
Valproate Attenuates Endoplasmic Reticulum Stress-Induced Apoptosis in SH-SY5Y Cells via the AKT/GSK3β Signaling Pathway


Abstract
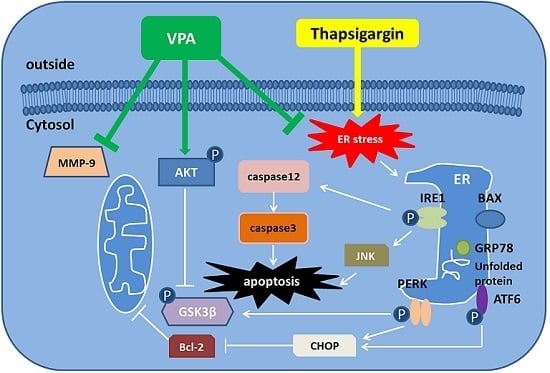
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
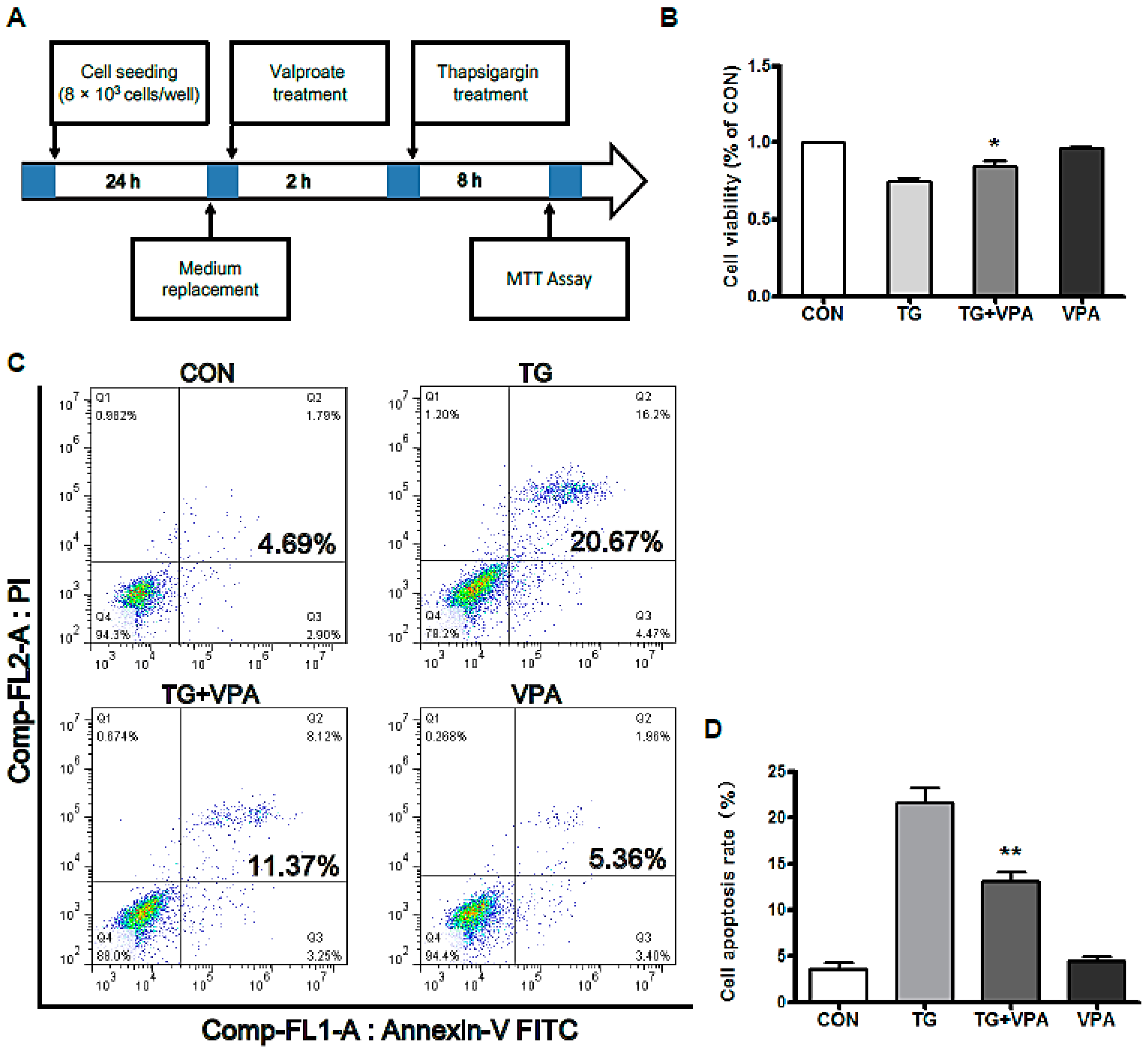
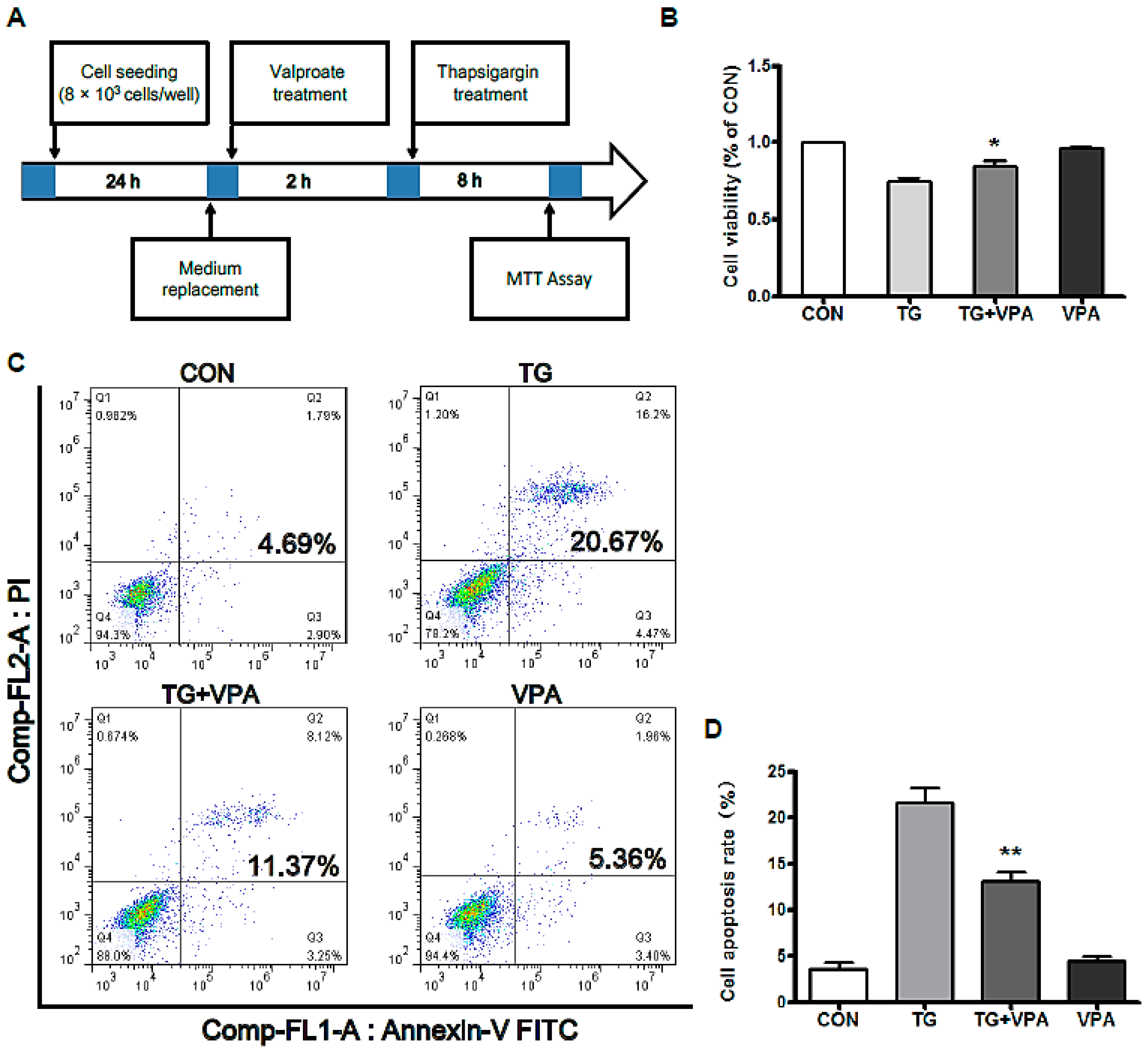
2.1. VPA Reduces TG-Induced Apoptosis in the SH-SY5Y Human Neuroblastoma Cell Line
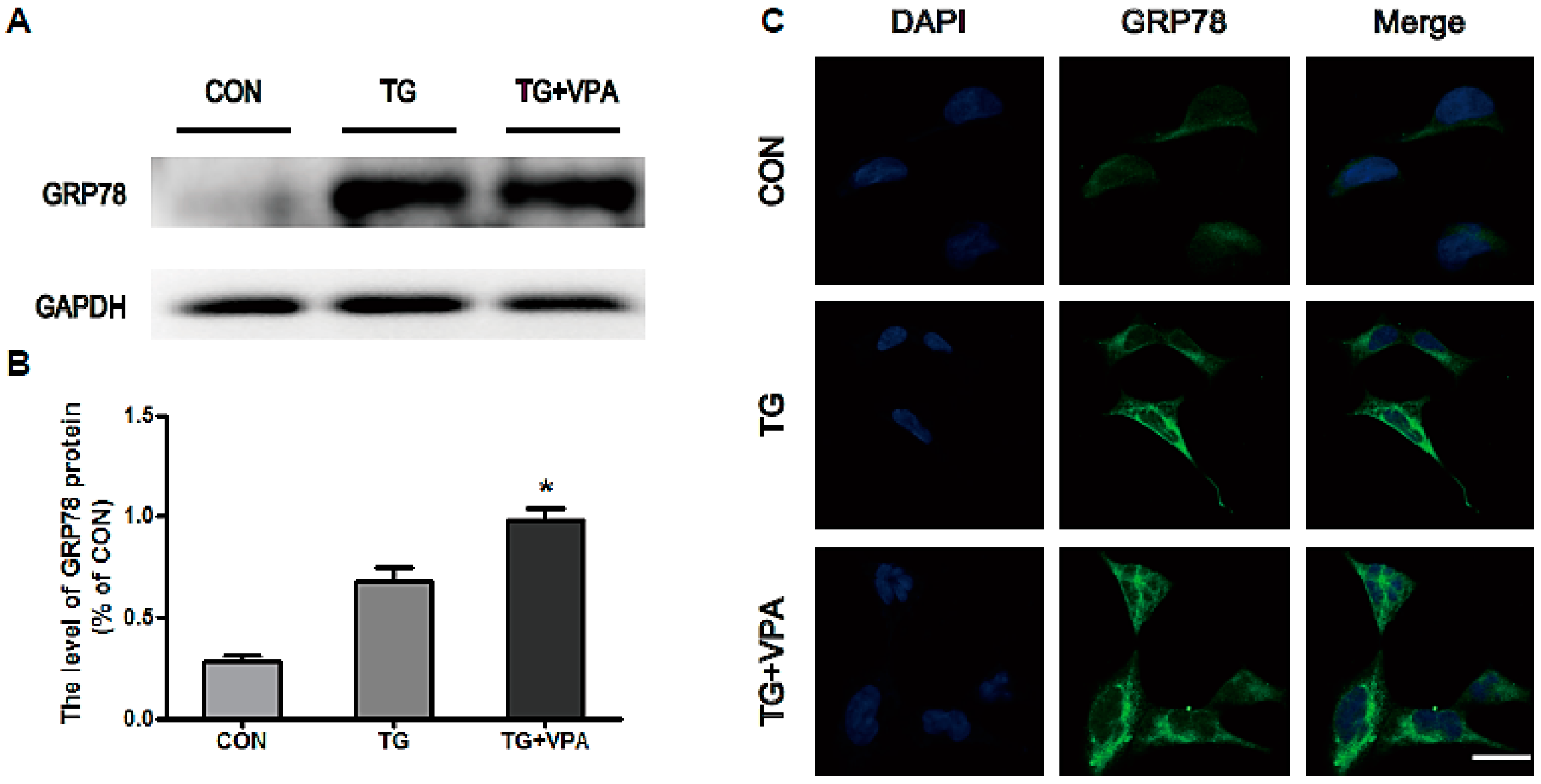
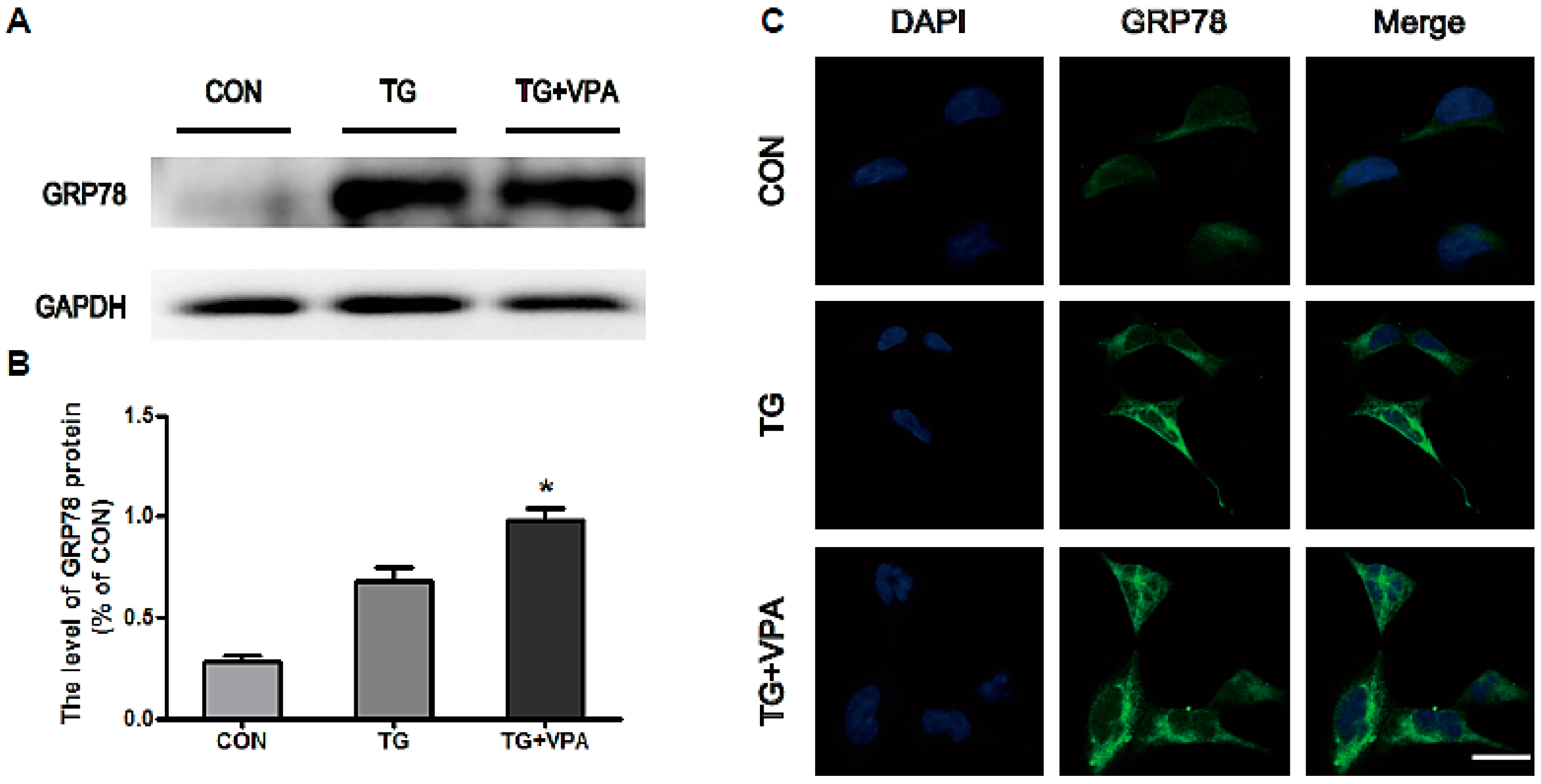
2.2. VPA Attenuates TG-Induced ER Stress in SH-SY5Y Cells
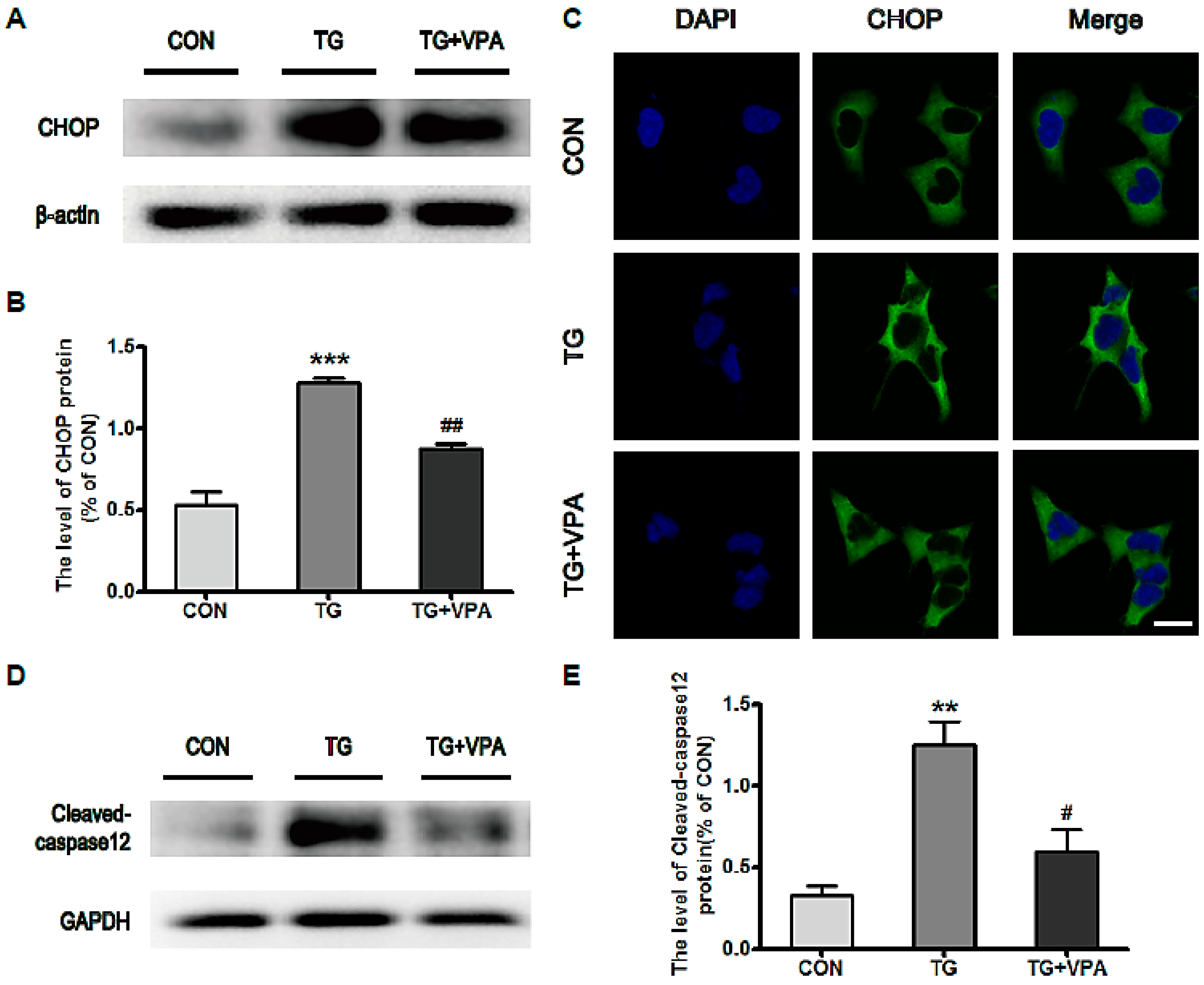
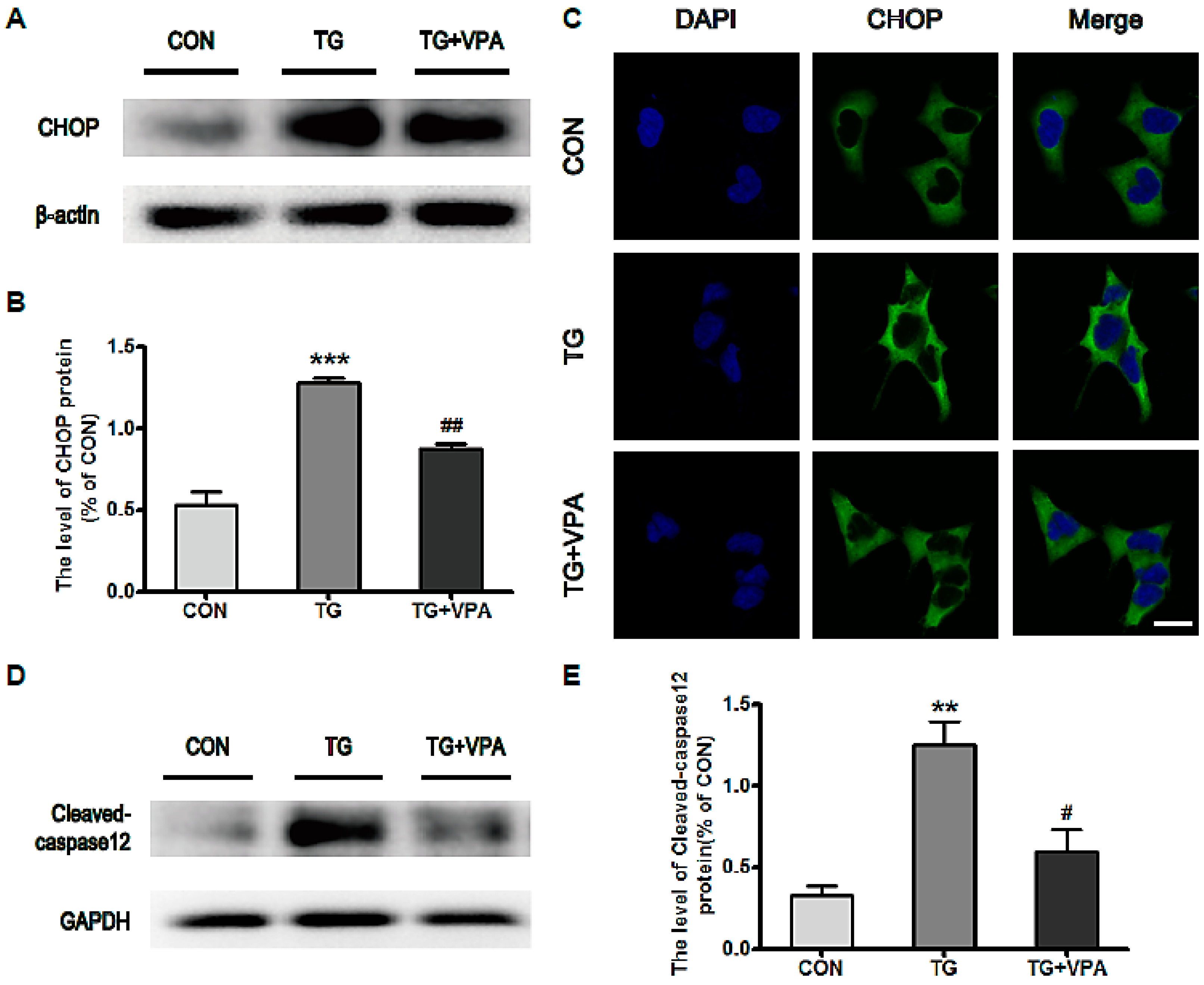
2.3. VPA Inhibits the Expression of C/EBP Homologous Protein (CHOP) and Caspase-12 in TG-Induced SH-SY5Y Cells
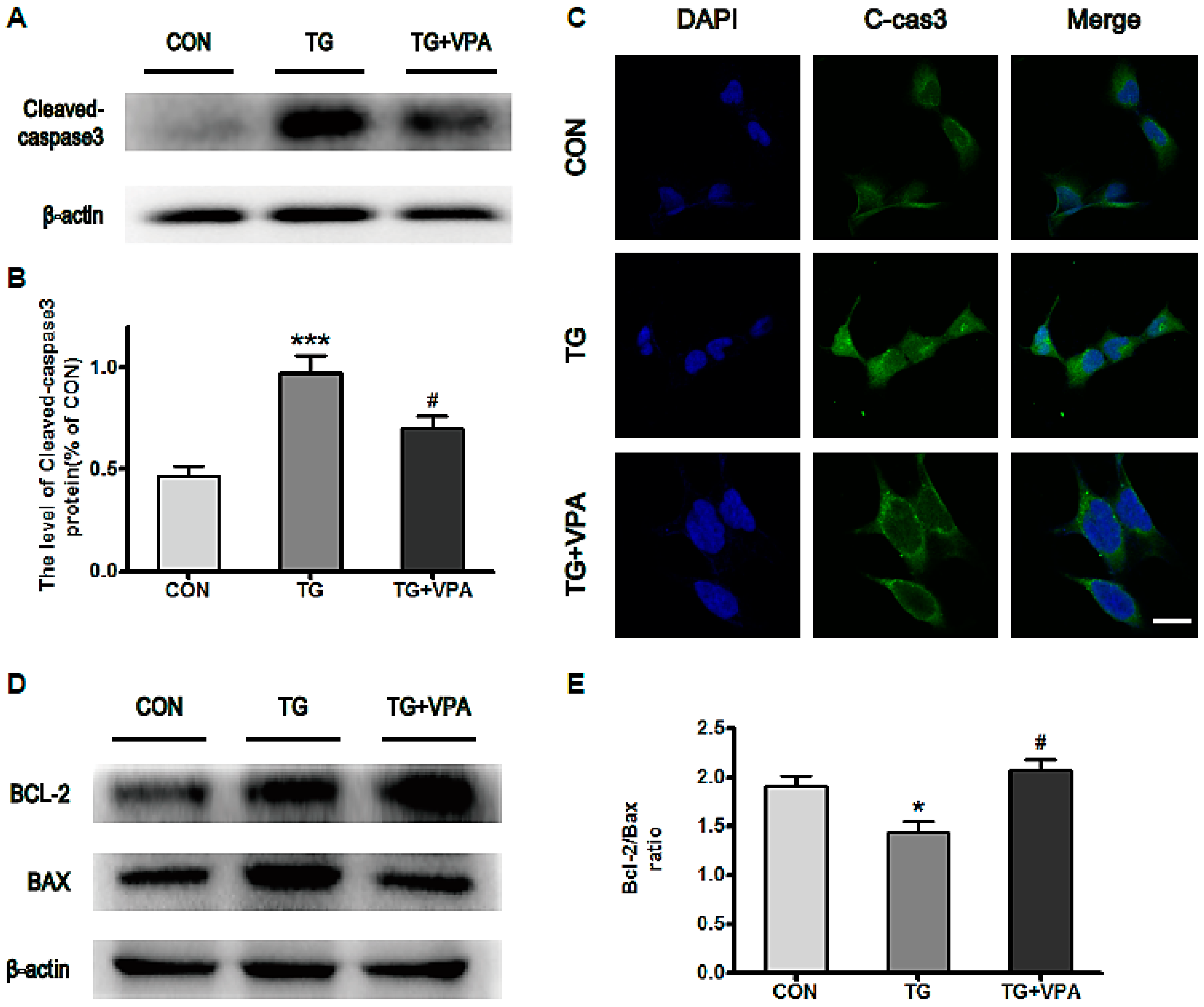
2.4. VPA Inhibits ER Stress-Induced Apoptosis and Up-Regulates the Ratio of Bcl-2/Bax in SH-SY5Y Cells
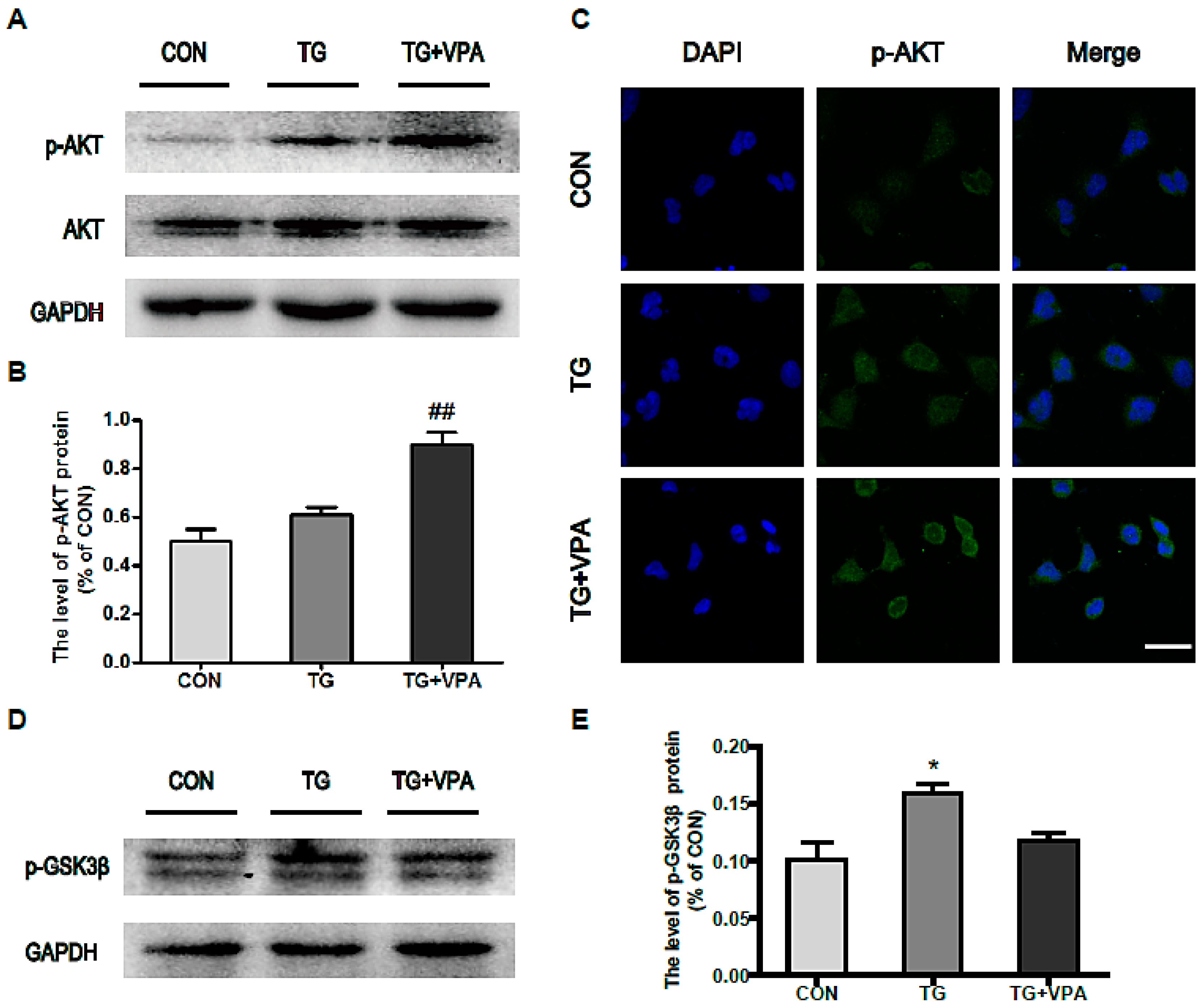
2.5. VPA Promotes Cell Proliferation through the PI3K/AKT/GSK3β Signaling Pathway in SH-SY5Y Cells
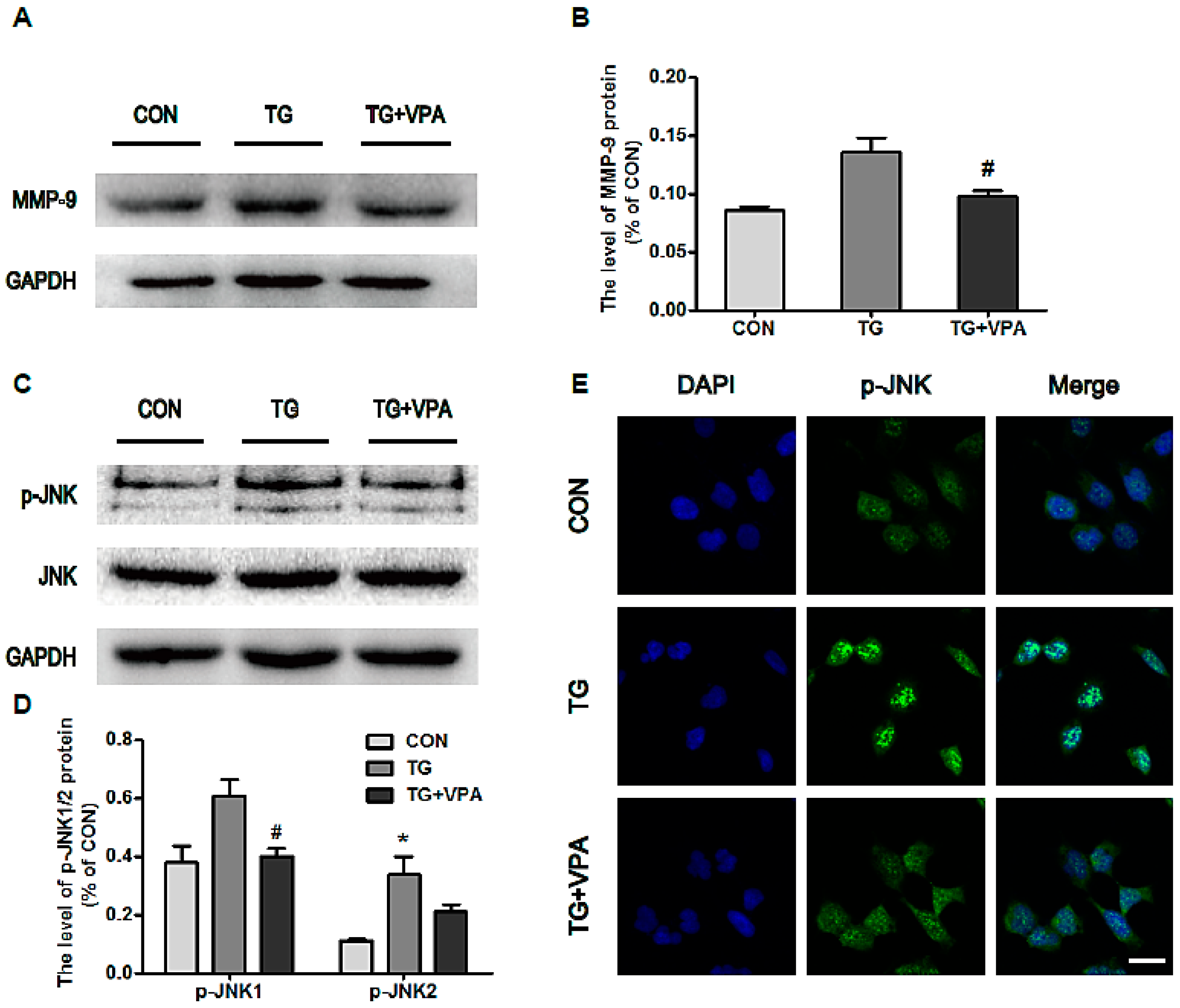
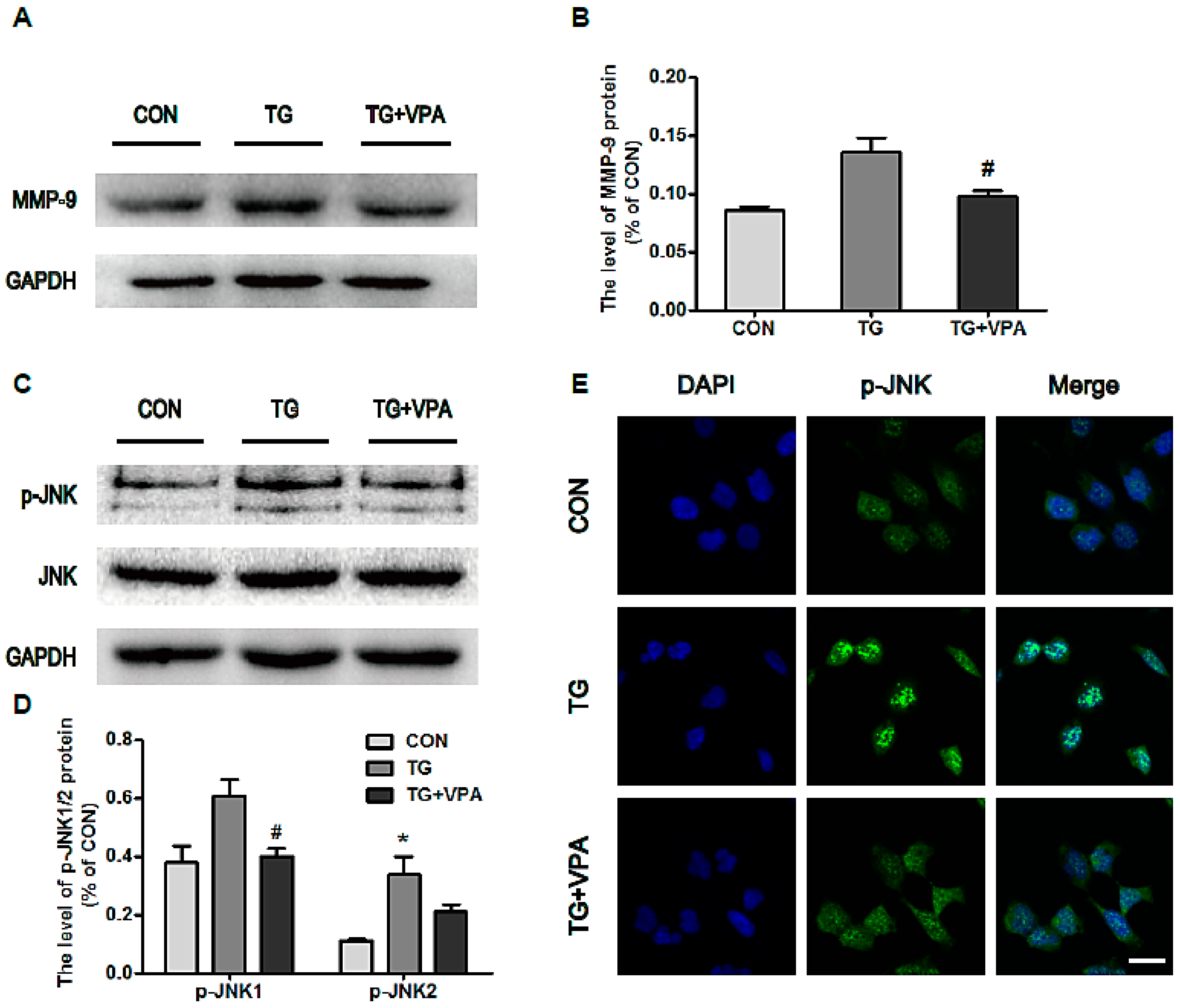
2.6. VPA Prevents Cell Apoptosis by Inhibiting MMP-9 Expression and c-Jun N-Terminal Kinase (JNK) Phosphorylation
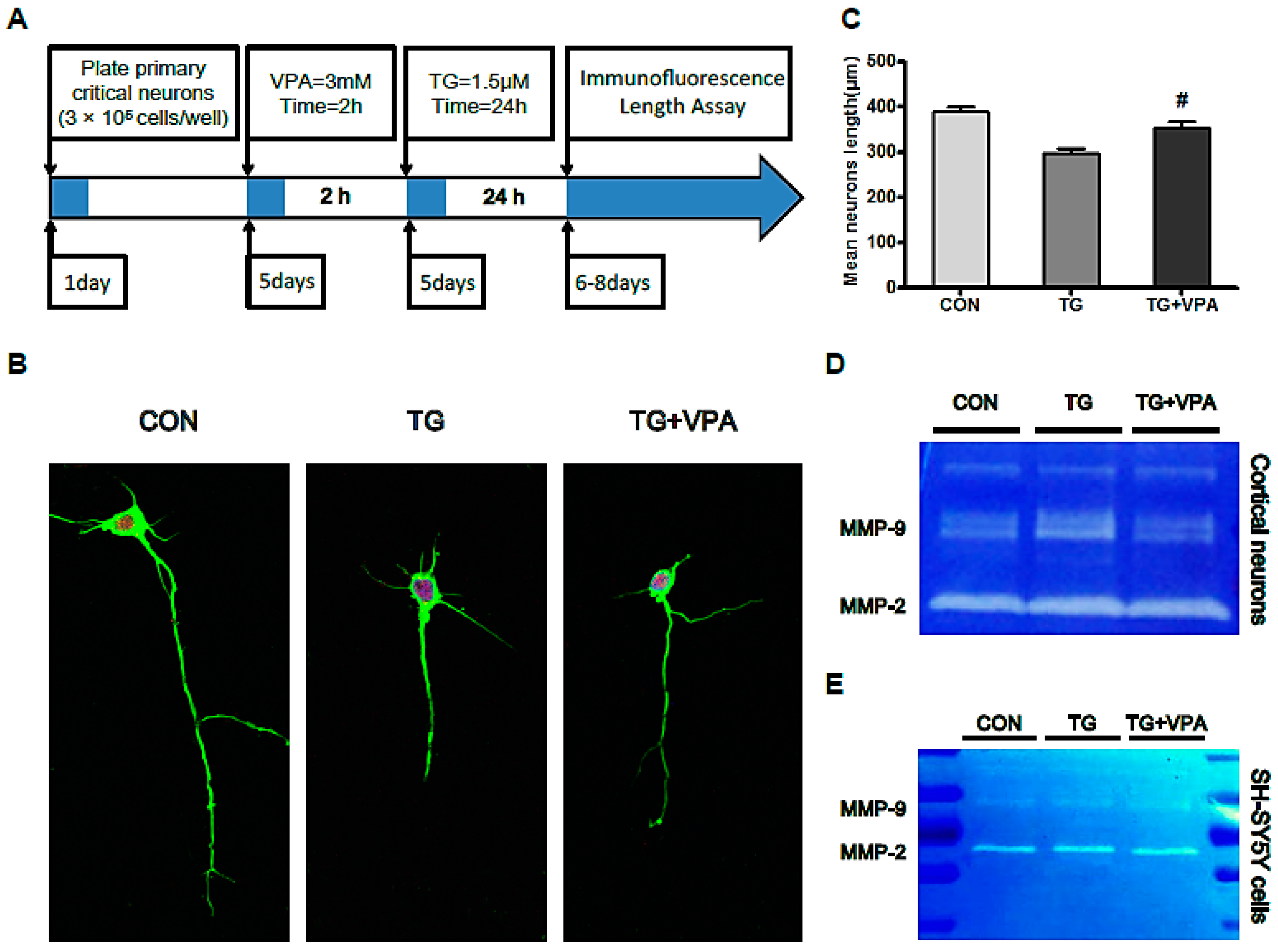
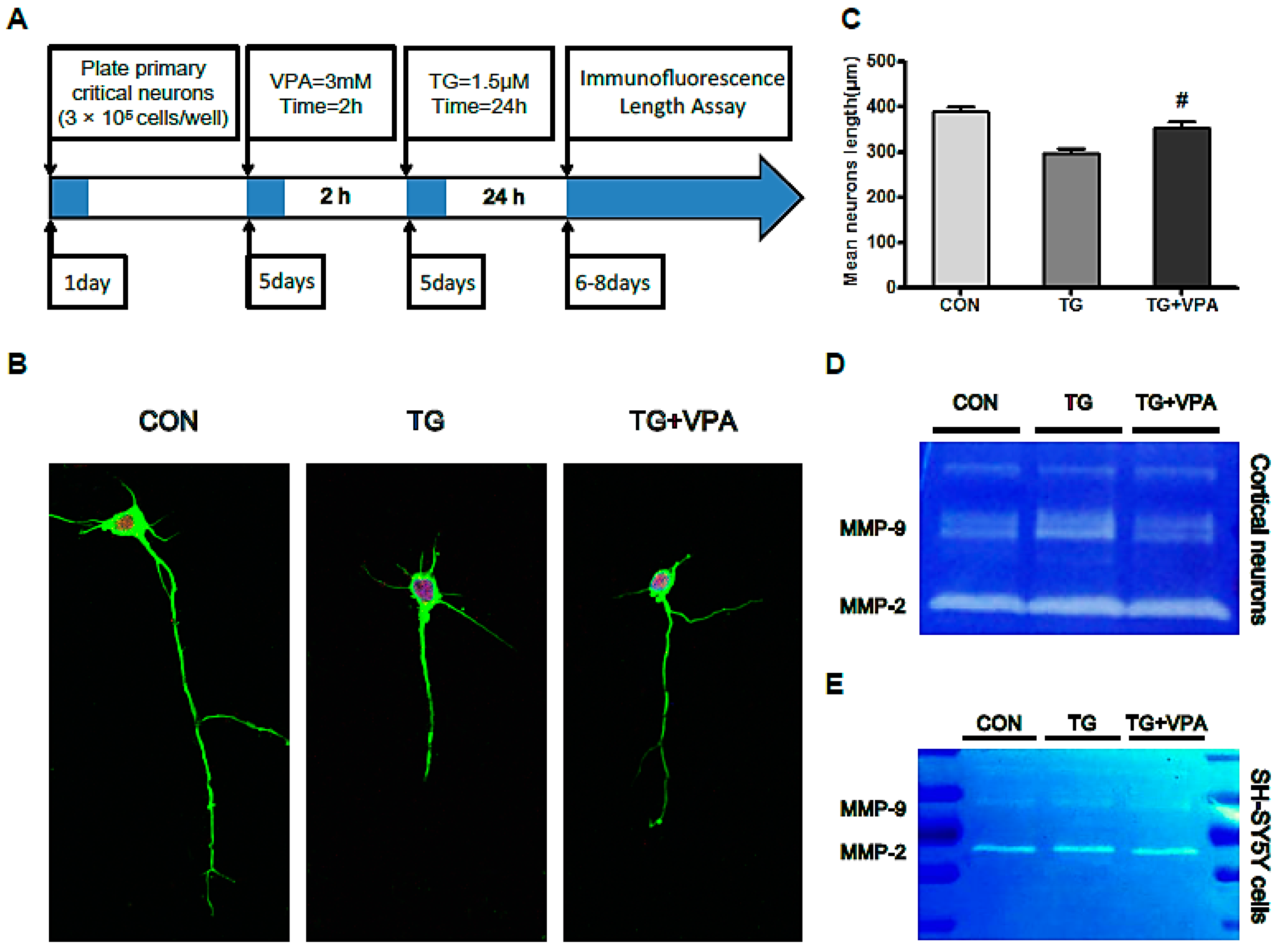
2.7. VPA Contributes to Axonal Maintenance in Primary Critical Neurons Treated with TG
3. Discussion
4. Materials and Methods
4.1. Primary Cortical Neurons Culture
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Cell Apoptosis Assay
4.5. Western Blot Analysis
4.6. Immunofluorescence Assay
4.7. Gelatin Zymography
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fu, S.; Watkins, S.M.; Hotamisligil, G.S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012, 15, 623–634. [Google Scholar] [CrossRef] [PubMed]
