
Cellular Stress and p53-Associated Apoptosis by Juniperus communis L. Berry Extract Treatment in the Human SH-SY5Y Neuroblastoma Cells

Abstract
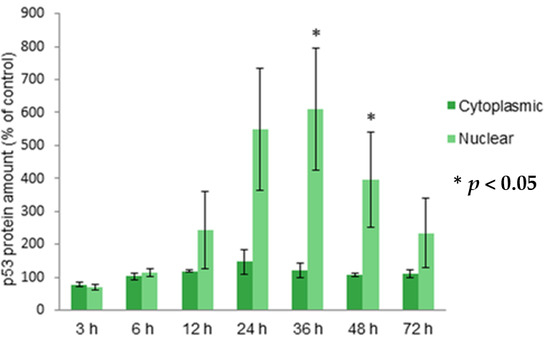
:
1. Introduction
2. Results
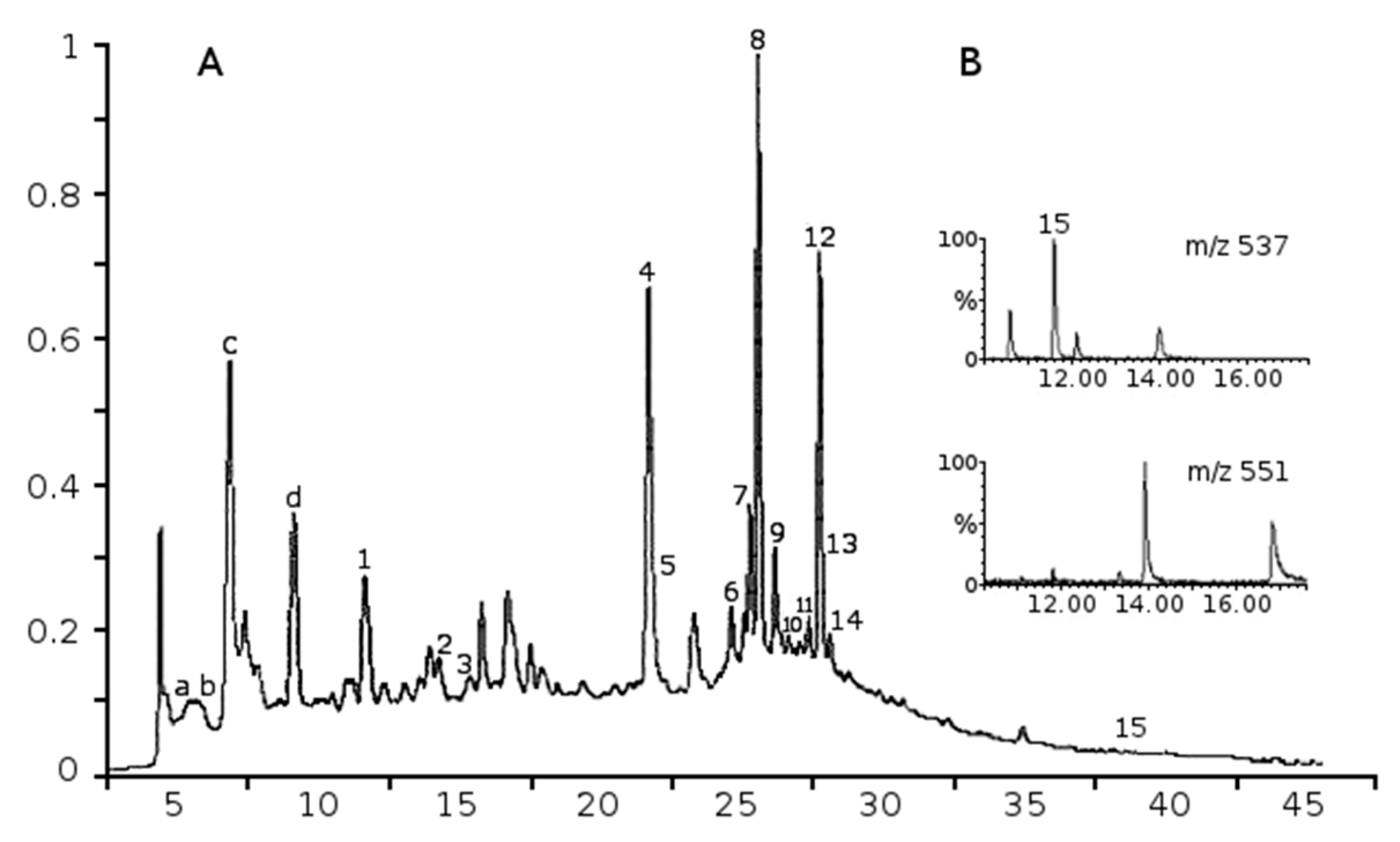
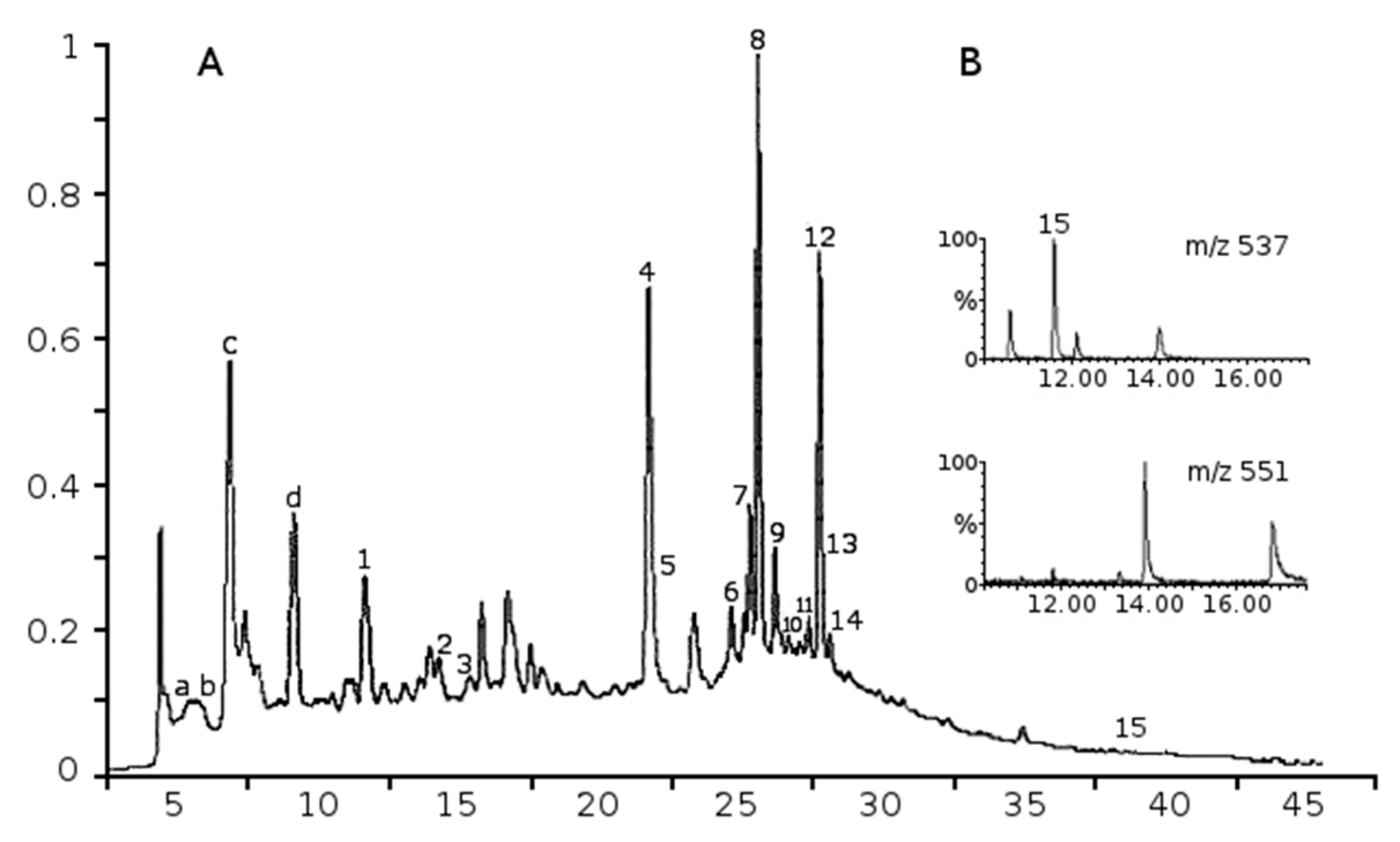
