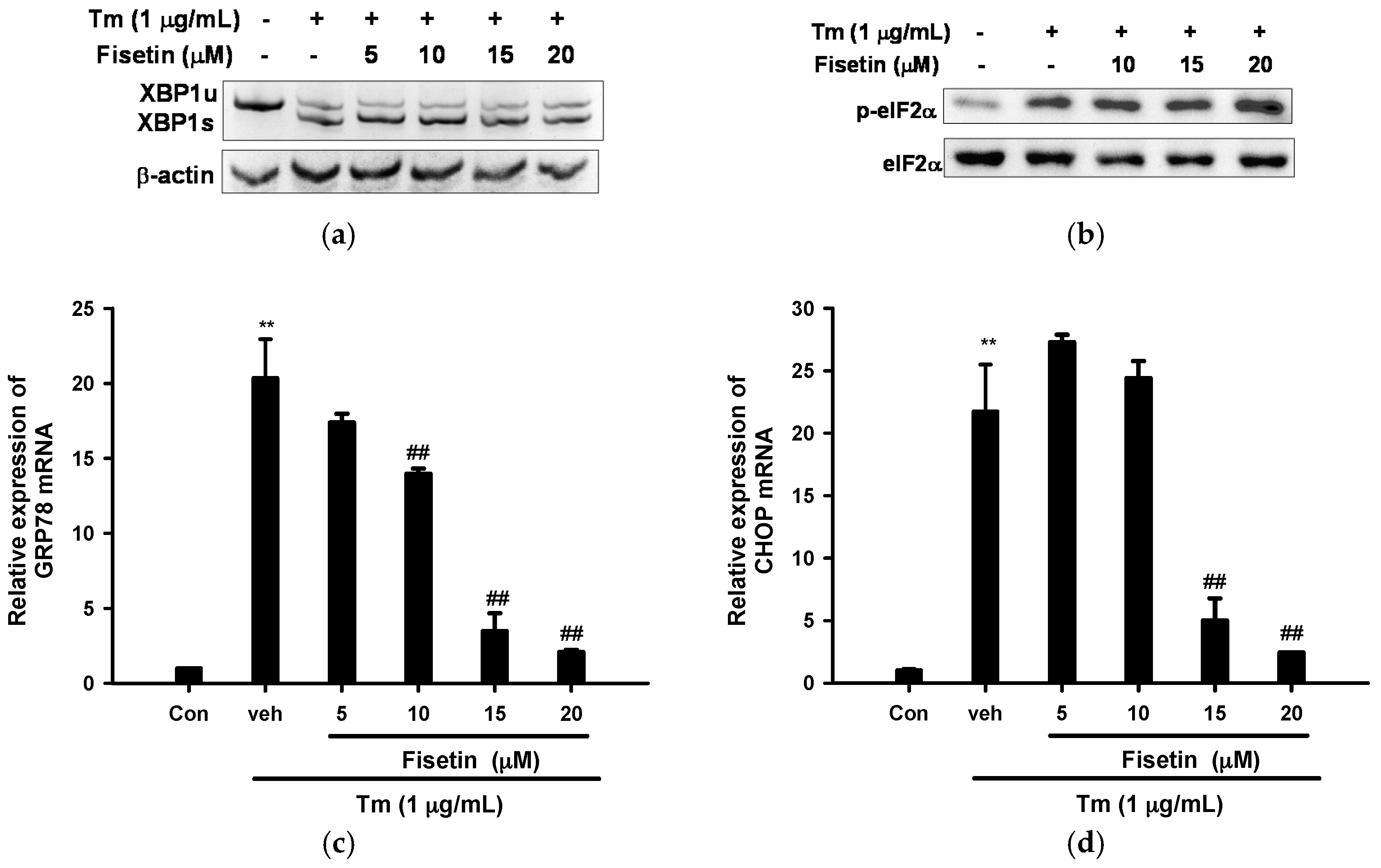
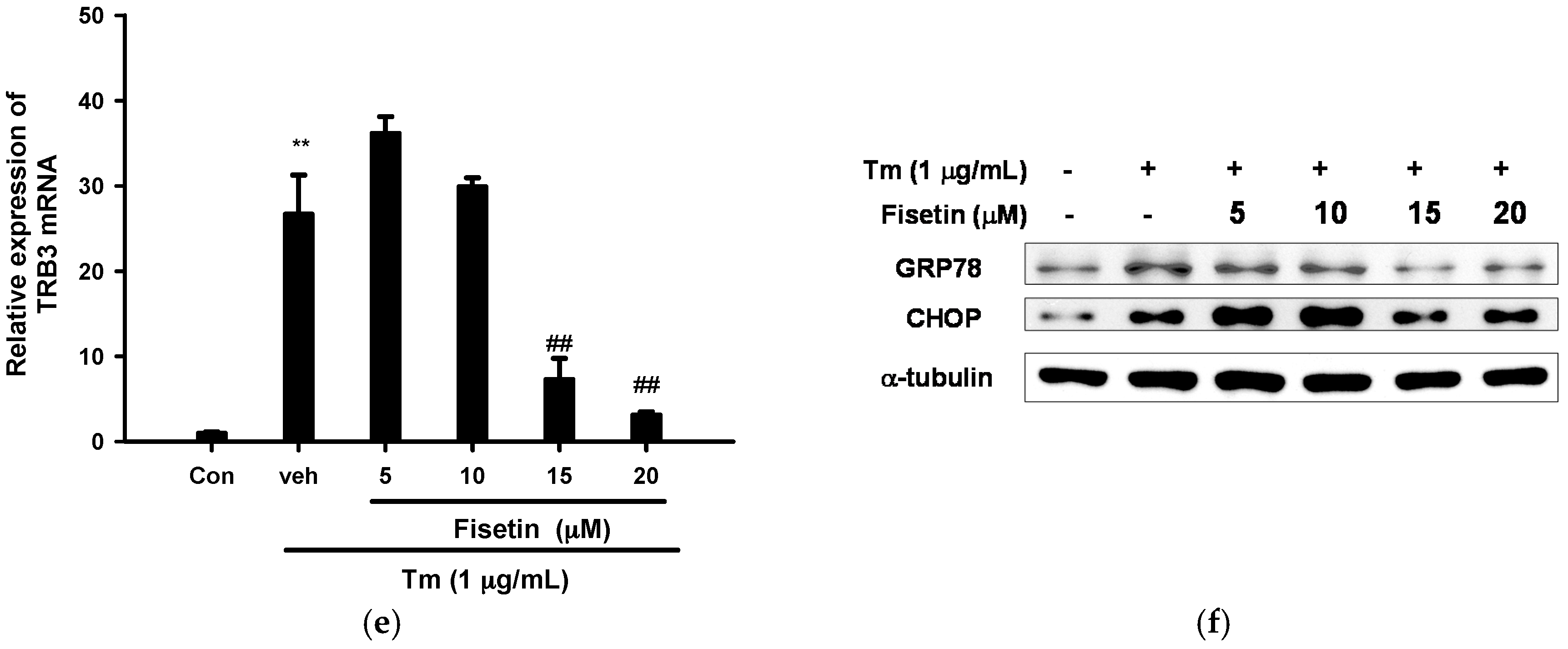
2.3. Fisetin Inhibits Tm-Mediated Endoplasmic Reticulum (ER) Stress Gene Expression
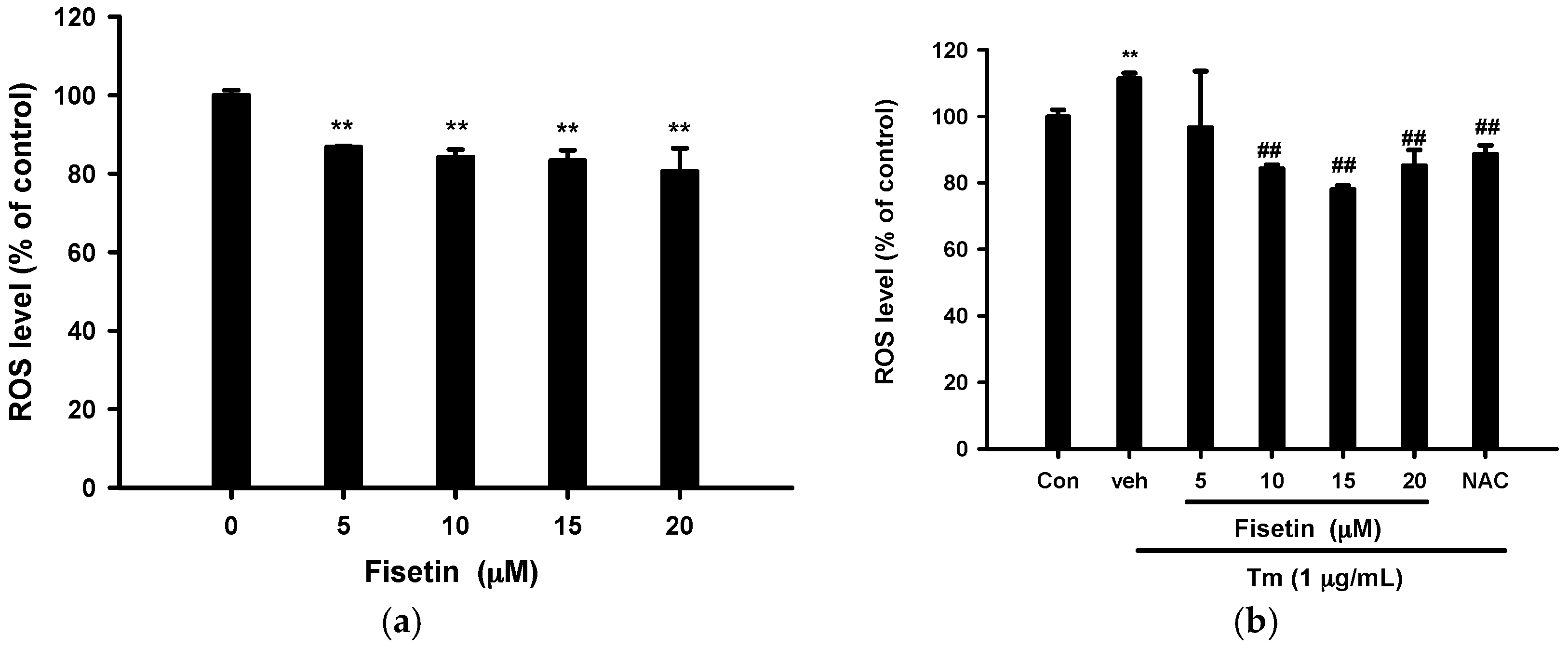
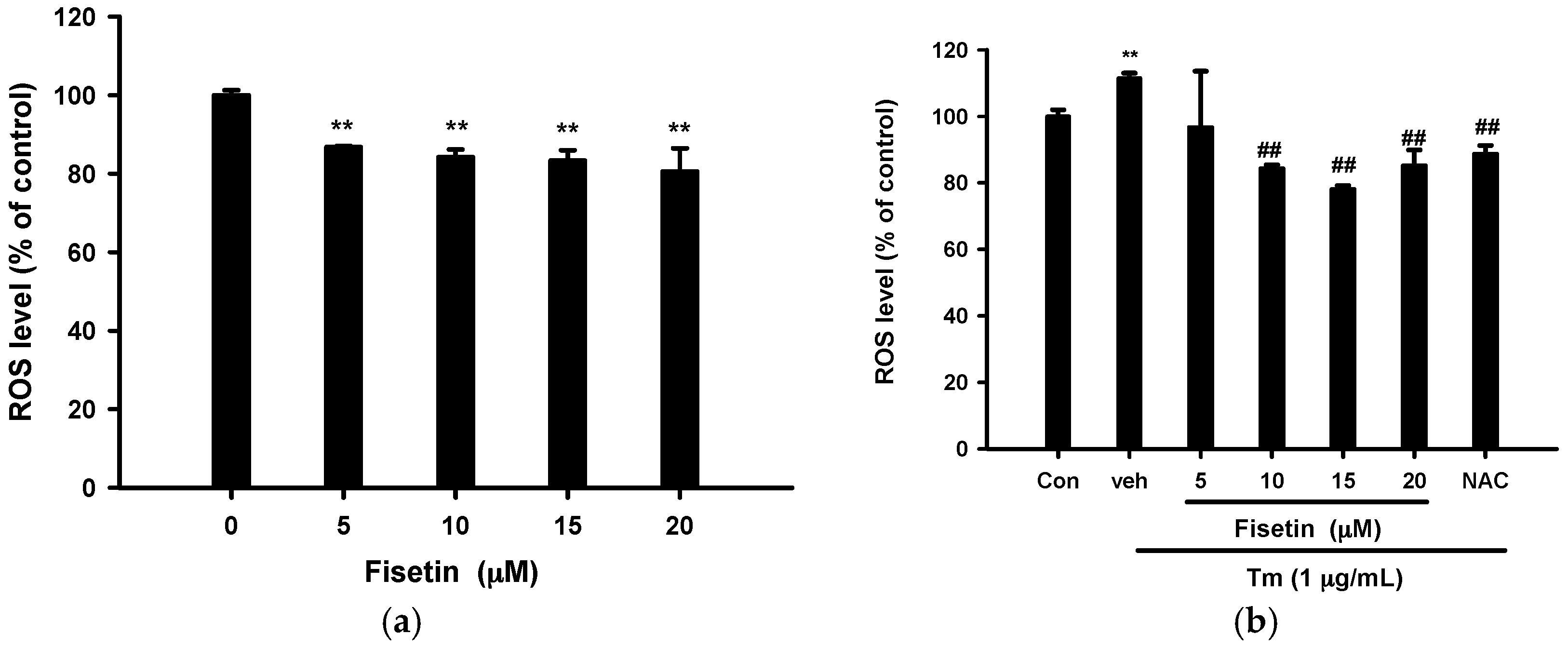
2.4. Fisetin Scavenges Reactive Oxygen Species (ROS) Production
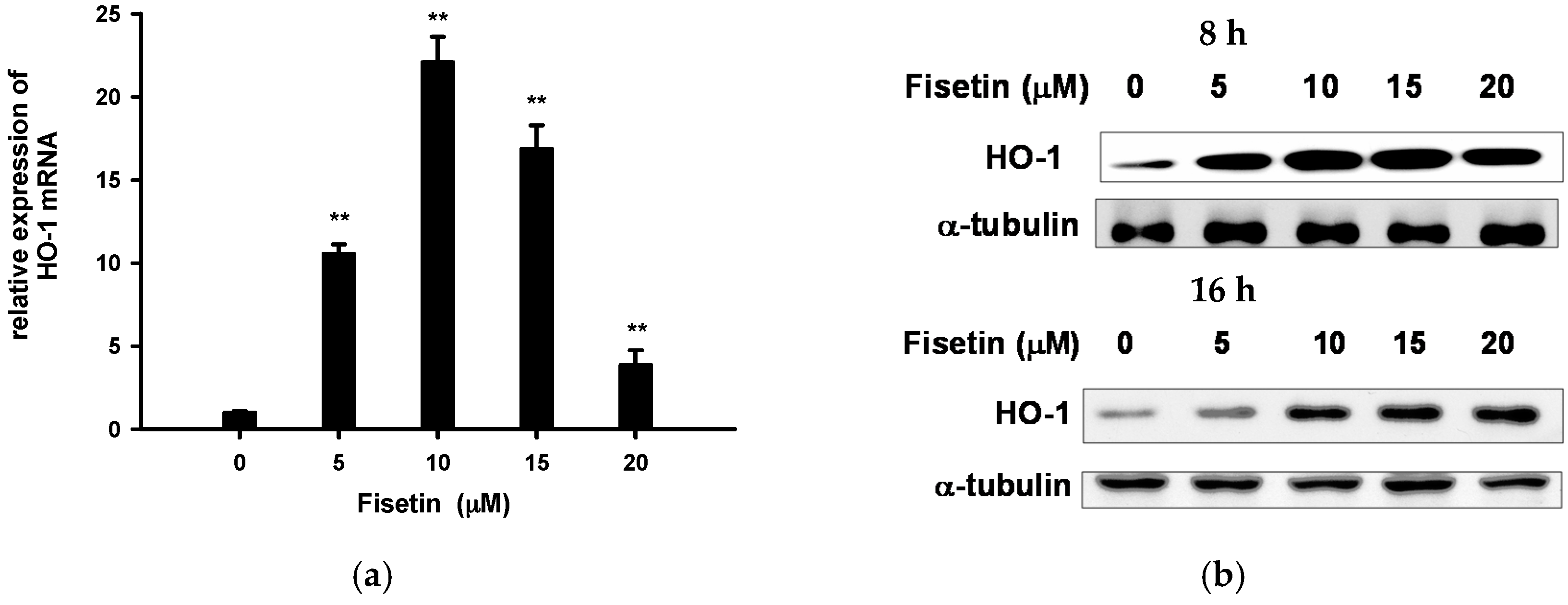
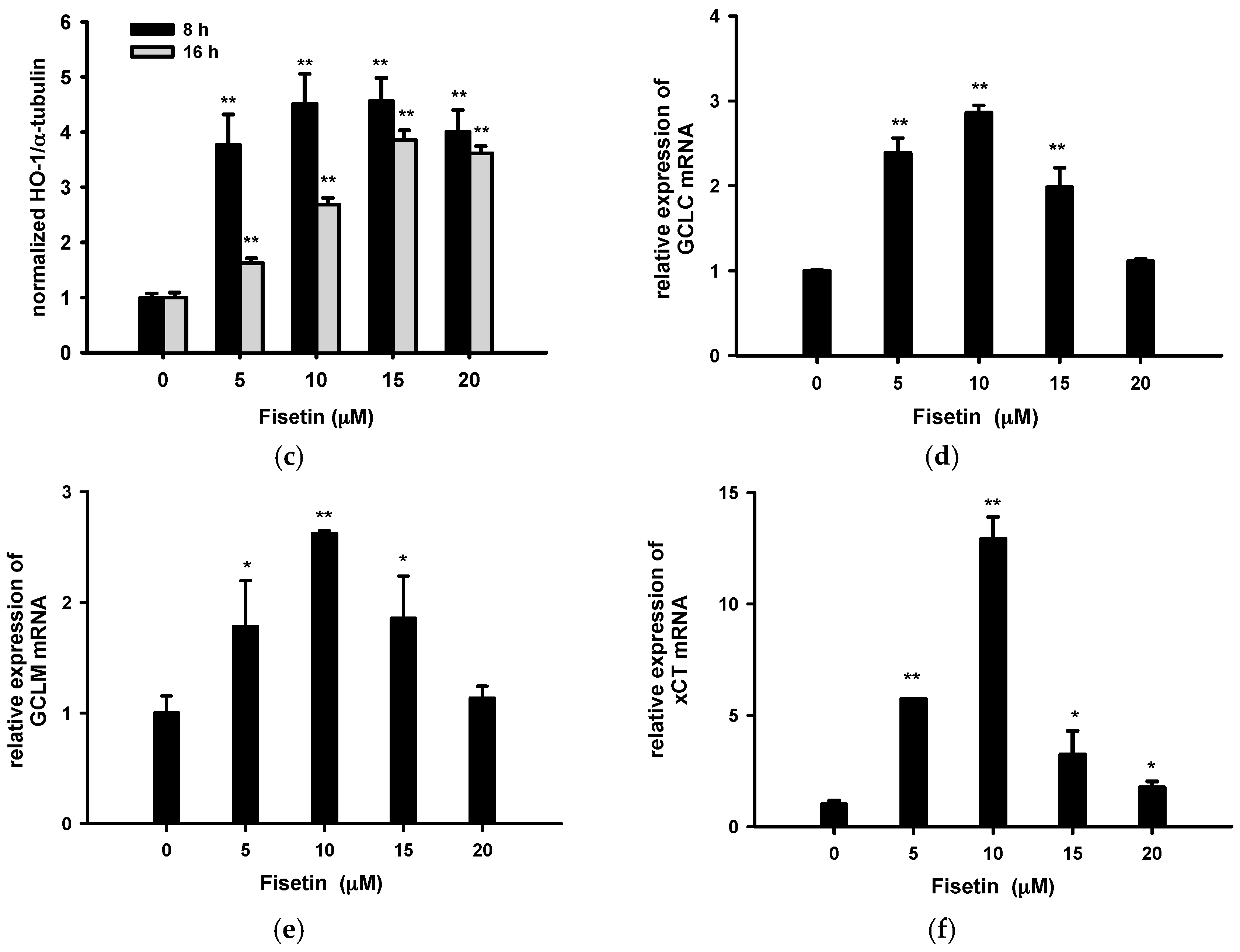
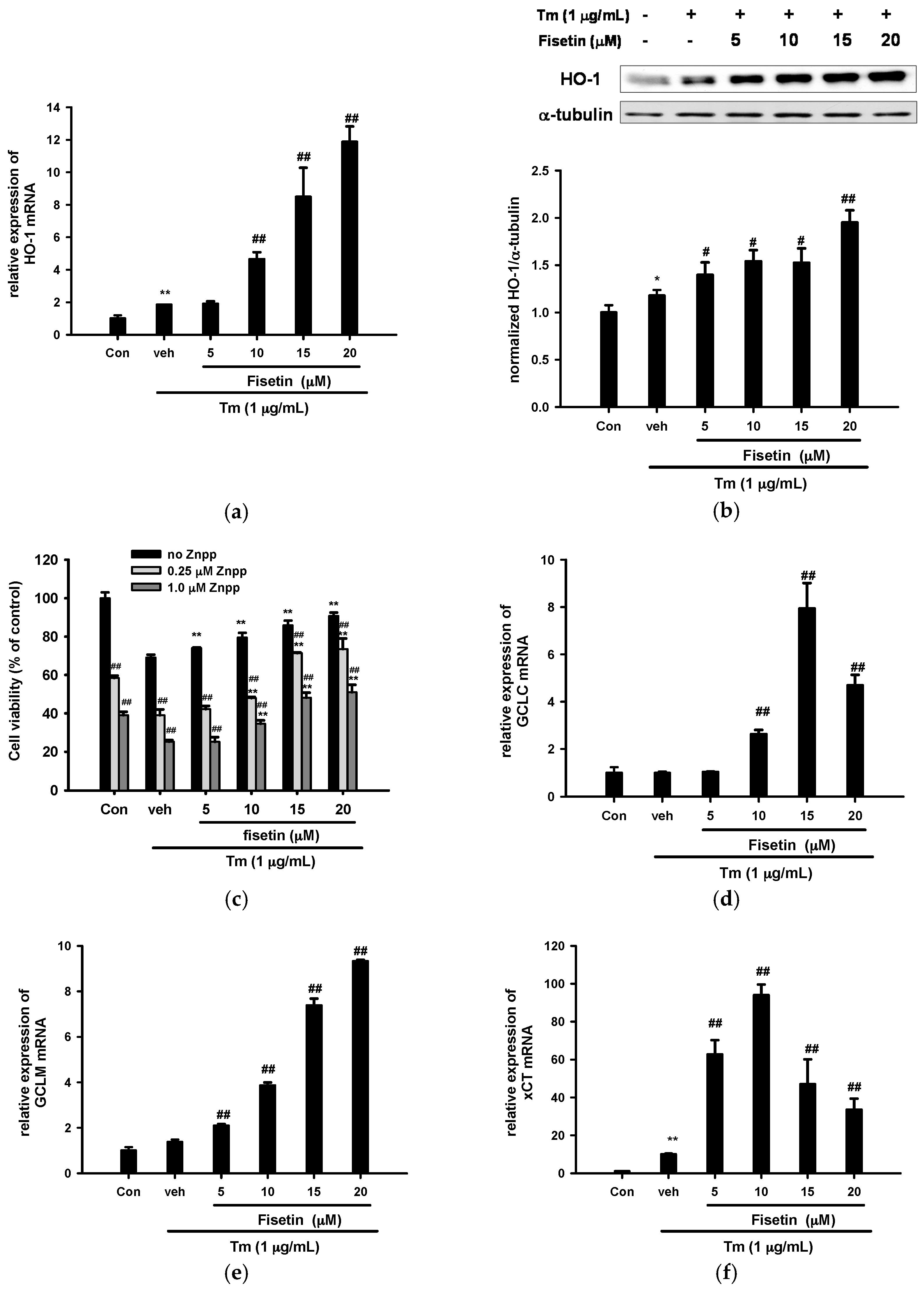
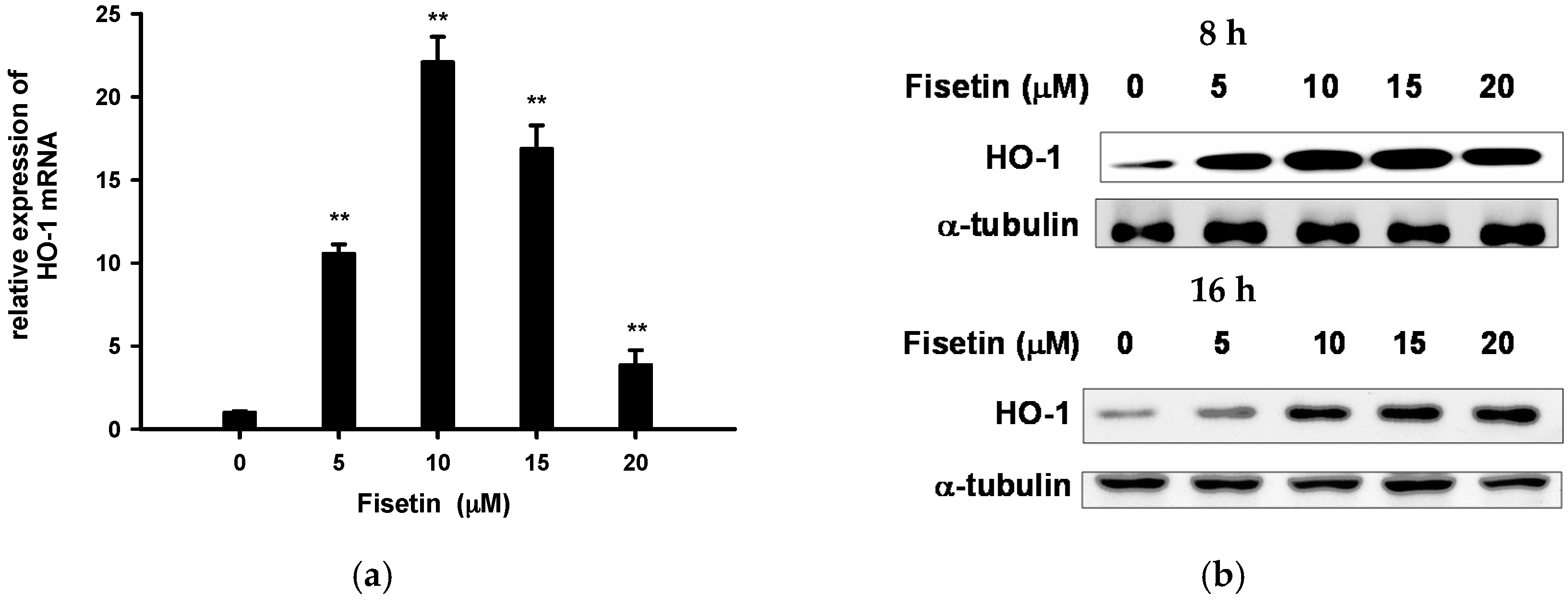
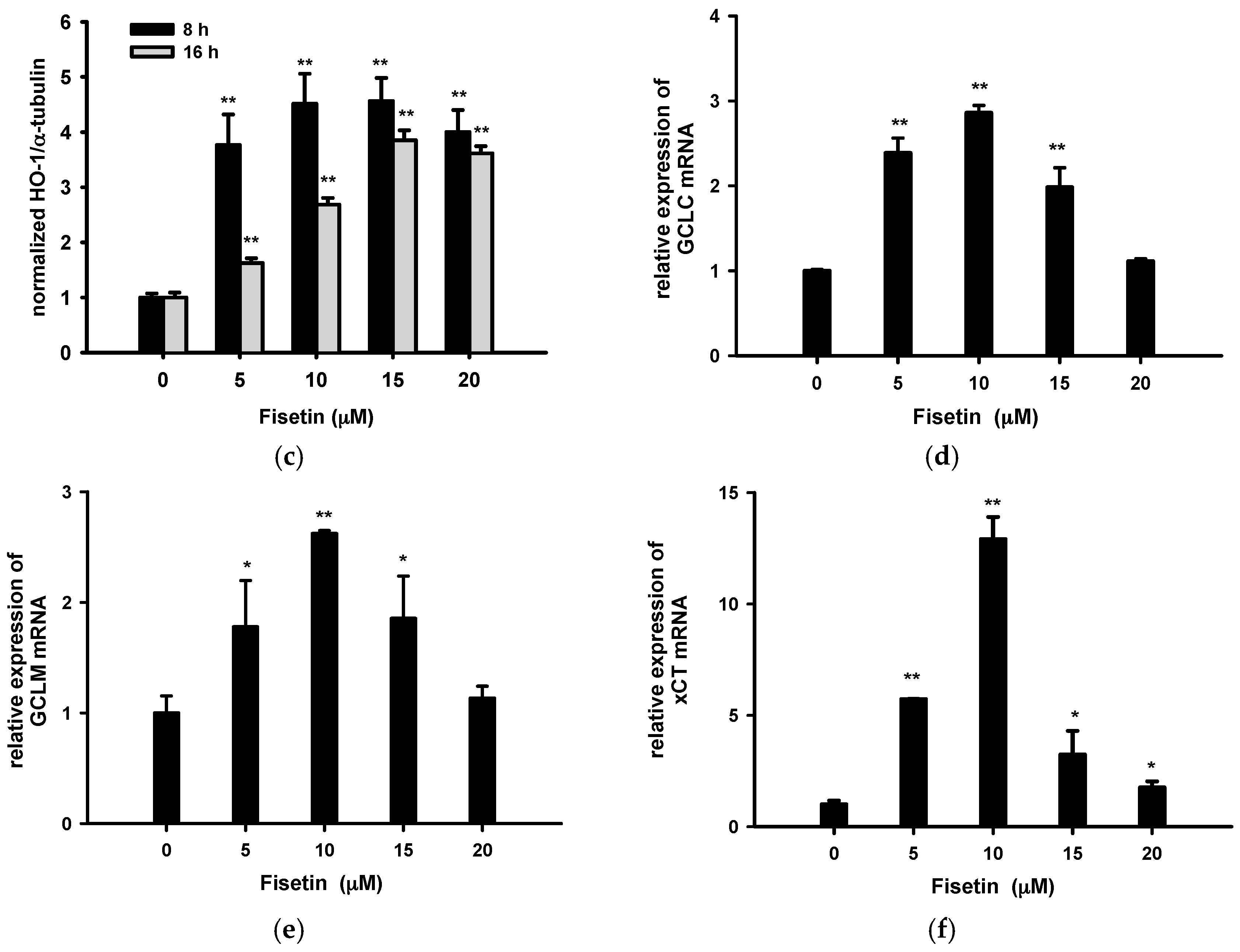
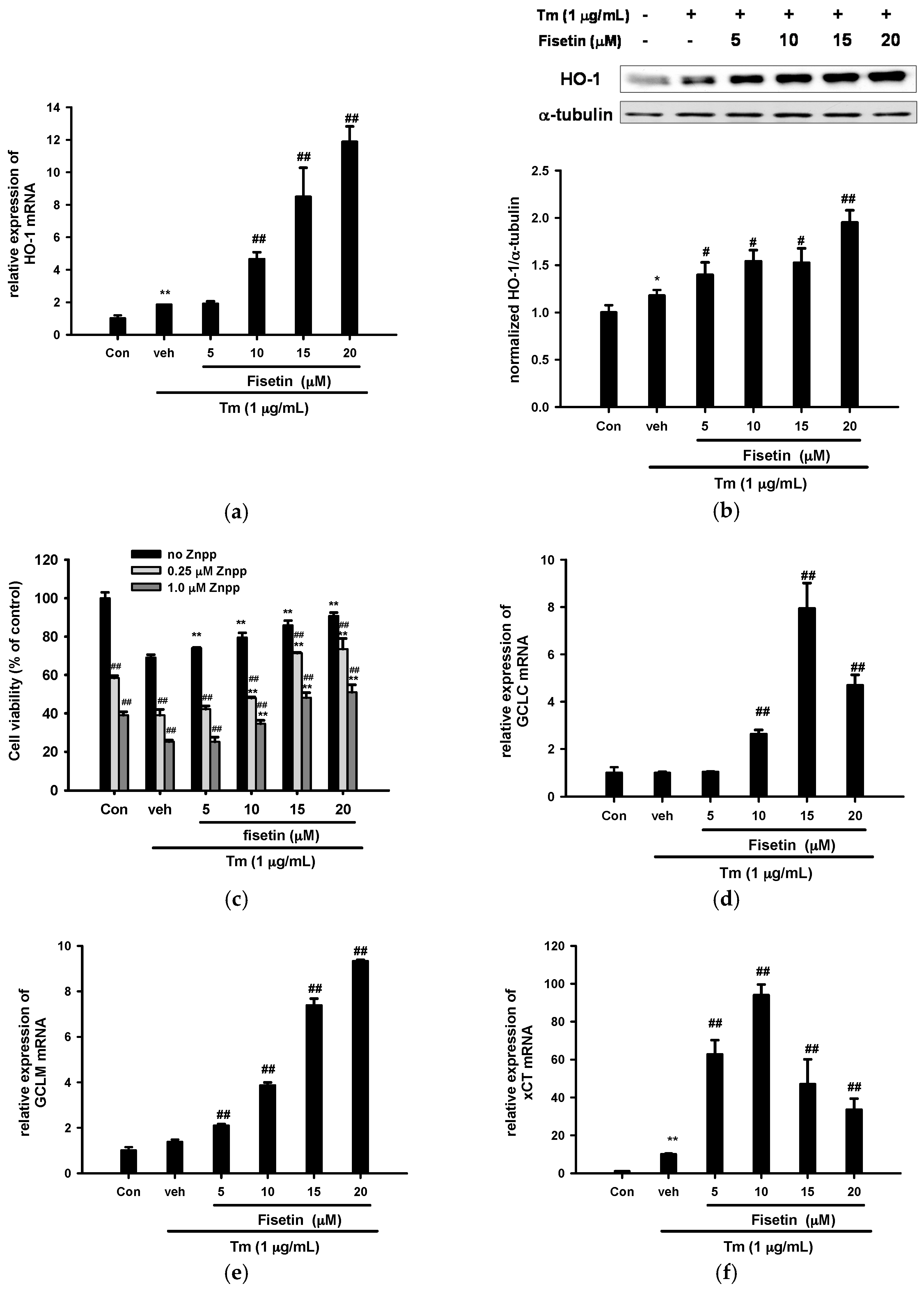
2.5. Fisetin Induces Nrf2-Driven Oxidative Stress Response Gene Expression
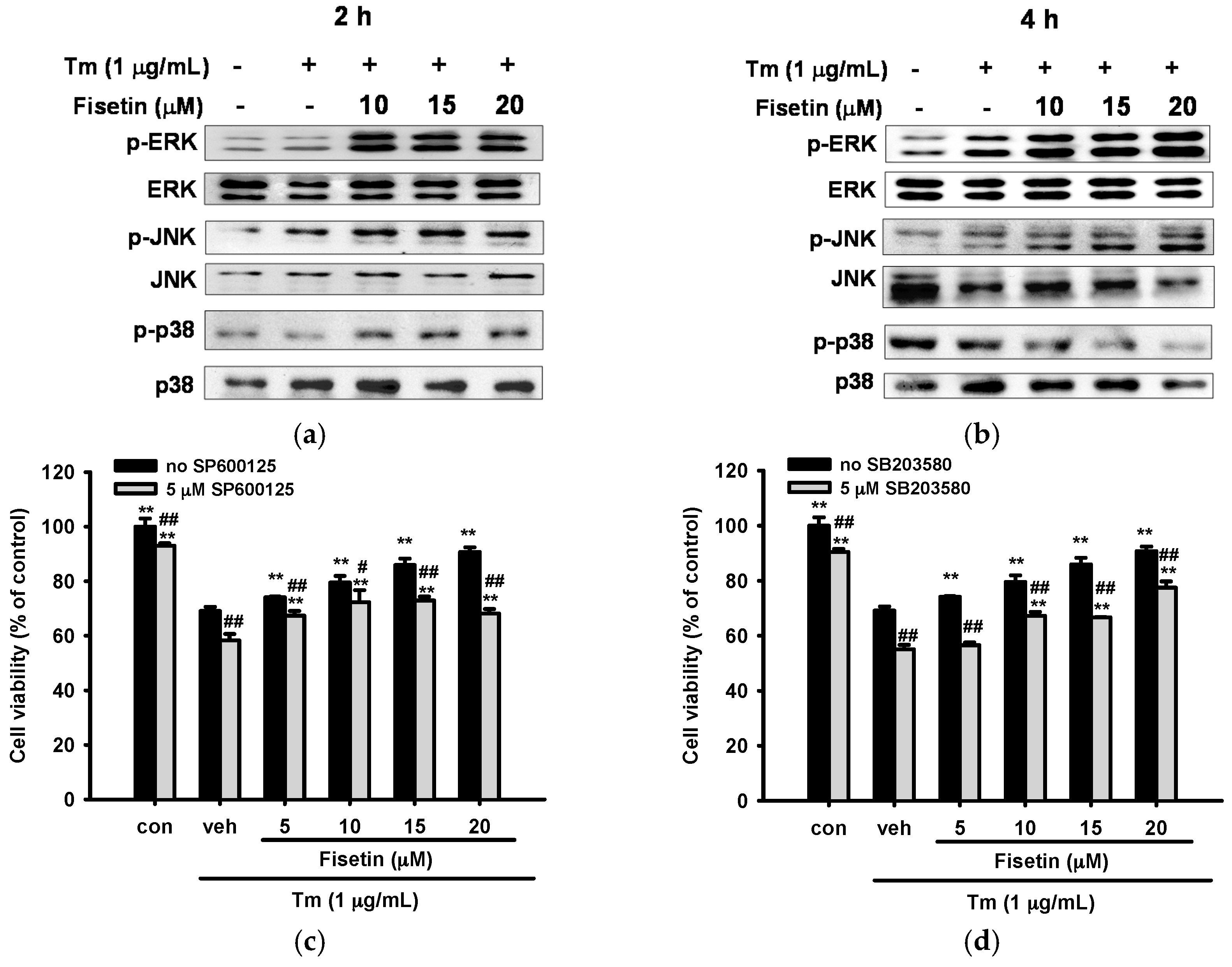
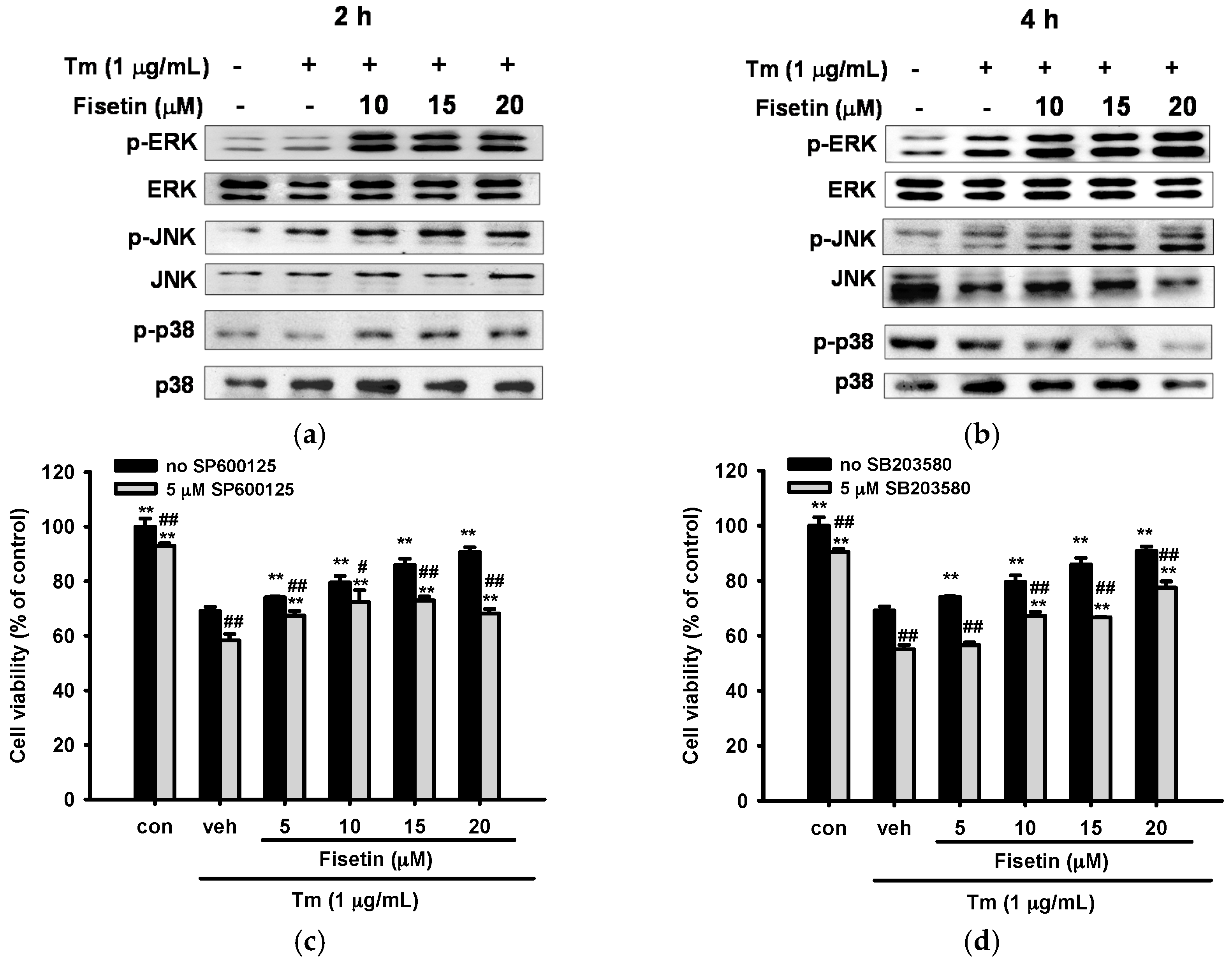
2.6. Fisetin Activates Mitogen-Activated Protein Kinase (MAPK) Signaling Pathways
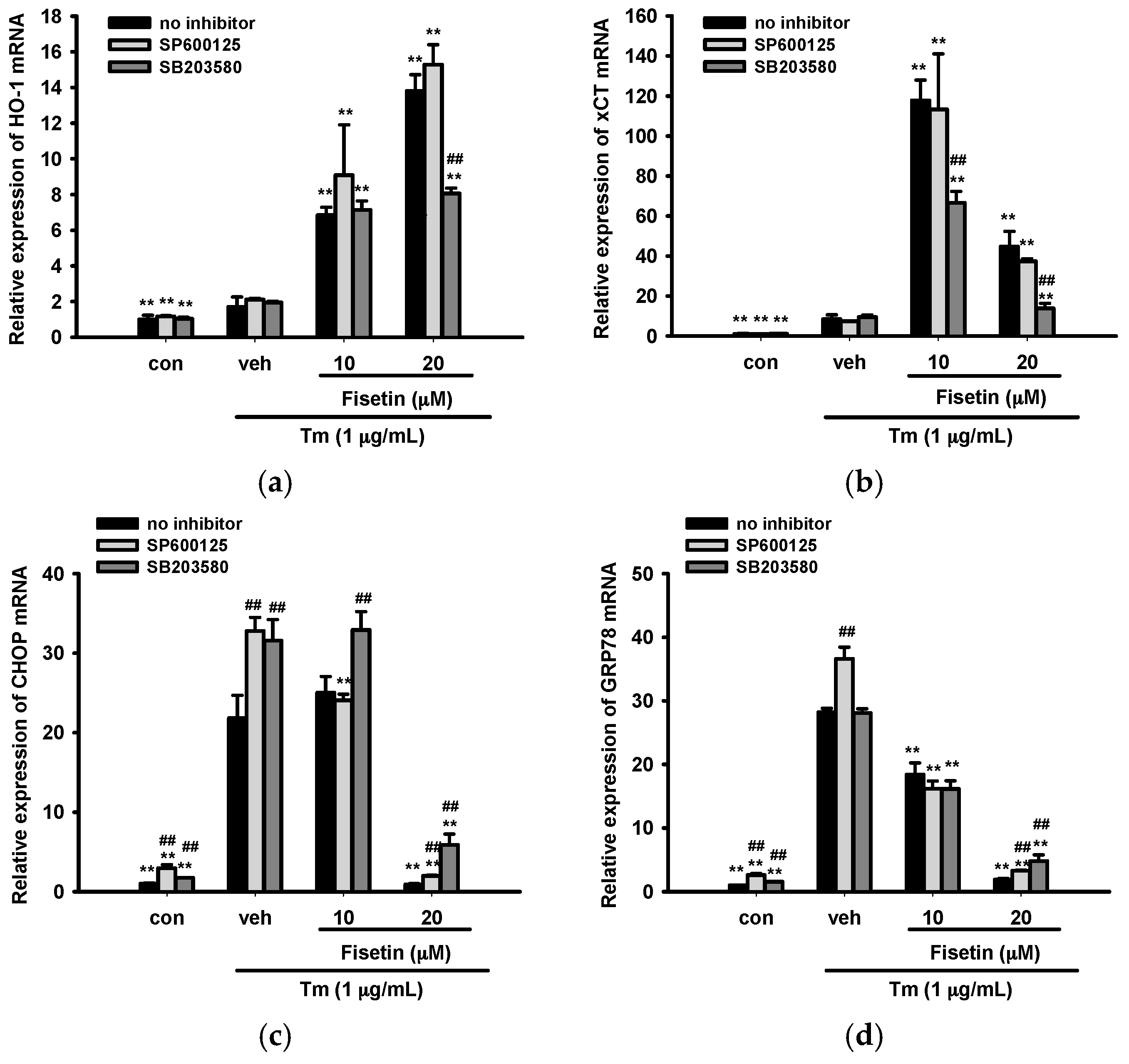
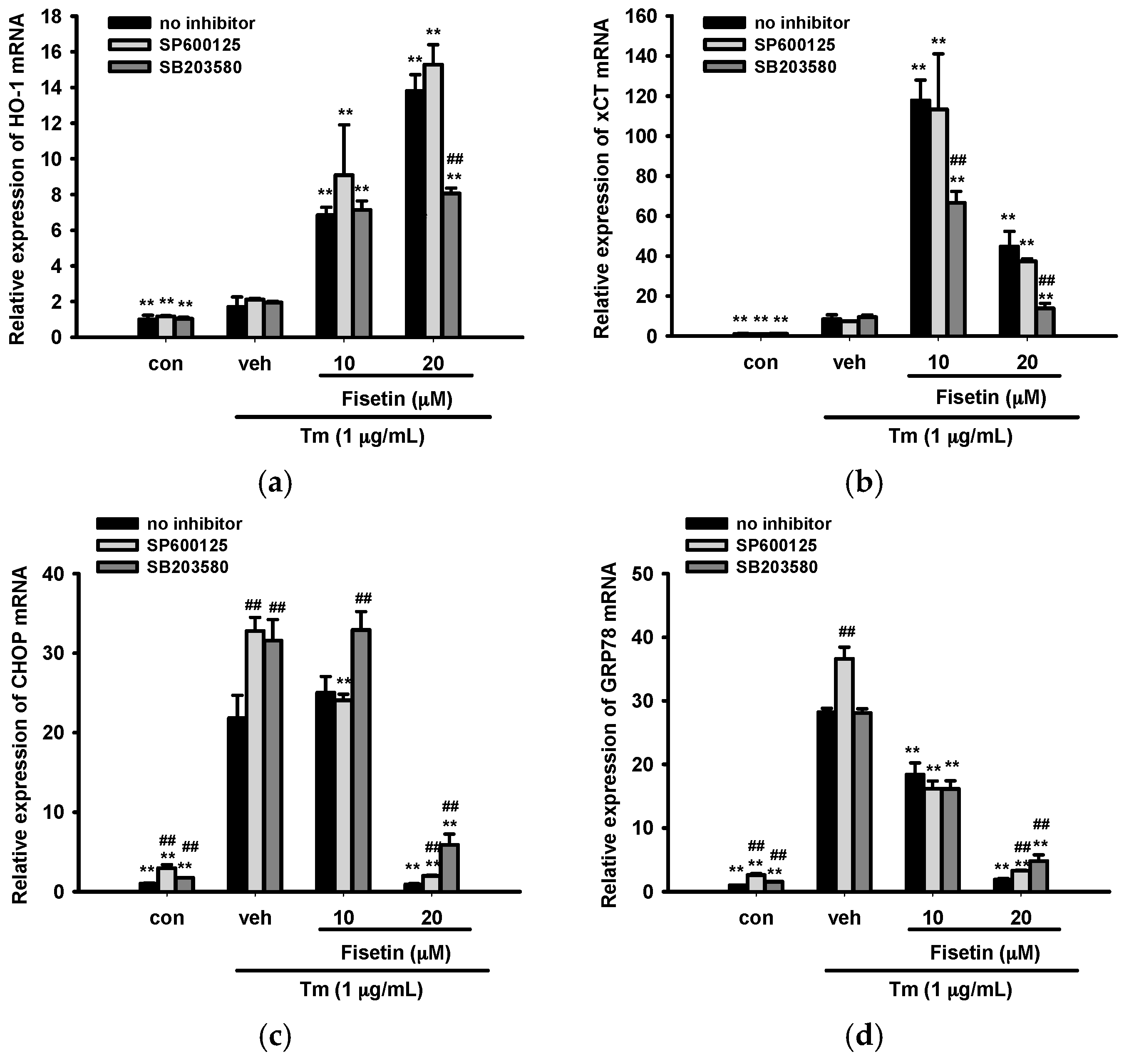
2.7. Contribution of JNK and p38 MAPK Pathways to ER Stress and Nrf2-Driven Gene Expression
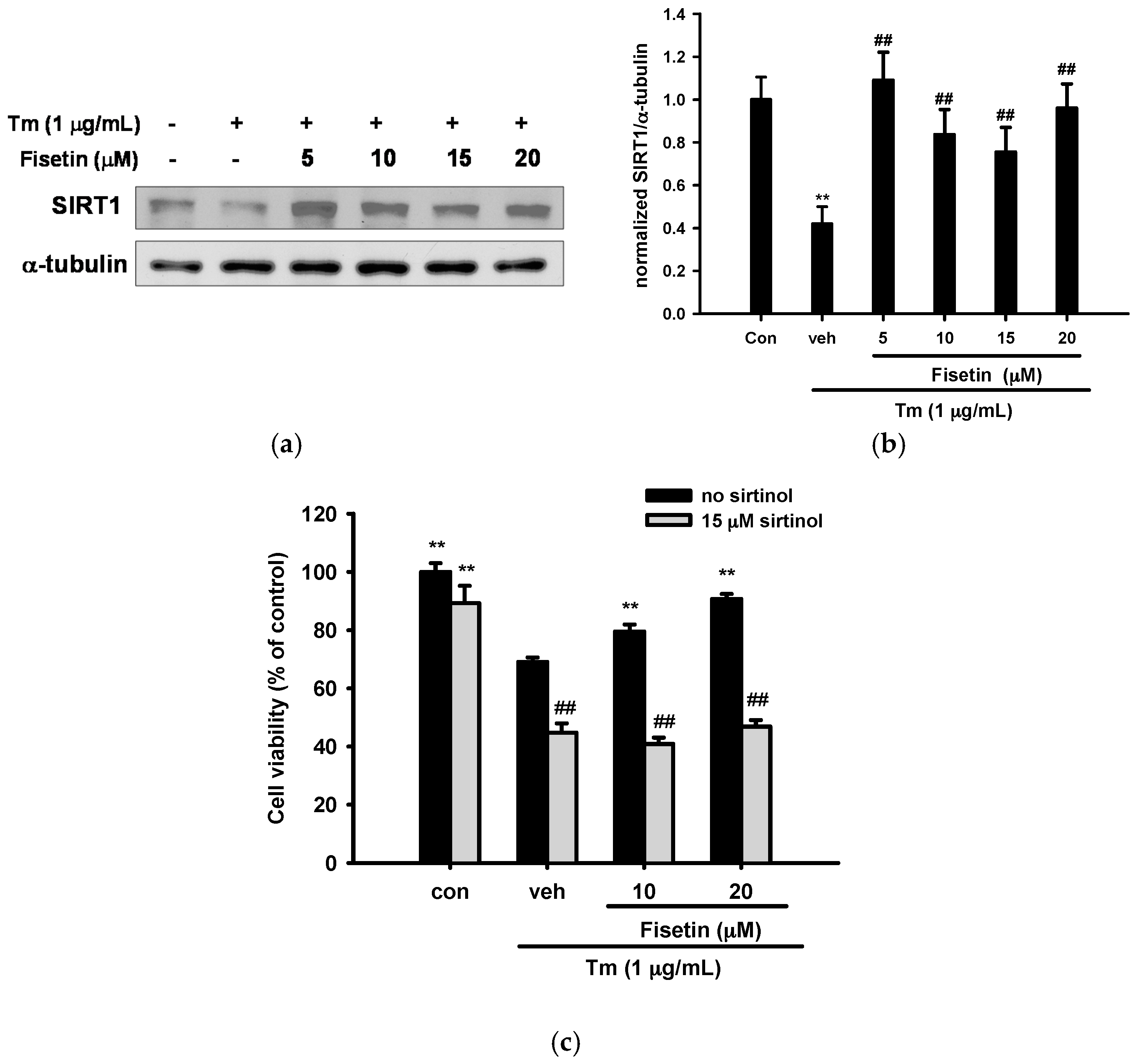
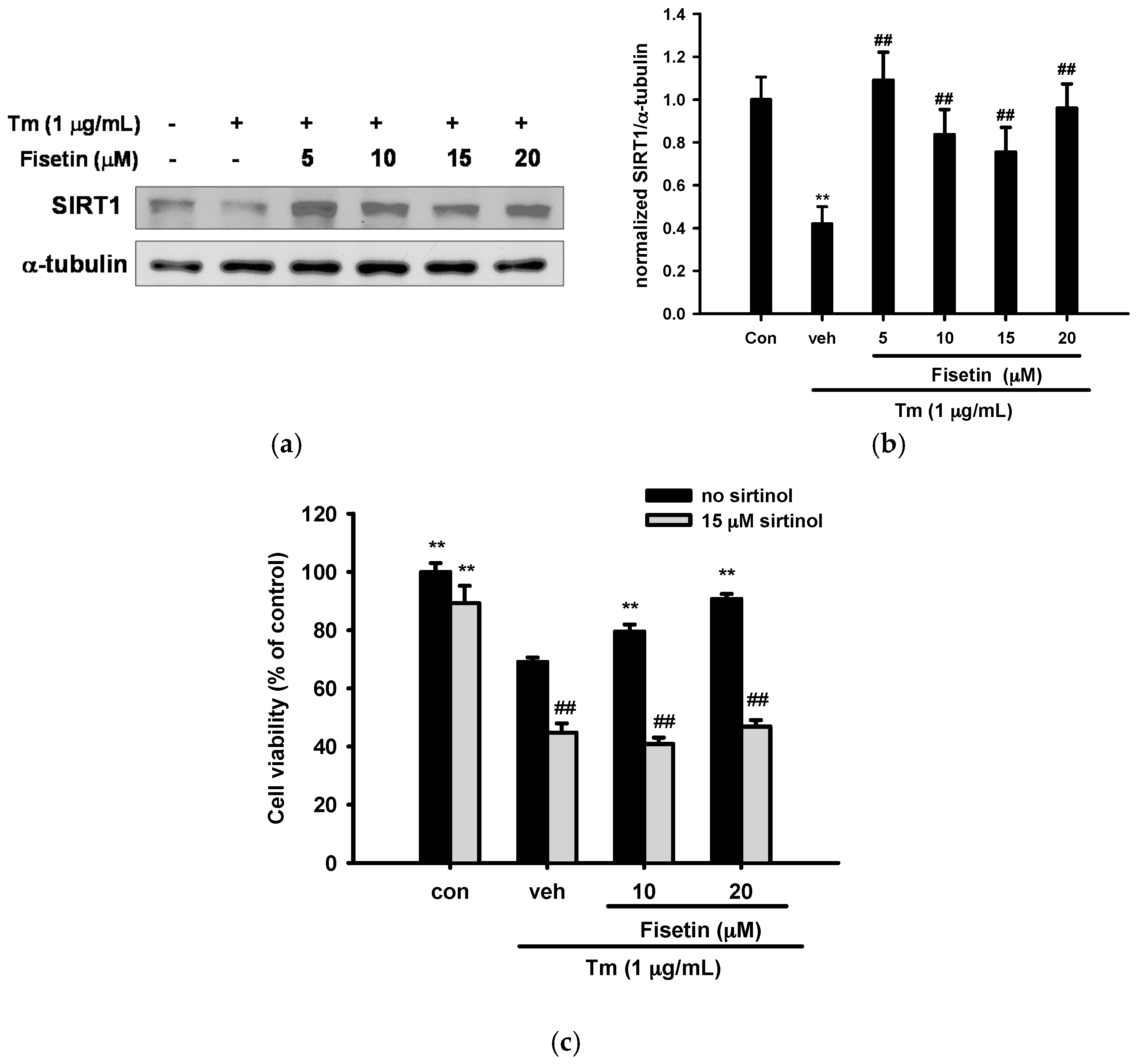
2.8. Effect of Fisetin on Sirtuin 1 (SIRT1) Expression
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. PC12 Cell Culture
4.3. Drug Treatments and Cell Viability Assay
4.4. Protein Extraction and Immunoblotting
4.5. RNA Extraction, Real-Time RT-PCR, and Semi-Quantitative RT-PCR
4.6. Intracellular ROS Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schroder, M.; Kaufman, R.J. Er stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.M.; Scheper, W. Endoplasmic reticulum stress in neurodegeneration. In Protein Folding and Misfolding: Neurodegenerative Diseases; Ovádi, J., Orosz, F., Eds.; Springer Netherlands: Dordrecht, The Netherlands, 2009; Volume 7, pp. 111–132. [Google Scholar]
- Heifetz, A.; Keenan, R.W.; Elbein, A.D. Mechanism of action of tunicamycin on the UDP-GlcNAc:dolichyl-phosphate Glc-NAc-1-phosphate transferase. Biochemistry 1979, 18, 2186–2192. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.; Teodoro, T.; Volchuk, A. Endoplasmic reticulum stress: Signaling the unfolded protein response. Physiology 2007, 22, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Kaufman, R.J. A trip to the ER: Coping with stress. Trends Cell Biol. 2004, 14, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Bernales, S.; Soto, M.M.; McCullagh, E. Unfolded protein stress in the endoplasmic reticulum and mitochondria: A role in neurodegeneration. Front. Aging Neurosci. 2012, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Rasheva, V.; Domingos, P. Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis 2009, 14, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Brewer, J.W.; Diehl, J.A.; Hendershot, L.M. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J. Mol. Biol. 2002, 318, 1351–1365. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Digaleh, H.; Kiaei, M.; Khodagholi, F. Nrf2 and Nrf1 signaling and ER stress crosstalk: Implication for proteasomal degradation and autophagy. Cell. Mol. Life Sci. 2013, 70, 4681–4694. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A.; Beier, V.; Franquelim, H.G.; Wollert, T. Molecular mechanism of autophagic membrane-scaffold assembly and disassembly. Cell 2014, 156, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H. Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem. 2013, 55, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Rendeiro, C.; Guerreiro, J.D.T.; Williams, C.M.; Spencer, J.P.E. Flavonoids as modulators of memory and learning: Molecular interactions resulting in behavioural effects. Proc. Nutr. Soc. 2012, 71, 246–262. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, B.P.; Sinclair, D.A. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol. Sci. 2014, 35, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Magesh, S.; Chen, Y.; Hu, L. Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Med. Res. Rev. 2012, 32, 687–726. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Cornelius, C.; Dinkova-Kostova, A.T.; Iavicoli, I.; di Paola, R.; Koverech, A.; Cuzzocrea, S.; Rizzarelli, E.; Calabrese, E.J. Cellular stress responses, hormetic phytochemicals and vitagenes in aging and longevity. Biochim. Biophys. Acta 2012, 1822, 753–783. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.P. Flavonoids and brain health: Multiple effects underpinned by common mechanisms. Genes Nutr. 2009, 4, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Yao, H.; Caito, S.; Hwang, J.W.; Arunachalam, G.; Rahman, I. Regulation of SIRT1 in cellular functions: Role of polyphenols. Arch. Biochem. Biophys. 2010, 501, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Qian, L.; Zhang, J.; Zhang, W.; Morrison, A.; Hayes, P.; Wilson, S.; Chen, T.; Zhao, J. SIRT1 overexpression in neurons promotes neurite outgrowth and cell survival through inhibition of the mtor signaling. J. Neurosci. Res. 2011, 89, 1723–1736. [Google Scholar] [CrossRef] [PubMed]
- Stiuso, P.; Bagarolo, M.L.; Ilisso, C.P.; Vanacore, D.; Martino, E.; Caraglia, M.; Porcelli, M.; Cacciapuoti, G. Protective effect of tyrosol and S-adenosylmethionine against ethanol-induced oxidative stress of Hepg2 cells involves Sirtuin 1, p53 and Erk1/2 signaling. Int. J. Mol. Sci. 2016, 17, 622. [Google Scholar] [CrossRef] [PubMed]
- Zenkov, N.K.; Menshchikova, E.B.; Tkachev, V.O. Keap1/Nrf2/are redox-sensitive signaling system as a pharmacological target. Biochemistry 2013, 78, 19–36. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.; Benzeroual, K. Neuroprotective effect of flavonoids, via up-regulating Nrf2-are pathway, in mpp+-induced PC12 cells, as a model of parkinson’s disease. FEBS J. 2015, 29. [Google Scholar]
- Maher, P. A comparison of the neurotrophic activities of the flavonoid fisetin and some of its derivatives. Free Radic. Res. 2006, 40, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Maher, P. The flavonoid fisetin promotes nerve cell survival from trophic factor withdrawal by enhancement of proteasome activity. Arch. Biochem. Biophys. 2008, 476, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Maher, P.; Dargusch, R.; Bodai, L.; Gerard, P.E.; Purcell, J.M.; Marsh, J.L. Erk activation by the polyphenols fisetin and resveratrol provides neuroprotection in multiple models of huntington’s disease. Hum. Mol. Genet. 2011, 20, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Ho, Y.R.; Wu, M.J.; Huang, S.P.; Chen, P.K.; Tai, M.H.; Ho, C.T.; Yen, J.H. Cytoprotective effects of fisetin against hypoxia-induced cell death in PC12 cells. Food Funct. 