- De Gracia, D.J.; Montie, H.L. Cerebral ischemia and the unfolded protein response. J. Neurochem. 2004, 91, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hasnain, S.Z.; Prins, J.B.; McGuckin, M.A. Oxidative and endoplasmic reticulum stress in β-cell dysfunction in diabetes. J. Mol. Endocrinol. 2016, 56, R33–R54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, F.; Kong, X.; Yang, J.; Chen, H.; Deng, L.; Cheng, Y.; Ye, L.; Zhu, S.; Zhang, X.; et al. Nerve growth factor improves functional recovery by inhibiting endoplasmic reticulum stress-induced neuronal apoptosis in rats with spinal cord injury. J. Transl. Med. 2014, 12, 130. [Google Scholar] [CrossRef] [PubMed]
- Rivas, A.; Vidal, R.L.; Hetz, C. Targeting the unfolded protein response for disease intervention. Expert Opin. Ther. Targets 2015, 19, 1203–1218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Wang, Z.G.; Lu, X.H.; Kong, X.X.; Wu, F.Z.; Lin, L.; Tan, X.; Ye, L.B.; Xiao, J. Endoplasmic reticulum stress: Relevance and therapeutics in central nervous system diseases. Mol. Neurobiol. 2015, 51, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Selimovic, D.; Hannig, M.; Haikel, Y.; Brodell, R.T.; Megahed, M. Endoplasmic reticulum stress-mediated pathways to both apoptosis and autophagy: Significance for melanoma treatment. World J. Exp. Med. 2015, 5, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, X.; Fang, F.; Jiang, D.; Tang, L. Down-regulation of GRP78 enhances apoptosis via CHOP pathway in retinal ischemia-reperfusion injury. Neurosci. Lett. 2014, 575, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes Dev. 1999, 13, 1211–1233. [Google Scholar] [CrossRef] [PubMed]
- Teske, B.F.; Wek, S.A.; Bunpo, P.; Cundiff, J.K.; McClintick, J.N.; Anthony, T.G.; Wek, R.C. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol. Biol. Cell 2011, 22, 4390–4405. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.M.; Yao, F.H.; Yao, Y.M.; Dong, N.; Yu, Y.; Sheng, Z.Y. Endoplasmic reticulum stress and its regulator XBP-1 contributes to dendritic cell maturation and activation induced by high mobility group BOX-1 protein. Int. J. Biochem. Cell Biol. 2012, 44, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yuan, X.; Hu, C.; Zhang, B.; Gao, H.; Wang, D.; Chi, J.; Jing, Q.; Wu, S.; Wu, C.L. Endoplasmic reticulum stress is involved in apoptosis of detrusor muscle in streptozocin-induced diabetic rats. Neurourol. Urodyn 2015. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Zhou, L.; Han, F.; Han, J.; Zhang, Y.; Sun, Z.; Zhao, W.; Wang, Z.; Zheng, L. β-Adrenoceptor activation by norepinephrine enhances lipopolysaccharide-induced matrix metalloproteinase-9 expression through the ERK/JNK-c-Fos pathway in human THP-1 cells. J. Atheroscler. Thromb. 2016. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.A.; Ubeda, A.; Moreno, J.; Trillo, M.A. Power frequency magnetic fields affect the p38 MAPK-mediated regulation of NB69 cell proliferation implication of free radicals. Int. J. Mol. Sci. 2016, 17, 510. [Google Scholar] [CrossRef] [PubMed]
- Tomicic, M.T.; Meise, R.; Aasland, D.; Berte, N.; Kitzinger, R.; Kramer, O.H.; Kaina, B.; Christmann, M. Apoptosis induced by temozolomide and nimustine in glioblastoma cells is supported by JNK/c-Jun-mediated induction of the BH3-only protein BIM. Oncotarget 2015, 6, 33755–33768. [Google Scholar] [PubMed]
- Zhu, H.; Zhang, Y.; Shi, Z.; Lu, D.; Li, T.; Ding, Y.; Ruan, Y.; Xu, A. The neuroprotection of liraglutide against ischaemia-induced apoptosis through the activation of the PI3K/AKT and MAPK pathways. Sci. Rep. 2016, 6, 26859. [Google Scholar] [CrossRef] [PubMed]
- Naganska, E.; Matyja, E.; Taraszewska, A.; Rafalowska, J. Protective effect of valproic acid on cultured motor neurons under glutamate excitotoxic conditions: Ultrastructural study. Folia Neuropathol. 2015, 53, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Penas, C.; Verdu, E.; Asensio-Pinilla, E.; Guzman-Lenis, M.S.; Herrando-Grabulosa, M.; Navarro, X.; Casas, C. Valproate reduces chop levels and preserves oligodendrocytes and axons after spinal cord injury. Neuroscience 2011, 178, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Ximenes, J.C.; Neves, K.R.; Leal, L.K.; do Carmo, M.R.; Brito, G.A.; Naffah-Mazzacoratti Mda, G.; Cavalheiro, E.A.; Viana, G.S. Valproic acid neuroprotection in the 6-OHDA model of Parkinson’s disease is possibly related to its anti-inflammatory and HDAC inhibitory properties. J. Neurodegener. Dis. 2015, 2015, 313702. [Google Scholar] [PubMed]
