Rebalancing β-Amyloid-Induced Decrease of ATP Level by Amorphous Nano/Micro Polyphosphate: Suppression of the Neurotoxic Effect of Amyloid β-Protein Fragment 25-35



 , , ,
, , , 

Abstract
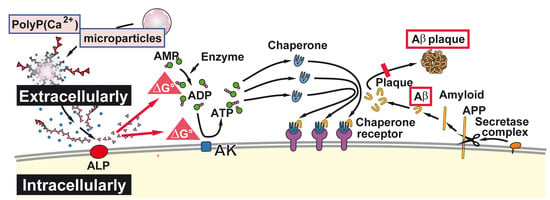
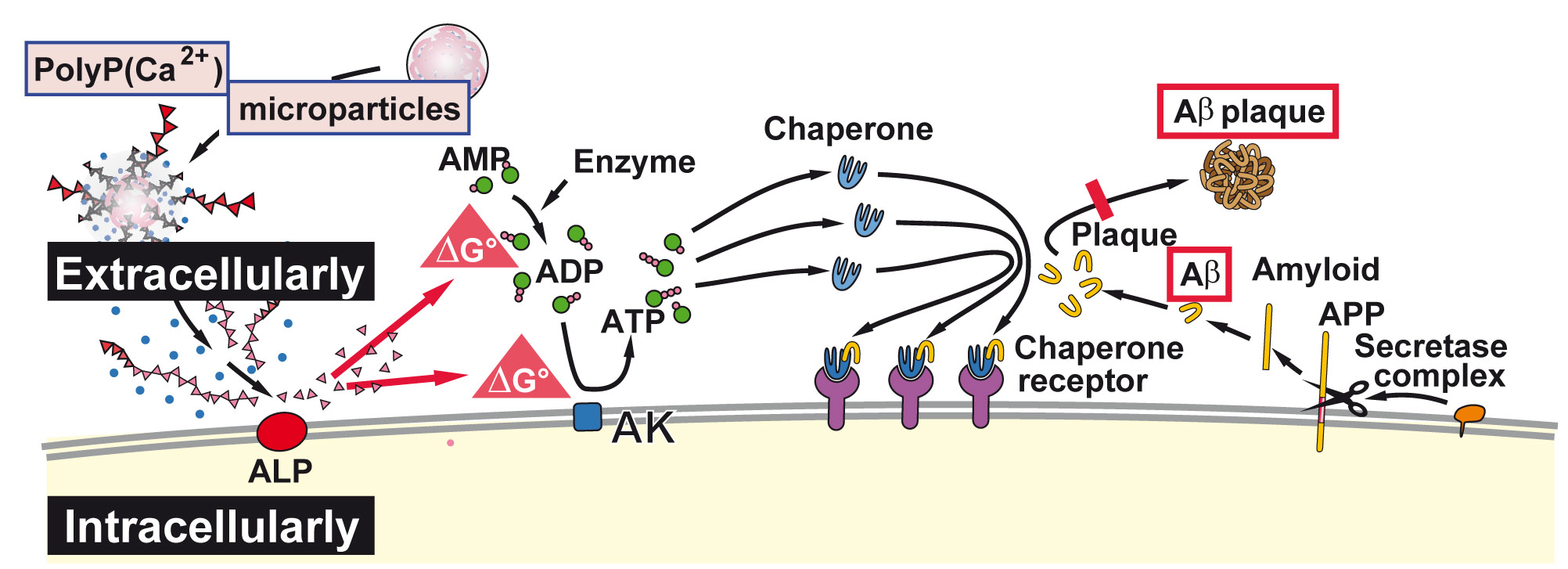{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Fabrication and Morphology of the Particles
2.2. Characterization by Fourier Transform Infrared and X-ray Diffraction
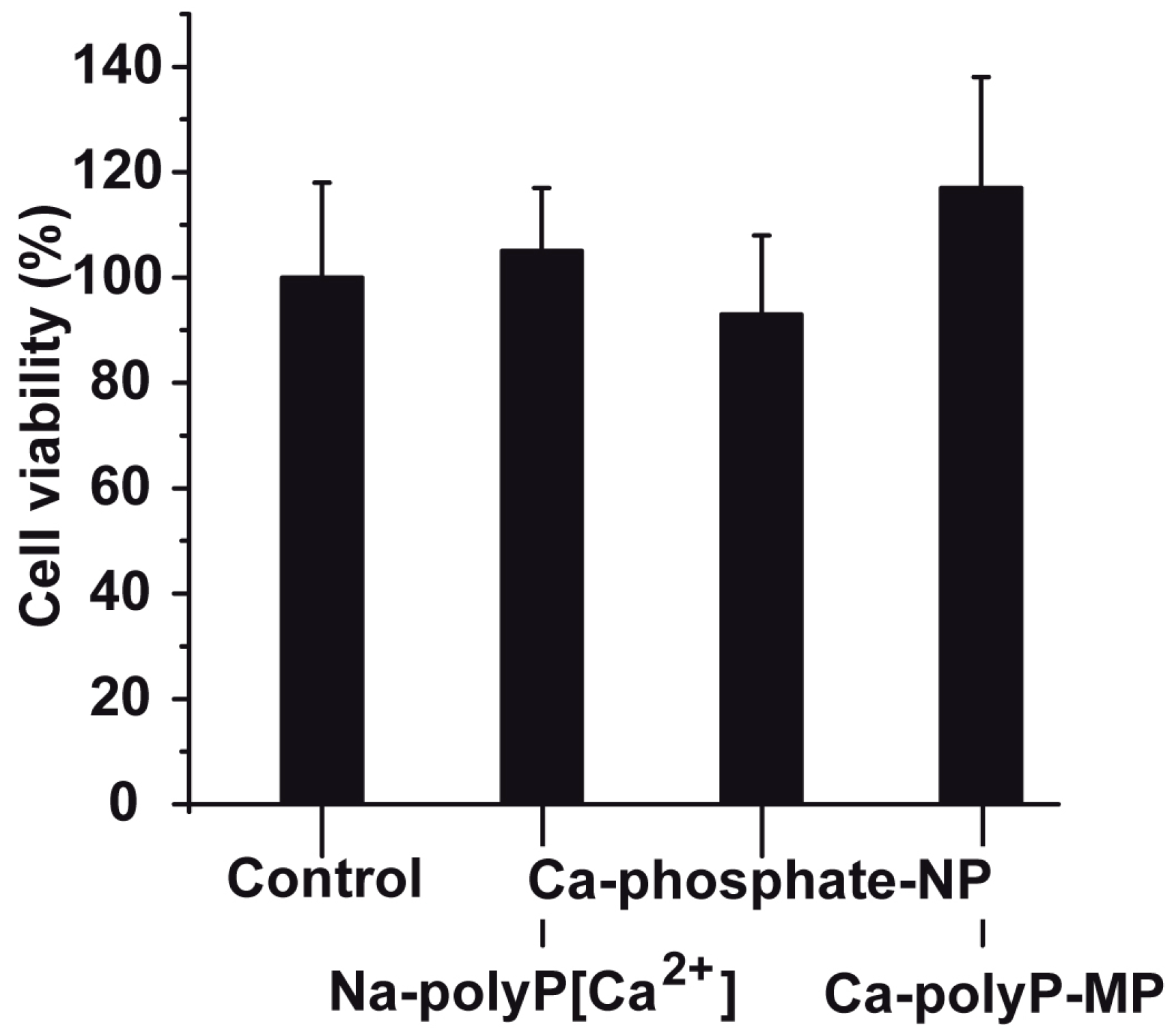
2.3. Cell viability after Exposure to Phosphate or polyP Preparations