2015, 6, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, M.; Leypoldt, F.; Lewerenz, J.; Birkenmayer, G.; Orozco, D.; Ludewig, P.; Thundyil, J.; Arumugam, T.V.; Gerloff, C.; Tolosa, E.; et al. The flavonoid fisetin attenuates postischemic immune cell infiltration, activation and infarct size after transient cerebral middle artery occlusion in mice. J. Cereb. Blood Flow Metab. 2012, 32, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Syed, D.N.; Lall, R.K.; Chamcheu, J.C.; Haidar, O.; Mukhtar, H. Involvement of ER stress and activation of apoptotic pathways in fisetin induced cytotoxicity in human melanoma. Arch. Biochem. Biophys. 2014, 563, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Piao, M.J.; Madduma Hewage, S.R.; Ryu, Y.S.; Oh, M.C.; Kwon, T.K.; Chae, S.; Hyun, J.W. Fisetin induces apoptosis and endoplasmic reticulum stress in human non-small cell lung cancer through inhibition of the mapk signaling pathway. Tumour Biol. 2016, 37, 9615–9624. [Google Scholar] [CrossRef] [PubMed]
- Su, C.H.; Kuo, C.L.; Lu, K.W.; Yu, F.S.; Ma, Y.S.; Yang, J.L.; Chu, Y.L.; Chueh, F.S.; Liu, K.C.; Chung, J.G. Fisetin-induced apoptosis of human oral cancer SCC-4 cells through reactive oxygen species production, endoplasmic reticulum stress, caspase-, and mitochondria-dependent signaling pathways. Environ. Toxicol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Shimoke, K.; Amano, H.; Kishi, S.; Uchida, H.; Kudo, M.; Ikeuchi, T. Nerve growth factor attenuates endoplasmic reticulum stress-mediated apoptosis via suppression of caspase-12 activity. J. Biochem. 2004, 135, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Sasaya, H.; Utsumi, T.; Shimoke, K.; Nakayama, H.; Matsumura, Y.; Fukunaga, K.; Ikeuchi, T. Nicotine suppresses tunicamycin-induced, but not thapsigargin-induced, expression of GRP78 during ER stress-mediated apoptosis in PC12 cells. J. Biochem. 2008, 144, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.H.; Yen, J.H.; Weng, C.Y.; Wang, L.; Ha, C.L.; Wu, M.J. Lipid peroxidation end product 4-hydroxy-trans-2-nonenal triggers unfolded protein response and heme oxygenase-1 expression in PC12 cells: Roles of ROS and MAPK pathways. Toxicology 2014, 315, 24–37. [Google Scholar] [CrossRef] [PubMed]