- Zhang, C.; Zhu, J.; Zhang, J.; Li, H.; Zhao, Z.; Liao, Y.; Wang, X.; Su, J.; Sang, S.; Yuan, X.; et al. Neuroprotective and anti-apoptotic effects of valproic acid on adult rat cerebral cortex through ERK and AKT signaling pathway at acute phase of traumatic brain injury. Brain Res. 2014, 1555, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kaliszczak, M.; Trousil, S.; Ali, T.; Aboagye, E.O. Akt activation controls cell survival in response to HDAC6 inhibition. Cell Death Dis. 2016, 7, e2286. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Hou, C.H.; Lin, F.L.; Tsao, Y.T.; Hou, S.M. Nimbolide induces ROS-regulated apoptosis and inhibits cell migration in osteosarcoma. Int. J. Mol. Sci. 2015, 16, 23405–23424. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Li, J.; Wang, K.; Hao, X.; Ge, R.; Li, Q. Isoquercitrin inhibits hydrogen peroxide-induced apoptosis of EA. hy926 cells via the PI3K/AKT/GSK3β signaling pathway. Molecules 2016, 21, 356. [Google Scholar] [CrossRef] [PubMed]
- Shigemi, Z.; Baba, Y.; Hara, N.; Matsuhiro, J.; Kagawa, H.; Watanabe, T.; Fujimuro, M. Effects of ER stress on unfolded protein responses, cell survival, and viral replication in primary effusion lymphoma. Biochem. Biophys. Res. Commun. 2016, 469, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Parra, V.; Moraga, F.; Kuzmicic, J.; Lopez-Crisosto, C.; Troncoso, R.; Torrealba, N.; Criollo, A.; Diaz-Elizondo, J.; Rothermel, B.A.; Quest, A.F.; et al. Calcium and mitochondrial metabolism in ceramide-induced cardiomyocyte death. Biochim. Biophys. Acta 2013, 1832, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, T.; Tang, S.; Zhao, D.; Zhang, C.; Zhang, S.; Deng, S.; Zhou, Y.; Xiao, X. Thapsigargin induces apoptosis when autophagy is inhibited in HepG2 cells and both processes are regulated by ROS-dependent pathway. Environ. Toxicol. Pharmacol. 