2.4. Induced Toxicity by the Amyloid β-Protein Peptide (Time-Dependent Pre-Incubation of Aβ25-35 in Water)
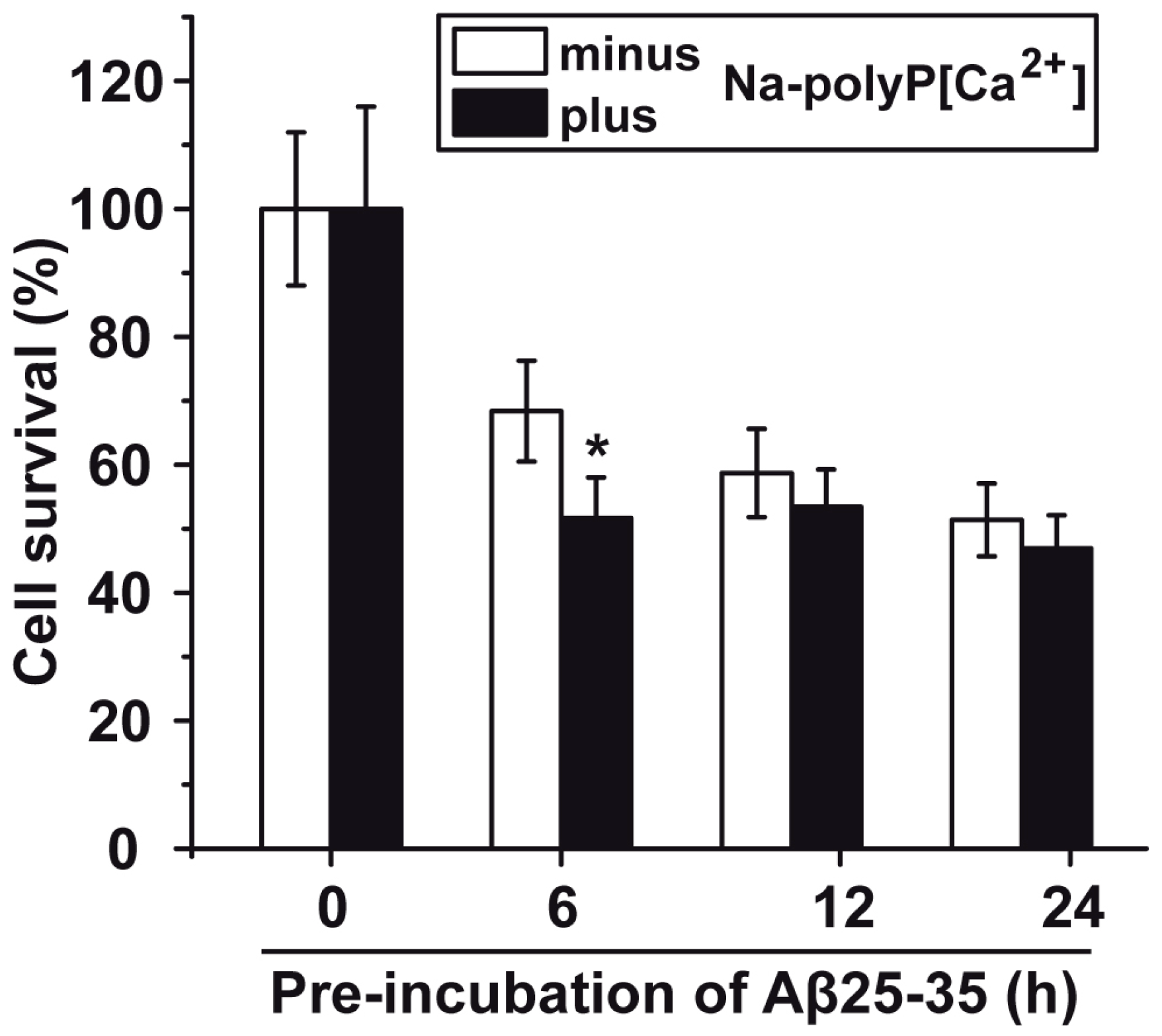
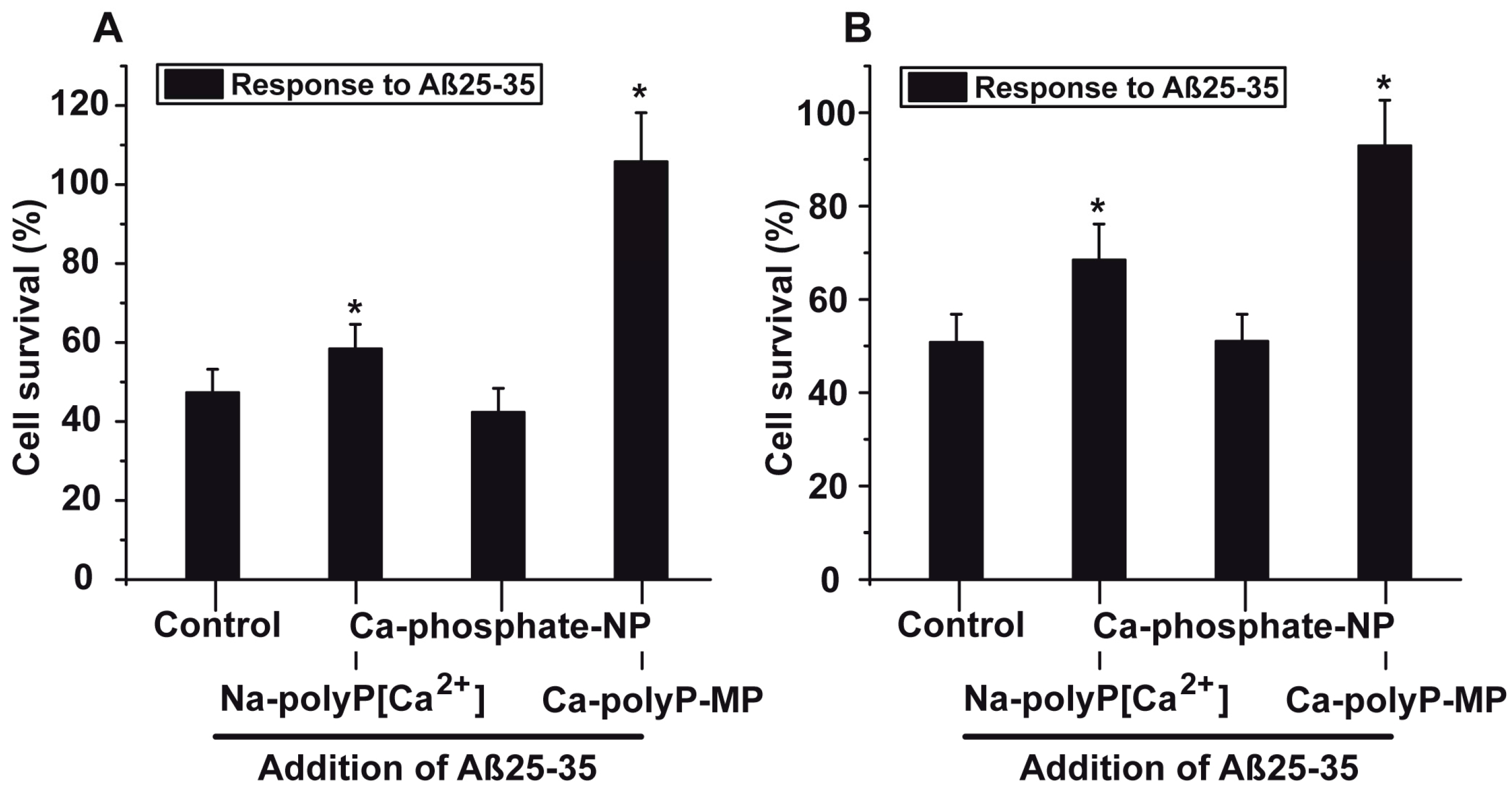
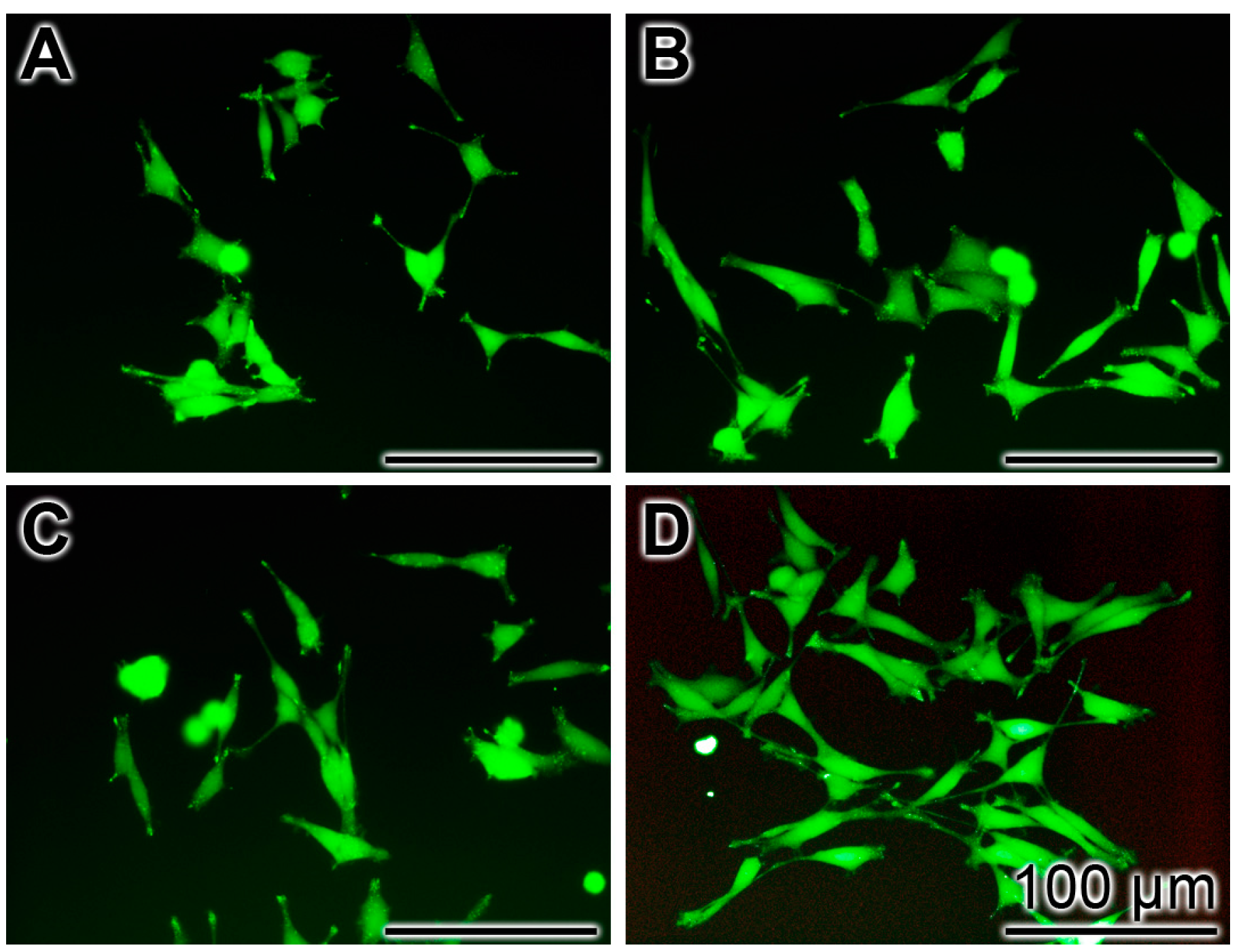
2.5. Protection against Aβ25-35-Caused Toxicity by polyP
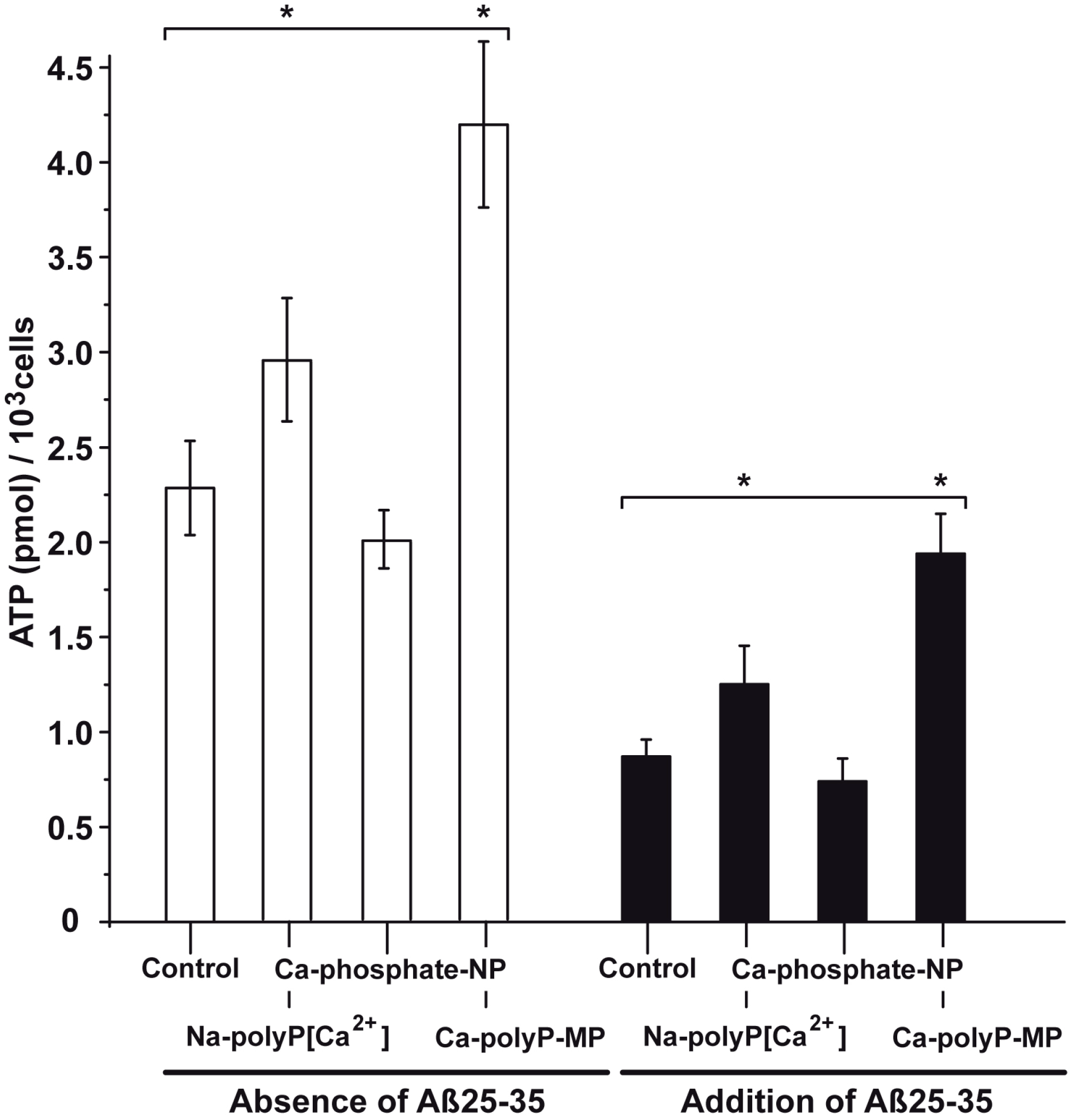
2.6. Modulation of the Intracellular ATP Pool in Cells in the Absence or Presence of Aβ25-35 and Phosphate/polyP
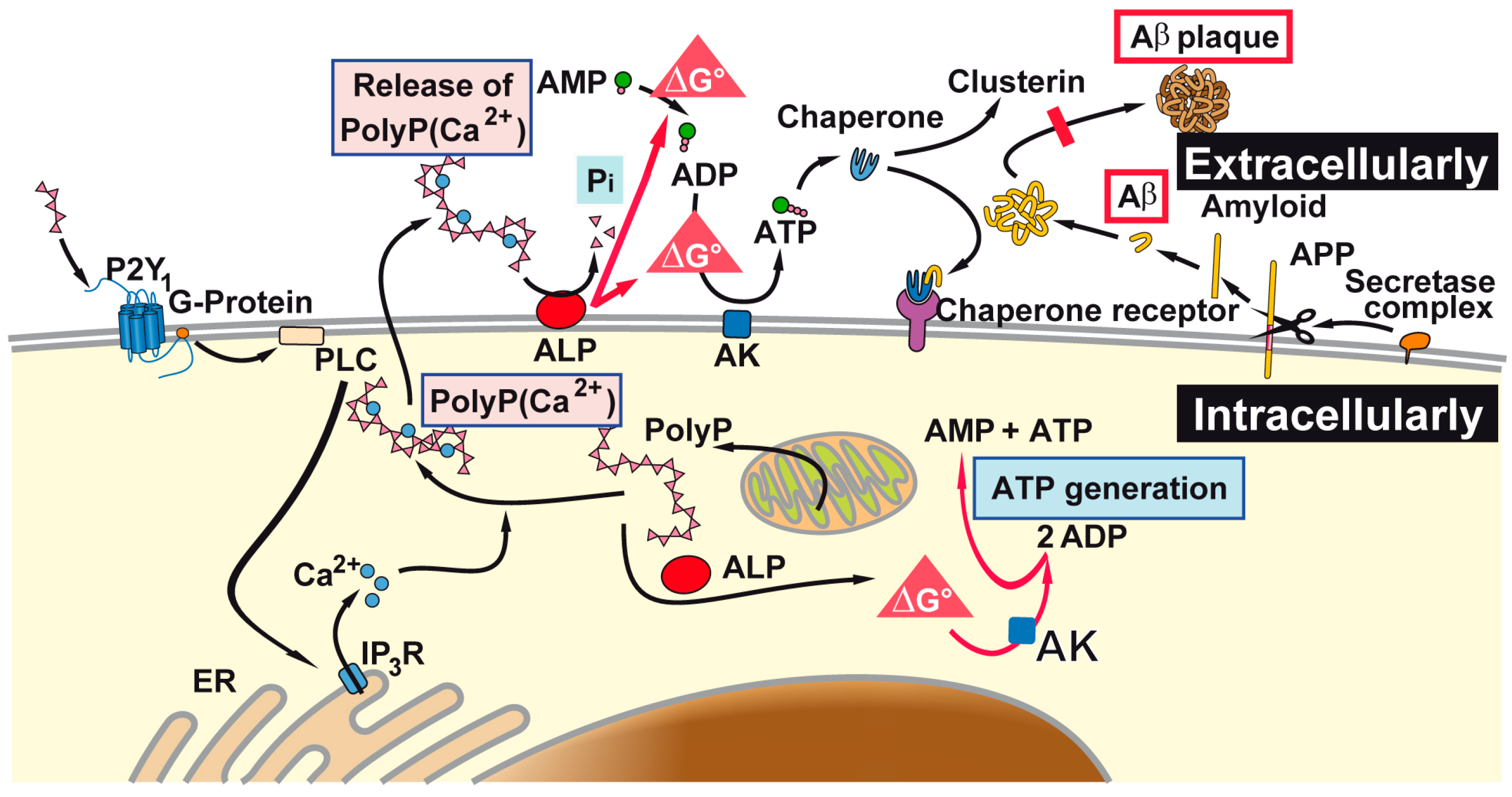
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Phosphate/Polyphosphate Sample Fabrication