- D’Amours, D.; Sallmann, F.R.; Dixit, V.M.; Poirier, G.G. Gain-of-function of poly(ADP-ribose) polymerase-1 upon cleavage by apoptotic proteases: Implications for apoptosis. J. Cell Sci. 2001, 114, 3771–3778. [Google Scholar] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12–Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.G.; Cao, X.Z.; Zhao, Y.S.; Gao, S.Y.; Li, S.D.; Liu, X.Y.; Zhang, Y.; Zhang, K.Q. The molecular mechanism of endoplasmic reticulum stress-induced apoptosis in PC-12 neuronal cells: The protective effect of insulin-like growth factor I. Endocrinology 2009, 150, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.J.; Heo, J.; Kim, Y.H. Tunicamycin promotes apoptosis in leukemia cells through ROS generation and downregulation of survivin expression. Apoptosis 2015, 20, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Ehren, J.L.; Maher, P. Concurrent regulation of the transcription factors Nrf2 and Atf4 mediates the enhancement of glutathione levels by the flavonoid fisetin. Biochem. Pharmacol. 2013, 85, 1816–1826. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Mimura, J.; Okada, T.; Sato, H.; Liu, T.; Maruyama, A.; Ohyama, C.; Itoh, K. Nrf2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol. Cell. Biol. 2014, 34, 3421–3434. [Google Scholar] [CrossRef] [PubMed]
- Mansuri, M.L.; Parihar, P.; Solanki, I.; Parihar, M.S. Flavonoids in modulation of cell survival signalling pathways. Genes Nutr. 2014, 9, 400. [Google Scholar] [CrossRef] [PubMed]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Kim, Y.H.; Son, S.W.; Moon, E.Y.; Pyo, S.; Um, S.H. Fisetin induces SIRT1 expression while inhibiting early adipogenesis in 3T3-L1 cells. Biochem. Biophys. Res. Commun. 2015, 467, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Gump, J.M.; Thorburn, A. Autophagy and apoptosis: What is the connection? Trends Cell Biol. 2011, 21, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Wang, L.; Wang, B.; Wang, T.; Yang, G.; Shen, L.; Wang, T.; Guo, X.; Liu, Y.; Xia, Y.; et al. Activation of volume-sensitive outwardly rectifying chloride channel by ROS contributes to ER stress and cardiac contractile dysfunction: Involvement of CHOP through wnt. Cell Death Dis. 2014, 5, e1528. [Google Scholar] [CrossRef] [PubMed]