2016, 41, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, A.; Navarria, L.; Zaltieri, M.; Falarti, E.; Bodei, S.; Sigala, S.; Battistin, L.; Spillantini, M.; Missale, C.; Spano, P. Induction of the unfolded protein response by α-synuclein in experimental models of Parkinson’s disease. J. Neurochem. 2011, 116, 588–605. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Won, S.M.; Suh, J.; Son, S.J.; Moon, G.J.; Park, U.J.; Gwag, B.J. Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Exp. Mol. Med. 2010, 42, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Kieran, D.; Woods, I.; Villunger, A.; Strasser, A.; Prehn, J.H. Deletion of the BH3-only protein puma protects motoneurons from ER stress-induced apoptosis and delays motoneuron loss in ALS mice. Proc. Natl. Acad. Sci. USA 2007, 104, 20606–20611. [Google Scholar] [CrossRef] [PubMed]
- Osada, N.; Kosuge, Y.; Ishige, K.; Ito, Y. Characterization of neuronal and astroglial responses to ER stress in the hippocampal CA1 area in mice following transient forebrain ischemia. Neurochem. Int. 2010, 57, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Kasahara, T.; Kametani, M.; Toyota, T.; Yoshikawa, T.; Kato, T. Aberrant endoplasmic reticulum stress response in lymphoblastoid cells from patients with bipolar disorder. Int. J. Neuropsychopharmacol. 2009, 12, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Pfaffenseller, B.; Wollenhaupt-Aguiar, B.; Fries, G.R.; Colpo, G.D.; Burque, R.K.; Bristot, G.; Ferrari, P.; Cereser, K.M.; Rosa, A.R.; Klamt, F.; et al. Impaired endoplasmic reticulum stress response in bipolar disorder: Cellular evidence of illness progression. Int. J. Neuropsychopharmacol. 2014, 17, 1453–1463. [Google Scholar] [CrossRef] [PubMed]
- Thotala, D.; Karvas, R.M.; Engelbach, J.A.; Garbow, J.R.; Hallahan, A.N.; DeWees, T.A.; Laszlo, A.; Hallahan, D.E. Valproic acid enhances the efficacy of radiation therapy by protecting normal hippocampal neurons and sensitizing malignant glioblastoma cells. Oncotarget 2015, 6, 35004–35022. [Google Scholar] [PubMed]