4.3. Fourier Transformed Infrared Spectroscopy and X-ray Diffraction
4.4. Microscopy
4.5. PC12 Cells
4.6. Cell Viability Assays
4.7. Aβ-Induced Cell Toxicity
4.8. Primary Culture of Cortical Neurons
4.9. Determination of the ATP Level in PC12 Cells
4.10. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Aβ | Amyloid β-peptides |
| Aβ25-35 | amyloid β-protein fragment 25-35 |
| ADP | adenosine diphosphate |
| AMP | adenosine monophosphate |
| ATP | adenosine triphosphate |
| Ca-polyP-MP | Ca-polyP microparticles |
| DMSO | dimethyl sulfoxide |
| ELISA | enzyme-linked immunosorbent assay |
| MTT | 3-[4,5-dimethyl thiazole-2S-yl]-2,5-diphenyl tetrazolium |
| Na-polyP | Sodium polyphosphate |
| Na-polyP[Ca2+] | Na+ salt of polyP complexed in a stoichiometric ratio to Ca2+ |
| PC12 cells | Pheochromocytoma cells |
| polyP | Polyphosphate |
| SEM | Scanning electron microscopy |
| TRPA1 | Transient receptor potential cation channel A/1 |
| TRPM8 | Cation channel subfamily M/8 |
References
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Alzheimer, A. Über eine eigenartige Erkrankung der Hirnrinde. Allg. Zschr. Psychiatr. Psych. Gerichtl. Med. 1907, 64, 146–148. [Google Scholar]
- Wolf, A.; Bauer, B.; Hartz, A.M. ABC transporters and the Alzheimer’s disease enigma. Front. Psychiatry 2012, 3, 54. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Chase, T.N. High-dose cholinesterase inhibitor treatment of Alzheimer’s disease. Alzheimers. Dement. 2015, 11, S466–S467. [Google Scholar] [CrossRef]
- Informed Health Online. Alzheimer’s Disease: Does Memantine Help? Available online: https://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0072540/ (accessed on 2 August 2016).