- Wardman, P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: Progress, pitfalls, and prospects. Free Radic. Biol. Med. 2007, 43, 995–1022. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Ybanez, M.D.; Ahmadi, S.; Yeh, K.; Kaplowitz, N. Redox regulation of tumor necrosis factor signaling. Antioxid. Redox Signal. 2009, 11, 2245–2263. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Oshima, Y.; Fujimura, A. An approach to elucidate potential mechanism of renal toxicity of arsenic trioxide. Exp. Hematol. 2007, 35, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.C.; Ho, I.C.; Lee, T.C. Oxidative stress mediates sodium arsenite-induced expression of heme oxygenase-1, monocyte chemoattractant protein-1, and interleukin-6 in vascular smooth muscle cells. Toxicol. Sci. 2005, 85, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Alam, J.; Wicks, C.; Stewart, D.; Gong, P.; Touchard, C.; Otterbein, S.; Choi, A.M.; Burow, M.E.; Tou, J. Mechanism of heme oxygenase-1 gene activation by cadmium in MCF-7 mammary epithelial cells. Role of p38 kinase and Nrf2 transcription factor. J. Biol. Chem. 2000, 275, 27694–27702. [Google Scholar] [CrossRef] [PubMed]
- Andreadi, C.K.; Howells, L.M.; Atherfold, P.A.; Manson, M.M. Involvement of Nrf2, p38, B-Raf, and nuclear factor-κB, but not phosphatidylinositol 3-kinase, in induction of hemeoxygenase-1 by dietary polyphenols. Mol. Pharmacol. 2006, 69, 1033–1040. [Google Scholar] [PubMed]
- Ogborne, R.M.; Rushworth, S.A.; O’Connell, M.A. α-lipoic acid-induced heme oxygenase-1 expression is mediated by nuclear factor erythroid 2-related factor 2 and p38 mitogen-activated protein kinase in human monocytic cells. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2100–2105. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Uzquiza, A.; Arechederra, M.; Bragado, P.; Aguirre-Ghiso, J.A.; Porras, A. P38α mediates cell survival in response to oxidative stress via induction of antioxidant genes: Effect on the P70S6K pathway. J. Biol. Chem. 2012, 287, 2632–2642. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, J.; DeGraff, W.G.; Gazdar, A.F.; Minna, J.D.; Mitchell, J.B. Evaluation of a tetrazolium-based semiautomated colorimetric assay: Assessment of chemosensitivity testing. Cancer Res. 1987, 47, 936–942. [Google Scholar] [PubMed]