- Song, B.S.; Yoon, S.B.; Sim, B.W.; Kim, Y.H.; Cha, J.J.; Choi, S.A.; Jeong, K.J.; Kim, J.S.; Huh, J.W.; Lee, S.R.; et al. Valproic acid enhances early development of bovine somatic cell nuclear transfer embryos by alleviating endoplasmic reticulum stress. Reprod. Fertil. Dev 2014, 26, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Dudley, S.C., Jr. Role for the unfolded protein response in heart disease and cardiac arrhythmias. Int. J. Mol. Sci. 2016, 17, 52. [Google Scholar] [CrossRef] [PubMed]
- Hardy, B.; Raiter, A. Peptide-binding heat shock protein GRP78 protects cardiomyocytes from hypoxia-induced apoptosis. J. Mol. Med. 2010, 88, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wei, Y.; Qiu, S.; Xu, Y.; Zhang, T.; Zhang, S. Propofol decreases endoplasmic reticulum stress-mediated apoptosis in retinal pigment epithelial cells. PLoS ONE 2016, 11, e0157590. [Google Scholar] [CrossRef] [PubMed]
- Esteves, S.; Duarte-Silva, S.; Naia, L.; Neves-Carvalho, A.; Teixeira-Castro, A.; Rego, A.C.; Silva-Fernandes, A.; Maciel, P. Limited effect of chronic valproic acid treatment in a mouse model of machado-joseph disease. PLoS ONE 2015, 10, e0141610. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Zhang, C.; Chai, M.; An, Y. Isoquercetin ameliorates tunicamycin-induced apoptosis in rat dorsal root ganglion neurons via suppressing ROS-dependent endoplasmic reticulum stress. Biomed. Pharmacother. 2016, 80, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Maeng, S.; Kang, S.R.; Choi, H.Y.; Oh, T.H.; Ju, B.G.; Yune, T.Y. Valproic acid protects motor neuron death by inhibiting oxidative stress and endoplasmic reticulum stress-mediated cytochrome c release after spinal cord injury. J. Neurotrauma 2014, 31, 582–594. [Google Scholar] [CrossRef] [PubMed]