2.1. Phenolic Composition of Aqueous Juniper Berry Extract
2.2. Identified Phenolics and Concentrations
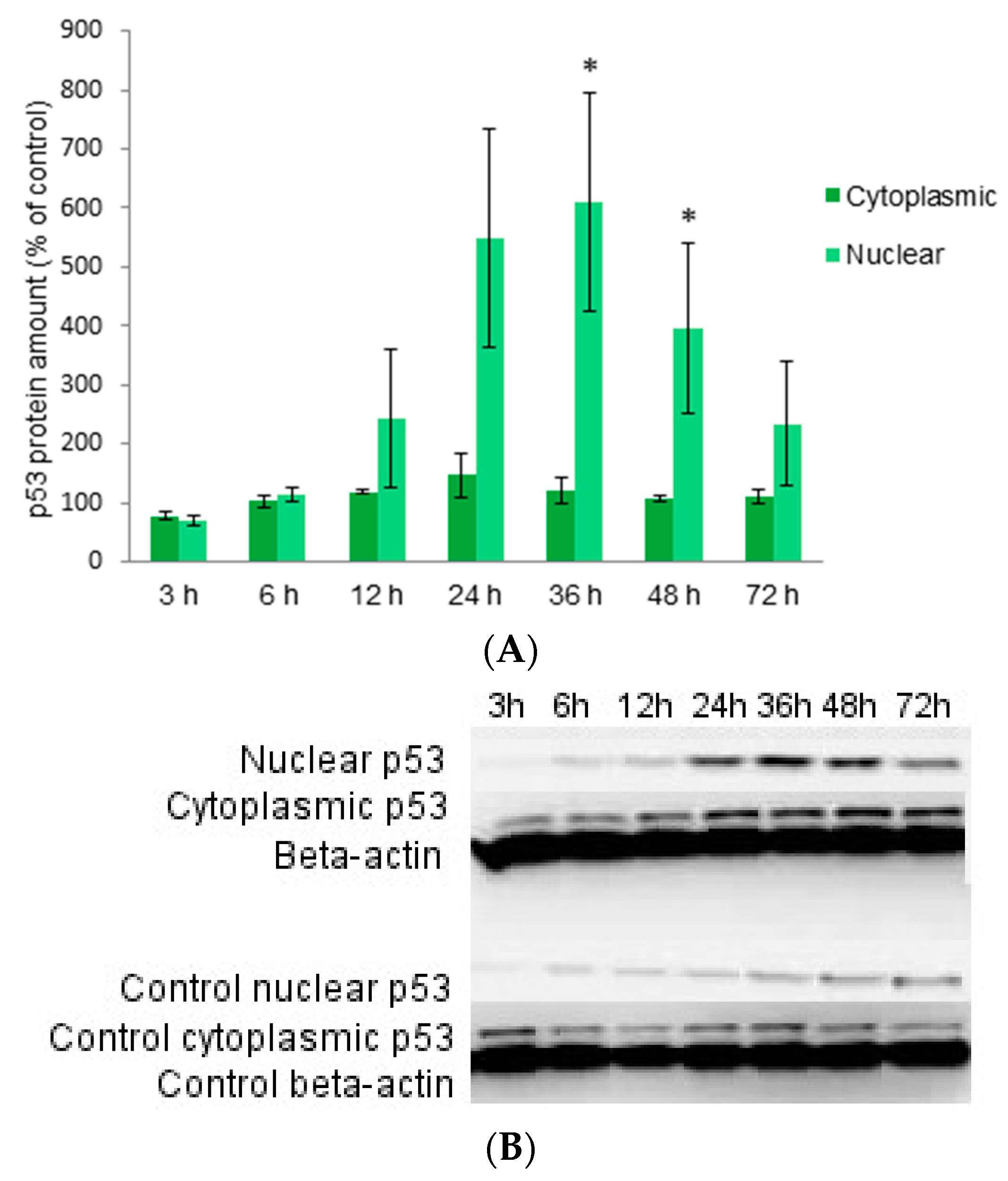
2.3. Localization of Protein p53 in Cytoplasm and Nucleus