- Rytik, P.G.; Eremin, V.F.; Kvacheva, Z.B.; Poleschuk, N.N.; Popov, A.S.; Schröder, H.C.; Weiler, B.E.; Bachmann, M.; Müller, W.E.G. Susceptibility of human astrocytes to human immunodeficiency virus infection in vitro; Anti-HIV activity of memantine. AIDS Res. Hum. Retrov. 1991, 7, 89–95. [Google Scholar]
- Müller, W.E.G.; Schröder, H.C.; Ushijima, H.; Dapper, J.; Bormann, J. Gp120 of HIV-1 induces apoptosis in rat cortical cell cultures: Prevention by memantine. Eur. J. Pharmacol. 1992, 226, 209–214. [Google Scholar] [CrossRef]
- Khatri, N.; Man, H.Y. Synaptic activity and bioenergy homeostasis: Implications in brain trauma and neurodegenerative diseases. Front. Neurol. 2013, 4, 199. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Yang, G.J.; Jin, L.; Yang, H.M.; Chen, J.; Chai, G.S.; Wang, L. Erythropoietin attenuates Alzheimer-like memory impairments and pathological changes induced by amyloid β42 in mice. Brain Res. 2015, 1618, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, B.; Schröder, H.C. Mammalian intestinal alkaline phosphatase acts as highly active exopolyphosphatase. Biochim. Biophys. Acta 2001, 1547, 254–261. [Google Scholar] [CrossRef]
- Lorenz, B.; Münkner, J.; Oliveira, M.P.; Kuusksalu, A.; Leitão, J.M.; Müller, W.E.G.; Schröder, H.C. Changes in metabolism of inorganic polyphosphate in rat tissues and human cells during development and apoptosis. Biochim. Biophys. Acta 1997, 1335, 51–60. [Google Scholar] [CrossRef]
- Morrissey, J.H.; Choi, S.H.; Smith, S.A. Polyphosphate, An ancient molecule that links platelets, coagulation, and inflammation. Blood 2012, 119, 5972–5979. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Marina, N.; Baev, A.Y.; Wood, N.W.; Gourine, A.V.; Abramov, A.Y. Signalling properties of inorganic polyphosphate in the mammalian brain. Nat. Commun. 2013, 4, 1362. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.E.G.; Tolba, E.; Schröder, H.C.; Wang, X.H. Polyphosphate: A morphogenetically active implant material serving as metabolic fuel for bone regeneration. Macromol. Biosci. 2015, 15, 1182–1197. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Schröder, H.C.; Müller, W.E.G. Polyphosphate as a metabolic fuel in Metazoa: A foundational breakthrough invention for biomedical applications. Biotechnol. J. 2016, 11, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Cremers, C.M.; Knoefler, D.; Gates, S.; Martin, N.; Dahl, J.U.; Lempart, J.; Xie, L.; Chapman, M.R.; Galvan, V.; Southworth, D.R.; et al. Polyphosphate: A Conserved modifier of amyloidogenic processes. Mol. Cell 2016, 63, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Fraley, C.; Diao, C.T.; Winkfein, R.; Colicos, M.A.; Duchen, M.R.; French, R.J.; Pavlov, E. Targeted polyphosphatase expression alters mitochondrial metabolism and inhibits calcium-dependent cell death. Proc. Natl. Acad. Sci. USA 2007, 104, 18091–18096. [Google Scholar] [CrossRef] [PubMed]
- Masilamoni, J.G.; Jesudason, E.P.; Jesudoss, K.S.; Murali, J.; Paul, S.F.D.; Yayakumar, R. Role of fibrillar Aβ25-35 in the inflammation induced rat model with respect to oxidative vulnerability. Free Rad. Res. 2005, 6, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.H.; Lee, J.B.; Shih, Y.C.; Wan, L.; Shieh, F.K.; Chen, C.Y. Location and conformation of amyloid β(25-35) peptide and its sequence-shuffled peptides within membranes: Implications for aggregation and toxicity in PC12 cells. ChemMedChem 2014, 9, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Xu, J.; Zheng, W. Artemisinin protects PC12 cells against β-amyloid-induced apoptosis through activation of the ERK1/2 signaling pathway. Redox Biol. 2017, 12, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.E.G.; Romero, F.J.; Perović, S.; Pergande, G.; Pialoglou, P. Protection of flupirtine on ß-amyloid-induced apoptosis in neuronal cells in vitro. Prevention of amyloid-induced glutathione depletion. J. Neurochem. 1997, 68, 2371–2377. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.E.G.; Pialoglou, P.; Romero, F.J.; Perovic, S.; Pergande, G. Protective effect of the drug flupirtine on β-amyloid-induced apoptosis in primary neuronal cells in vitro. J. Brain Res. 1996, 37, 575–577. [Google Scholar] [CrossRef]
- Dinarvand, P.; Hassanian, S.M.; Qureshi, S.H.; Manithody, C.; Eissenberg, J.C.; Yang, L.; Rezaie, A.R. Polyphosphate amplifies proinflammatory responses of nuclear proteins through interaction with receptor for advanced glycation end products and P2Y1 purinergic receptor. Blood 2014, 123, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, M.M.; Gamelas, J.A.F.; Martins, A.G. Characterization of Bone and Bone-Based Graft Materials Using FTIR Spectroscopy. In Infrared Spectroscopy—Life and Biomedical Sciences; Theophanides, T., Ed.; InTech: Rijeka, Croatia, 2012; pp. 315–338. Available online: https://www.intechopen.com/books/infrared-spectroscopy-life-and-biomedical-sciences/characterization-of-bone-and-bone-based-graft-materials-using-ftir-spectroscopy (accessed on 1 June 2017). [CrossRef]
- Khoshmanesh, A.; Cook, P.L.; Wood, B.R. Quantitative determination of polyphosphate in sediments using Attenuated Total Reflectance-Fourier Transform Infrared (ATR-FTIR) spectroscopy and partial least squares regression. Analyst 2012, 137, 3704–3709. [Google Scholar] [CrossRef] [PubMed]
- Landi, E. Carbonated hydroxyapatite as bone substitute. J. Eur. Ceram. Soc. 2003, 23, 2931–2937. [Google Scholar] [CrossRef]
- Fleet, M.E. Infrared spectra of carbonate apatites: ν2-region bands. Biomaterials 2009, 30, 1473–1481. [Google Scholar] [CrossRef] [PubMed]