- Rouaux, C.; Panteleeva, I.; Rene, F.; Gonzalez de Aguilar, J.L.; Echaniz-Laguna, A.; Dupuis, L.; Menger, Y.; Boutillier, A.L.; Loeffler, J.P. Sodium valproate exerts neuroprotective effects in vivo through creb-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. J. Neurosci. 2007, 27, 5535–5545. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Dong, Y.; Zhao, J.; Yin, Y.; Zheng, Y. The CLC-2 chloride channel modulates ECM synthesis, differentiation, and migration of human conjunctival fibroblasts via the PI3K/AKT signaling pathway. Int. J. Mol. Sci. 2016, 17, 910. [Google Scholar] [CrossRef] [PubMed]
- Golpich, M.; Amini, E.; Hemmati, F.; Ibrahim, N.M.; Rahmani, B.; Mohamed, Z.; Raymond, A.A.; Dargahi, L.; Ghasemi, R.; Ahmadiani, A. Glycogen synthase kinase-3 β (GSK-3β) signaling: Implications for Parkinson’s disease. Pharmacol. Res. 2015, 97, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.; Zhou, H.; Lu, L.; Kong, X.; Wang, T.; Pan, B.; Feng, S. Valproic acid-mediated neuroprotection and neurogenesis after spinal cord injury: From mechanism to clinical potential. Regen. Med. 2015, 10, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Ying, G.Y.; Jing, C.H.; Li, J.R.; Wu, C.; Yan, F.; Chen, J.Y.; Wang, L.; Dixon, B.J.; Chen, G. Neuroprotective effects of valproic acid on blood-brain barrier disruption and apoptosis-related early brain injury in rats subjected to subarachnoid hemorrhage are modulated by heaet shock protein 70/matrix metalloproteinases and heat shock protein 70/AKT pathways. Neurosurgery 2016. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, W.; Pei, F.; Ma, W.; Wang, Y. Downregulation of matrix metalloproteinases contributes to the inhibition of cell migration and invasion in HepG2 cells by sodium valproate. Oncol. Lett. 2015, 10, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.N.; Kim, M.K.; Cho, K.S.; Choi, C.S.; Park, S.H.; Yang, S.I.; Joo, S.H.; Park, J.H.; Bahn, G.; Shin, C.Y.; et al. Valproic acid regulates α-synuclein expression through JNK pathway in rat primary astrocytes. Biomol. Ther. 2013, 21, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Bhatia, H.S.; Kumar, A.; de