- Pedrosa, R.; Soares-da-Silva, P. Oxidative and non-oxidative mechanisms of neuronal cell death and apoptosis by l-3,4-dihydroxyphenylalanine (l-dopa) and dopamine. Br. J. Pharmacol. 2002, 137, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.S.; Yen, J.H.; Kou, M.C.; Wu, M.J. Luteolin and apigenin attenuate 4-hydroxy-2-nonenal-mediated cell death through modulation of upr, Nrf2-are and mapk pathways in PC12 cells. PLoS ONE 2015, 10, e0130599. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Cismowski, M.J.; Toyota, E.; Smrcka, A.V.; Lucchesi, P.A.; Chilian, W.M.; Lanier, S.M. Identification of a receptor-independent activator of G protein signaling (AGS8) in ischemic heart and its interaction with Gβγ. Proc. Natl. Acad. Sci. USA 2006, 103, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Su, J.D.; Yen, J.H.; Li, S.; Weng, C.Y.; Lin, M.H.; Ho, C.T.; Wu, M.J. 3′,4′-didemethylnobiletin induces phase II detoxification gene expression and modulates PI3K/Akt signaling in PC12 cells. Free Radic. Biol. Med. 2012, 52, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Rojo, A.I.; Salinas, M.; Diaz, R.; Gallardo, G.; Alam, J.; De Galarreta, C.M.; Cuadrado, A. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J. Biol. Chem. 2004, 279, 8919–8929. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Company | Catalog Number |
|---|---|---|
| α-tubulin | Sigma | T 6199 |
| β-actin | GeneTex (Hsinchu, Taiwan) | GTX629630 |
| PARP-1 | Santa Cruz | H-250 |
| LC3B | Gene Tex | GTX127375 |
| Atg12 | Gene Tex | GTX124181 |
| eIF2α | Gene Tex | GTX101241 |
| p-eIF2α | Gene Tex | GTX61039 |
| GRP78 | BD (Franklin Lakes, NJ, USA) | 610978 |
| CHOP | Santa Cruz | sc-7351 |
| HO-1 | Enzo Life Sciences (Farmingdale, NY, USA) | SPA-895 |
| ERK | Cell Signaling (Danvers, MA, USA) | 4695 |
| p-ERK | Cell Signaling | 4370 |
| p38 | Cell Signaling | 9212 |
| p-p38 | Cell Signaling | 9215 |
| JNK | Cell Signaling | 9258 |
| p-JNK | Cell Signaling | 4668 |