- Mohmmad Abdul, H.; Butterfield, D.A. Protection against amyloid beta-peptide (1-42)-induced loss of phospholipid asymmetry in synaptosomal membranes by tricyclodecan-9-xanthogenate (D609) and ferulic acid ethyl ester: Implications for Alzheimer’s disease. Biochim. Biophys. Acta 2005, 1741, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Swomley, A.M.; Sultana, R. Amyloid β-peptide (1-42)-induced oxidative stress in Alzheimer disease: Importance in disease pathogenesis and progression. Antioxid. Redox Signal. 2013, 19, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Babić, L.M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Tau protein hyperphosphorylation and aggregation in Alzheimer’s Disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Leroy, K.; Ando, K.; Laporte, V.; Dedecker, R.; Suain, V.; Authelet, M.; Héraud, C.; Pierrot, N.; Yilmaz, Z.; Octave, J.N.; et al. Lack of tau proteins rescues neuronal cell death and decreases amyloidogenic processing of APP in APP/PS1 mice. Am. J. Pathol. 2012, 181, 1928–1940. [Google Scholar] [CrossRef] [PubMed]
- Behl, C. Apoptosis and Alzheimer’s disease. J. Neural. Transm. 2000, 107, 1325–1344. [Google Scholar] [CrossRef] [PubMed]
- Niikura, T.; Tajima, H.; Kita, Y. Neuronal cell death in Alzheimer’s disease and a neuroprotective factor, humanin. Curr. Neuropharmacol. 2006, 4, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Aisen, P.S.; DuBois, B.; Frölich, L.; Jack, C.R., Jr.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug development in Alzheimer’s disease: The path to 2025. Alzheimer’s Res. Ther. 2016, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Perović, S.; Schleger, C.; Pergande, G.; Iskric, S.; Ushijima, H.; Rytik, P.; Müller, W.E.G. The triaminopyridine Flupirtine prevents cell death in rat cortical cells induced by N-methyl-d-aspartate and gp120 of HIV-1. Eur. J. Pharmacol. 1994, 288, 27–33. [Google Scholar] [CrossRef]
- Kornhuber, J.; Bleich, S.; Wiltfang, J.; Maler, M.; Parsons, C.G. Flupirtine shows functional NMDA receptor antagonism by enhancing Mg2+ block via activation of voltage independent potassium channels. Rapid communication. J. Neural. Transm. 1999, 106, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A. The molecular basis of memantine action in Alzheimer’s disease and other neurologic disorders: Low-affinity, uncompetitive antagonism. Curr. Alzheimer Res. 2005, 2, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Dickey, C.A.; Kamal, A.; Lundgren, K.; Klosak, N.; Bailey, R.M.; Dunmore, J.; Ash, P.; Shoraka, S.; Zlatkovic, J.; Eckman, C.B.; et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Investig. 2007, 117, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cao, Z.; Klein, W.L.; Luo, Y. Heat shock treatment reduces beta amyloid toxicity in vivo by diminishing oligomers. Neurobiol. Aging 2010, 31, 1055–1058. [Google Scholar] [CrossRef] [PubMed]
- Mallouk, Y.; Vayssier-Taussat, M.; Bonventre, J.V.; Polla, B.S. Heat shock protein 70 and ATP as partners in cell homeostasis (Review). Int. J. Mol. Med. 1999, 4, 463–474. [Google Scholar] [CrossRef] [PubMed]
- De Maio, A.; Vazquez, D. Extracellular heat shock proteins: A new location, a new function. Shock 2013, 40, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Yalak, G.; Ehrlich, Y.H.; Olsen, B.R. Ecto-protein kinases and phosphatases: An emerging field for translational medicine. J. Transl. Med. 2014, 12, 165. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.E.G.; Tolba, E.; Schröder, H.C.; Wang, S.; Glaßer, G.; Muñoz-Espí, R.; Link, T.; Wang, X.H. A new polyphosphate calcium material with morphogenetic activity. Mater. Lett. 2015, 148, 163–166. [Google Scholar] [CrossRef]
- Gabel, N.W.; Thomas, V. Evidence for the occurrence and distribution of inorganic polyphosphates in vertebrate tissues. J. Neurochem. 1971, 18, 1229–1242. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Baev, A.Y.; Berezhnov, A.V.; Abramov, A.Y. Role of inorganic polyphosphate in mammalian cells: From signal transduction and mitochondrial metabolism to cell death. Biochem. Soc. Trans. 2016, 44, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 2010, 1802, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Weiner, S.; Mahamid, J.; Politi, Y.; Ma, Y.; Addadi, L. Overview of the amorphous precursor phase strategy in biomineralization. Front. Mater. Sci. China 2009, 3, 104–108. [Google Scholar] [CrossRef]
- Wang, X.H.; Schröder, H.C.; Müller, W.E.G. Enzyme-based biosilica and biocalcite: Biomaterials for the future in regenerative medicine. Trends Biotechnol. 2014, 32, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.E.G.; Neufurth, M.; Schlossmacher, U.; Schröder, H.C.; Pisignano, D.; Wang, X.H. The sponge silicatein-interacting protein silintaphin-2 blocks calcite formation of calcareous sponge spicules at the vaterite stage. RSC Adv. 2014, 4, 2577–2585. [Google Scholar] [CrossRef]
- Butterworth, P.J. Alkaline phosphatase. Biochemistry of mammalian alkaline phosphatases. Cell Biochem. Funct. 1983, 1, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.E.G.; Wang, X.H.; Diehl-Seifert, B.; Kropf, K.; Schloßmacher, U.; Lieberwirth, I.; Glasser, G.; Wiens, M.; Schröder, H.C. Inorganic polymeric phosphate/polyphosphate as an inducer of alkaline phosphatase and a modulator of intracellular Ca2+ level in osteoblasts (SaOS-2 cells) in vitro. Acta Biomater. 2011, 7, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Tarozzi, A.; Morroni, F.; Merlicco, A.; Bolondi, C.; Teti, G.; Falconi, M.; Cantelli-Forti, G.; Hrelia, P. Neuroprotective effects of cyanidin 3-O-glucopyranoside on amyloid beta (25–35) oligomer-induced toxicity. Neurosci. Lett. 2010, 473, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Baev, A.Y.; Negoda, A.; Abramov, A.Y. Modulation of mitochondrial ion transport by inorganic polyphosphate—Essential role in mitochondrial permeability transition pore. J. Bioenerg. Biomembr. 