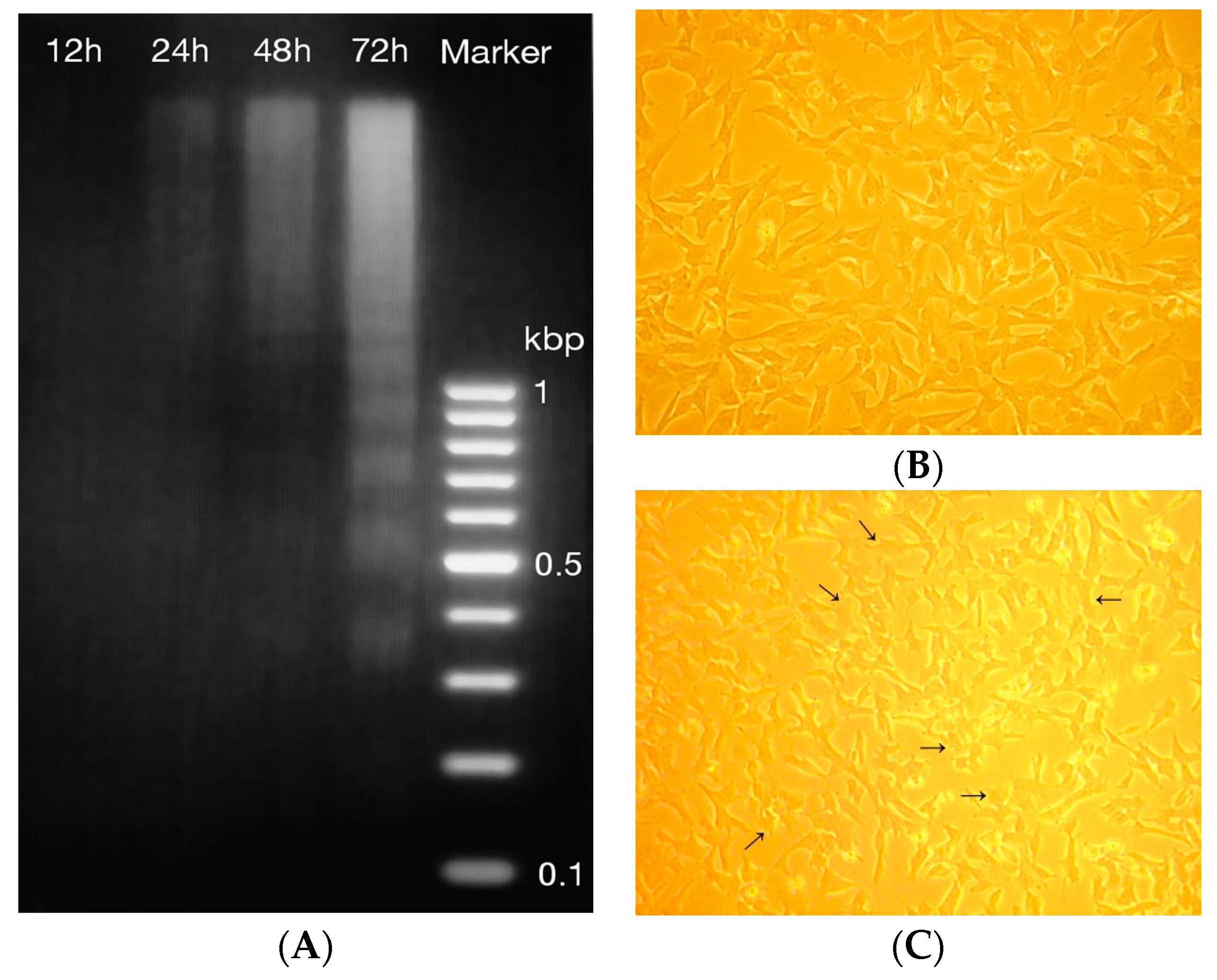
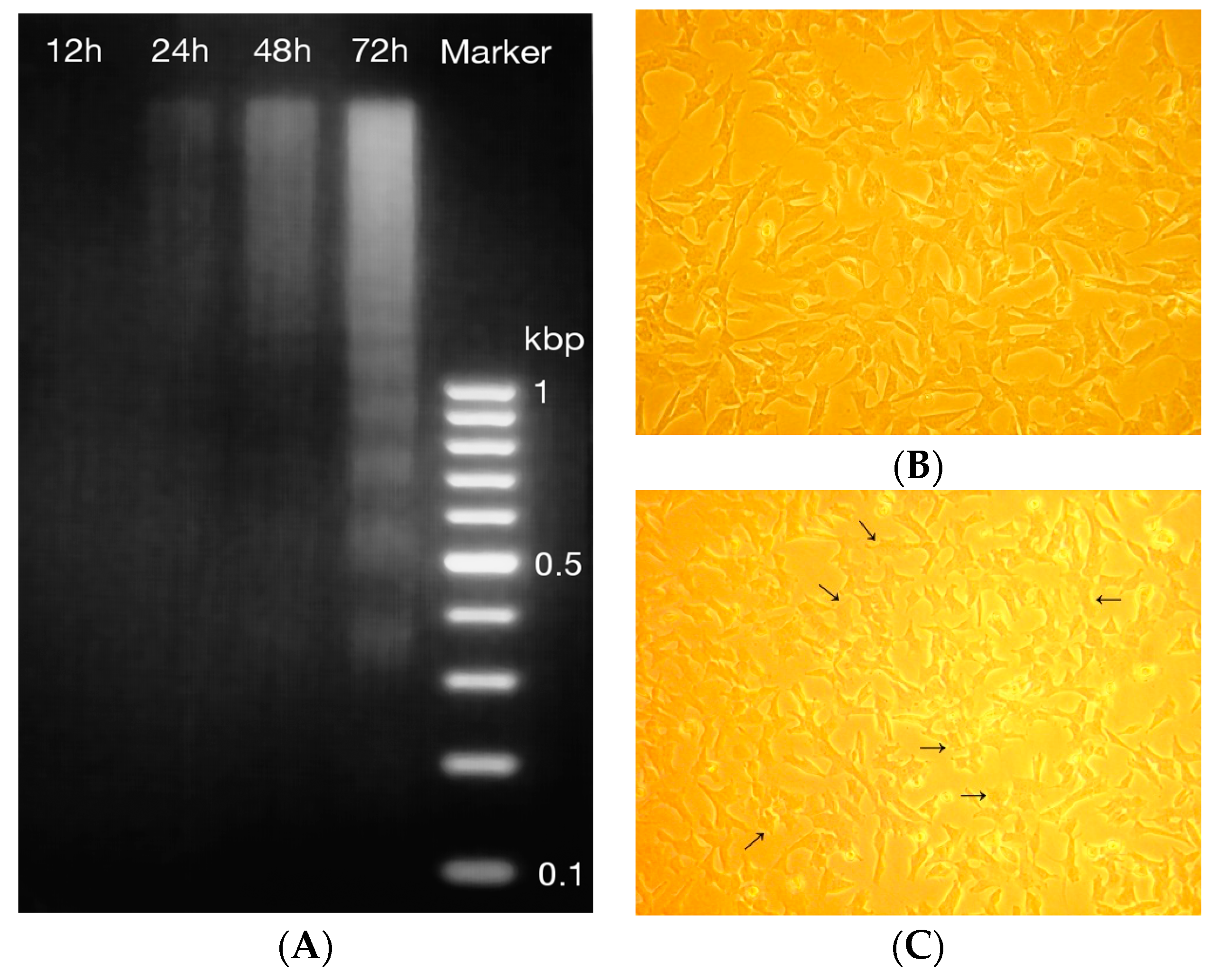
2.4. DNA Fragmentation