| SIRT1 | Cell Signaling | 8469 |
| Gene | Primers | Amplicon (bp) |
|---|---|---|
| β-actin [61] | (F) CCTCTGAACCCTAAGGCCAA (R) AGCCTGGATGGCTACGTACA | 90 |
| ATF4 [39] | (F) CTTCTCCAGGTGTTCCTCGT (R) TGCTCAGCCCTCTTCTTCTG | 163 |
| CHOP [35] | (F) AAGAATCAAAAACCTTCACTACTCTTGACC (R) TGGGAGGTGCTTGTGACCTCTGC | 91 |
| GCLC [62] | (F) TGGCCAGCCGTACGGAGGAA (R) CAGGGAGCCTAGCCTGGGA | 143 |
| GCLM [62] | (F) CTTTCCTTGGAGCATTTGCAGCCTT (R[35]) ACCTGTGCCCACTGGTACAGCTG | 131 |
| GRP78 [35] | (F) CAACTCACGTCCAACCCGGAGAA (R) TGTCTTGGTTTGCCCACCTCCG | 171 |
| HO-1 [63] | (F) GCCTGCTAGCCTGGTTCAAG (R) AGCGGTGTCTGGGATGAACTA | 87 |
| TRB3 [39] | (F) GGACAAGATGCGAGCCACAT (R) CCACAGCAGGTGACAAGTCT | 179 |
| xCT [60] | (F) GACAGTGTGTGCATCCCCTT (R) GCATGCATTTCTTGCACAGTTC | 110 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yen, J.-H.; Wu, P.-S.; Chen, S.-F.; Wu, M.-J. Fisetin Protects PC12 Cells from Tunicamycin-Mediated Cell Death via Reactive Oxygen Species Scavenging and Modulation of Nrf2-Driven Gene Expression, SIRT1 and MAPK Signaling in PC12 Cells. Int. J. Mol. Sci. 2017, 18, 852. https://doi.org/10.3390/ijms18040852
Yen J-H, Wu P-S, Chen S-F, Wu M-J. Fisetin Protects PC12 Cells from Tunicamycin-Mediated Cell Death via Reactive Oxygen Species Scavenging and Modulation of Nrf2-Driven Gene Expression, SIRT1 and MAPK Signaling in PC12 Cells. International Journal of Molecular Sciences. 2017; 18(4):852. https://doi.org/10.3390/ijms18040852
Chicago/Turabian StyleYen, Jui-Hung, Pei-Shan Wu, Shu-Fen Chen, and Ming-Jiuan Wu. 2017. "Fisetin Protects PC12 Cells from Tunicamycin-Mediated Cell Death via Reactive Oxygen Species Scavenging and Modulation of Nrf2-Driven Gene Expression, SIRT1 and MAPK Signaling in PC12 Cells" International Journal of Molecular Sciences 18, no. 4: 852. https://doi.org/10.3390/ijms18040852
APA StyleYen, J.-H., Wu, P.-S., Chen, S.-F., & Wu, M.-J. (2017). Fisetin Protects PC12 Cells from Tunicamycin-Mediated Cell Death via Reactive Oxygen Species Scavenging and Modulation of Nrf2-Driven Gene Expression, SIRT1 and MAPK Signaling in PC12 Cells. International Journal of Molecular Sciences, 18(4), 852. https://doi.org/10.3390/ijms18040852