2017, 49, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.E.G.; Tolba, E.; Feng, Q.; Schröder, H.C.; Markl, J.S.; Kokkinopoulou, M.; Wang, X.H. Amorphous Ca2+ polyphosphate nanoparticles regulate the ATP level in bone-like SaOS-2 cells. J. Cell Sci. 2015, 128, 2202–2207. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.E.G.; Wang, S.; Neufurth, M.; Kokkinopoulou, M.; Feng, Q.; Schröder, H.C.; Wang, X.H. Polyphosphate as donor of high-energy phosphate for the synthesis of ADP and ATP. J. Cell Sci. 2017, 130, 2747–2756. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, Y.; Takenaka, H.; Sumida, M.; Oka, K.; Hamada, M.; Kuby, S.A. Multiforms of mammalian adenylate kinase and its monoclonal antibody against AK1. Enzyme 1990, 43, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espí, R.; Mastai, Y.; Gross, S.; Landfester, K. Colloidal systems for crystallization processes from liquid phase. CrystEngComm 2013, 15, 2175–2191. [Google Scholar] [CrossRef]
- Woo, Y.I.; Park, B.J.; Kim, H.L.; Lee, M.H.; Kim, J.; Yang, Y.I.; Kim, J.K.; Tsubaki, K.; Han, D.W.; Park, J.C. The biological activities of (1,3)-(1,6)-β-d-glucan and porous electrospun PLGA membranes containing β-glucan in human dermal fibroblasts and adipose tissue-derived stem cells. Biomed. Mater. 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Hogins, J.; Crawford, D.C.; Zorumski, C.F.; Mennerick, S. Excitotoxicity triggered by Neurobasal culture medium. PLoS ONE 2011, 6, e25633. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Mori, T.; Sumii, T.; Lo, E.H. Hemoglobin-induced cytotoxicity in rat cerebral cortical neurons: Caspase activation and oxidative stress. Stroke 2002, 33, 1882–1888. [Google Scholar] [CrossRef] [PubMed]
- Stanley, P.E. Extraction of adenosine triphosphate from microbial and somatic cells. Methods Enzymol. 1986, 133, 14–22. [Google Scholar] [PubMed]
- Marcaida, G.; Miñana, M.D.; Grisolía, S.; Felipo, V. Determination of intracellular ATP in primary cultures of neurons. Brain Res. Brain Res. Protoc. 1997, 1, 75–78. [Google Scholar] [CrossRef]
- Moriwaki, T.; Kato, S.; Kato, Y.; Hosoki, A.; Zhang-Akiyama, Q.M. Extension of lifespan and protection against oxidative stress by an antioxidant herb mixture complex (KPG-7) in Caenorhabditis elegans. J. Clin. Biochem. Nutr. 2013, 53, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, P.W. Optimization of the tetrazolium dye (MTT) colorimetric assay for cellular growth and viability. Methods Mol. Biol. 2011, 716, 157–168. [Google Scholar] [PubMed]
- Petrie, A.; Watson, P. Statistics for Veterinary and Animal Science; Wiley-Blackwell: Oxford, UK, 2013; pp. 85–99. [Google Scholar]
- Granata, D.; Baftizadeh, F.; Habchi, J.; Galvagnion, C.; De Simone, A.; Camilloni, C.; Laio, A.; Vendruscolo, M. The inverted free energy landscape of an intrinsically disordered peptide by simulations and experiments. Sci. Rep. 2015, 5, 15449. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, J.K.; Wong, K.; Fritz, I.B.; Griswold, M.D. Structural analysis of sulphated glycoprotein 2 from amino acid sequence. Relationship to clusterin and serum protein 40,40. Biochem. J. 1990, 268, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, C.; Praça, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-mediated brain drug delivery: Overcoming blood-brain barrier to treat neurodegenerative diseases. J. Control. Release 2016, 235, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Grabrucker, A.M.; Ruozi, B.; Belletti, D.; Pederzoli, F.; Forni, F.; Vandelli, M.A.; Tosi, G. Nanoparticle transport across the blood brain barrier. Tissue Barriers 2016, 4, e1153568. [Google Scholar] [CrossRef] [PubMed]
- Gowert, N.S.; Donner, L.; Chatterjee, M.; Eisele, Y.S.; Towhid, S.T.; Münzer, P.; Walker, B.; Ogorek, I.; Borst, O.; Grandoch, M.; et al. Blood platelets in the progression of Alzheimer’s disease. PLoS ONE 2014, 9, e90523. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Sowade, R.F.; Jahn, T.R. Seed-induced acceleration of amyloid-β mediated neurotoxicity in vivo. Nat. Commun. 2017. [Google Scholar] [CrossRef] [PubMed]








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, W.E.G.; Wang, S.; Ackermann, M.; Neufurth, M.; Steffen, R.; Mecja, E.; Muñoz-Espí, R.; Feng, Q.; Schröder, H.C.; Wang, X. Rebalancing β-Amyloid-Induced Decrease of ATP Level by Amorphous Nano/Micro Polyphosphate: Suppression of the Neurotoxic Effect of Amyloid β-Protein Fragment 25-35. Int. J. Mol. Sci. 2017, 18, 2154. https://doi.org/10.3390/ijms18102154
Müller WEG, Wang S, Ackermann M, Neufurth M, Steffen R, Mecja E, Muñoz-Espí R, Feng Q, Schröder HC, Wang X. Rebalancing β-Amyloid-Induced Decrease of ATP Level by Amorphous Nano/Micro Polyphosphate: Suppression of the Neurotoxic Effect of Amyloid β-Protein Fragment 25-35. International Journal of Molecular Sciences. 2017; 18(10):2154. https://doi.org/10.3390/ijms18102154
Chicago/Turabian StyleMüller, Werner E. G., Shunfeng Wang, Maximilian Ackermann, Meik Neufurth, Renate Steffen, Egherta Mecja, Rafael Muñoz-Espí, Qingling Feng, Heinz C. Schröder, and Xiaohong Wang. 2017. "Rebalancing β-Amyloid-Induced Decrease of ATP Level by Amorphous Nano/Micro Polyphosphate: Suppression of the Neurotoxic Effect of Amyloid β-Protein Fragment 25-35" International Journal of Molecular Sciences 18, no. 10: 2154. https://doi.org/10.3390/ijms18102154
APA StyleMüller, W. E. G., Wang, S., Ackermann, M., Neufurth, M., Steffen, R., Mecja, E., Muñoz-Espí, R., Feng, Q., Schröder, H. C., & Wang, X. (2017). Rebalancing β-Amyloid-Induced Decrease of ATP Level by Amorphous Nano/Micro Polyphosphate: Suppression of the Neurotoxic Effect of Amyloid β-Protein Fragment 25-35. International Journal of Molecular Sciences, 18(10), 2154. https://doi.org/10.3390/ijms18102154