Oliveira, A.C.; Fiebich, B.L. Histone deacetylase inhibitors valproic acid and sodium butyrate enhance prostaglandins release in lipopolysaccharide-activated primary microglia. Neuroscience 2014, 265, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Carriere, C.H.; Kang, N.H.; Niles, L.P. Neuroprotection by valproic acid in an intrastriatal rotenone model of parkinson’s disease. Neuroscience 2014, 267, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qin, X.; Tong, N.; Zhao, X.; Gong, Y.; Shi, Y.; Wu, X. Valproic acid-mediated neuroprotection in retinal ischemia injury via histone deacetylase inhibition and transcriptional activation. Exp. Eye Res. 2012, 94, 98–108. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Wu, F.; Zhang, X.; Chai, Y.; Chen, D.; Yang, Y.; Xu, K.; Yin, J.; Li, R.; Shi, H.; et al. Valproate Attenuates Endoplasmic Reticulum Stress-Induced Apoptosis in SH-SY5Y Cells via the AKT/GSK3β Signaling Pathway. Int. J. Mol. Sci. 2017, 18, 315. https://doi.org/10.3390/ijms18020315
Li Z, Wu F, Zhang X, Chai Y, Chen D, Yang Y, Xu K, Yin J, Li R, Shi H, et al. Valproate Attenuates Endoplasmic Reticulum Stress-Induced Apoptosis in SH-SY5Y Cells via the AKT/GSK3β Signaling Pathway. International Journal of Molecular Sciences. 2017; 18(2):315. https://doi.org/10.3390/ijms18020315
Chicago/Turabian StyleLi, Zhengmao, Fenzan Wu, Xie Zhang, Yi Chai, Daqing Chen, Yuetao Yang, Kebin Xu, Jiayu Yin, Rui Li, Hongxue Shi, and et al. 2017. "Valproate Attenuates Endoplasmic Reticulum Stress-Induced Apoptosis in SH-SY5Y Cells via the AKT/GSK3β Signaling Pathway" International Journal of Molecular Sciences 18, no. 2: 315. https://doi.org/10.3390/ijms18020315
APA StyleLi, Z., Wu, F., Zhang, X., Chai, Y., Chen, D., Yang, Y., Xu, K., Yin, J., Li, R., Shi, H., Wang, Z., Li, X., Xiao, J., & Zhang, H. (2017). Valproate Attenuates Endoplasmic Reticulum Stress-Induced Apoptosis in SH-SY5Y Cells via the AKT/GSK3β Signaling Pathway. International Journal of Molecular Sciences, 18(2), 315. https://doi.org/10.3390/ijms18020315