and Morphology
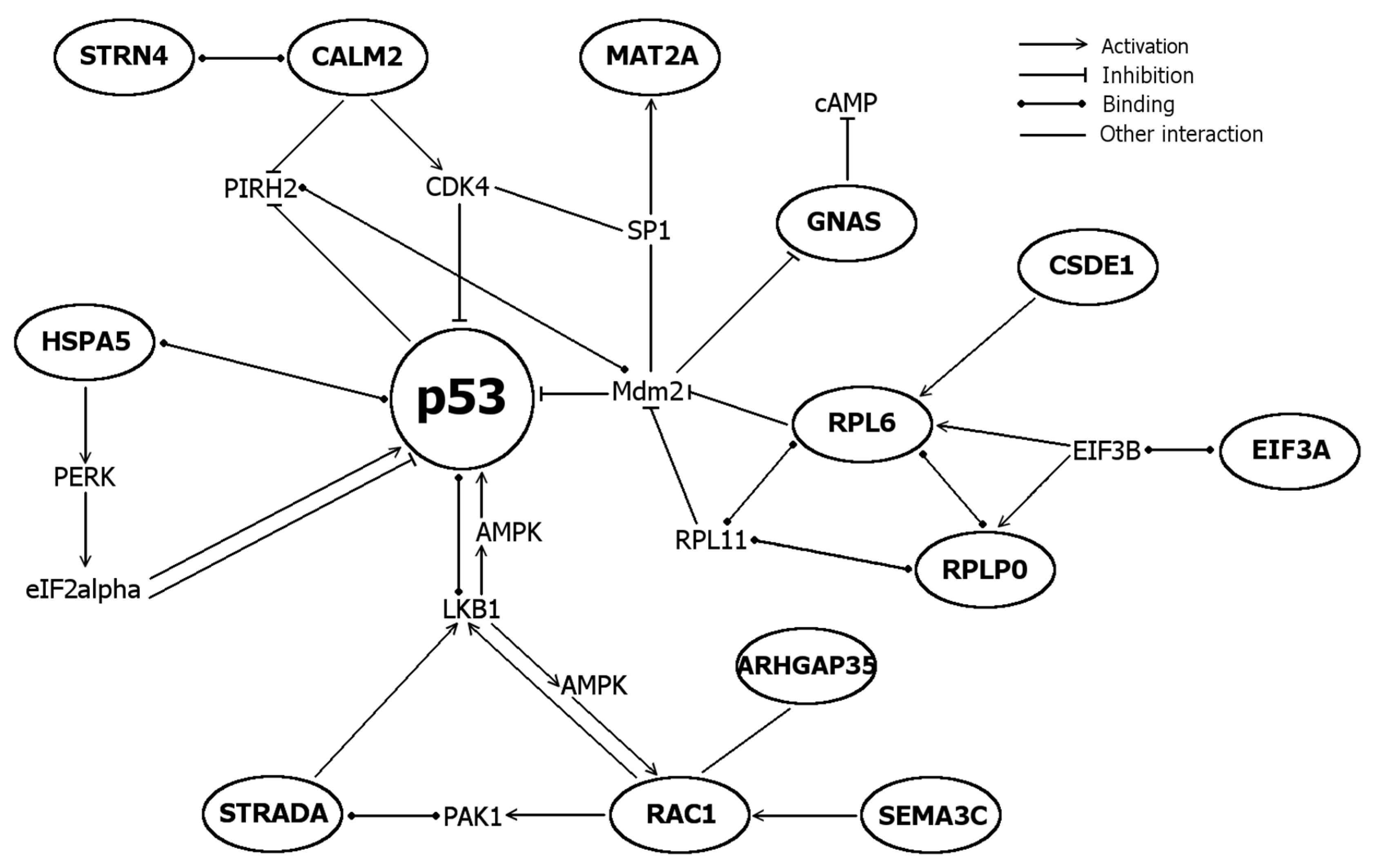
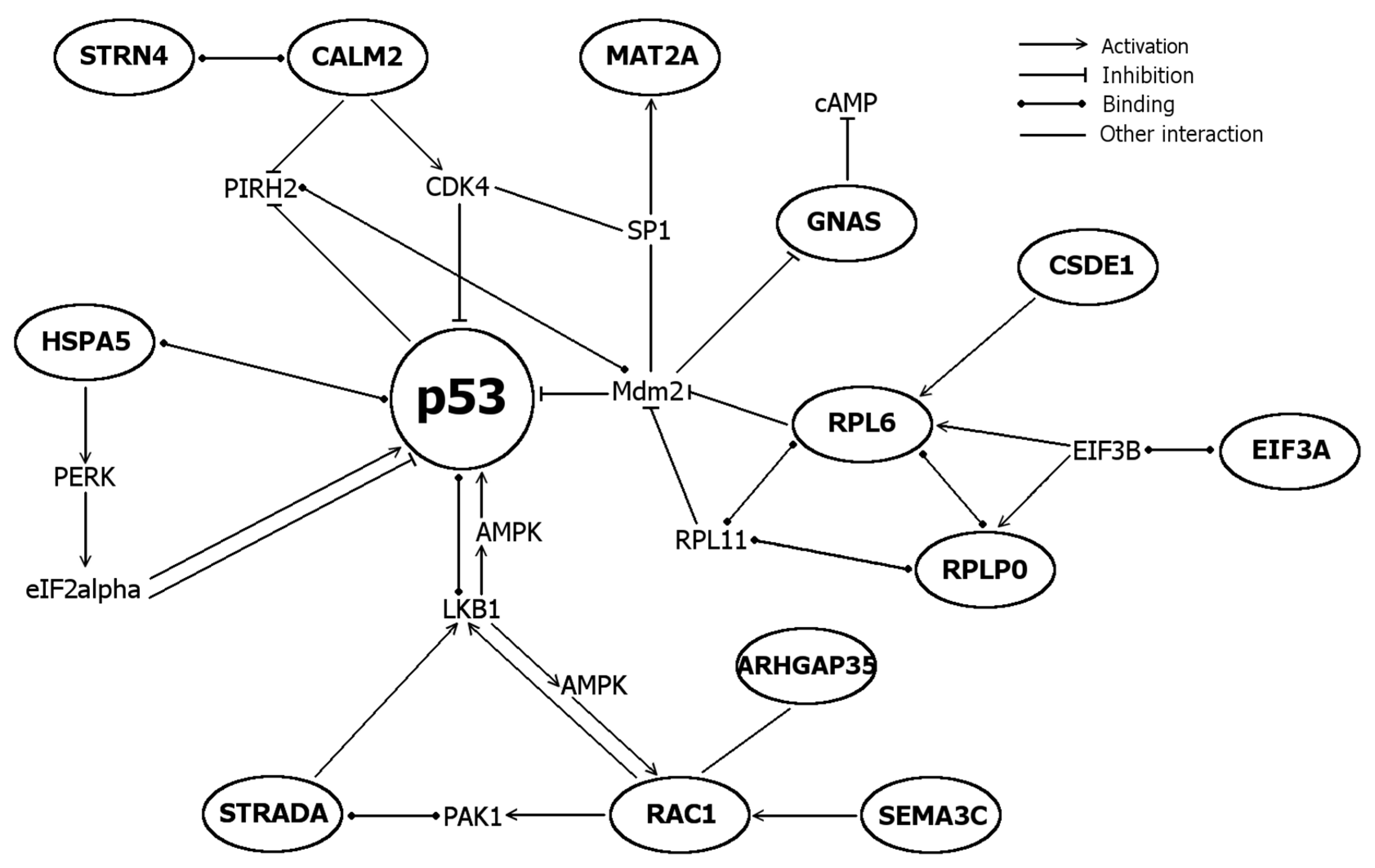
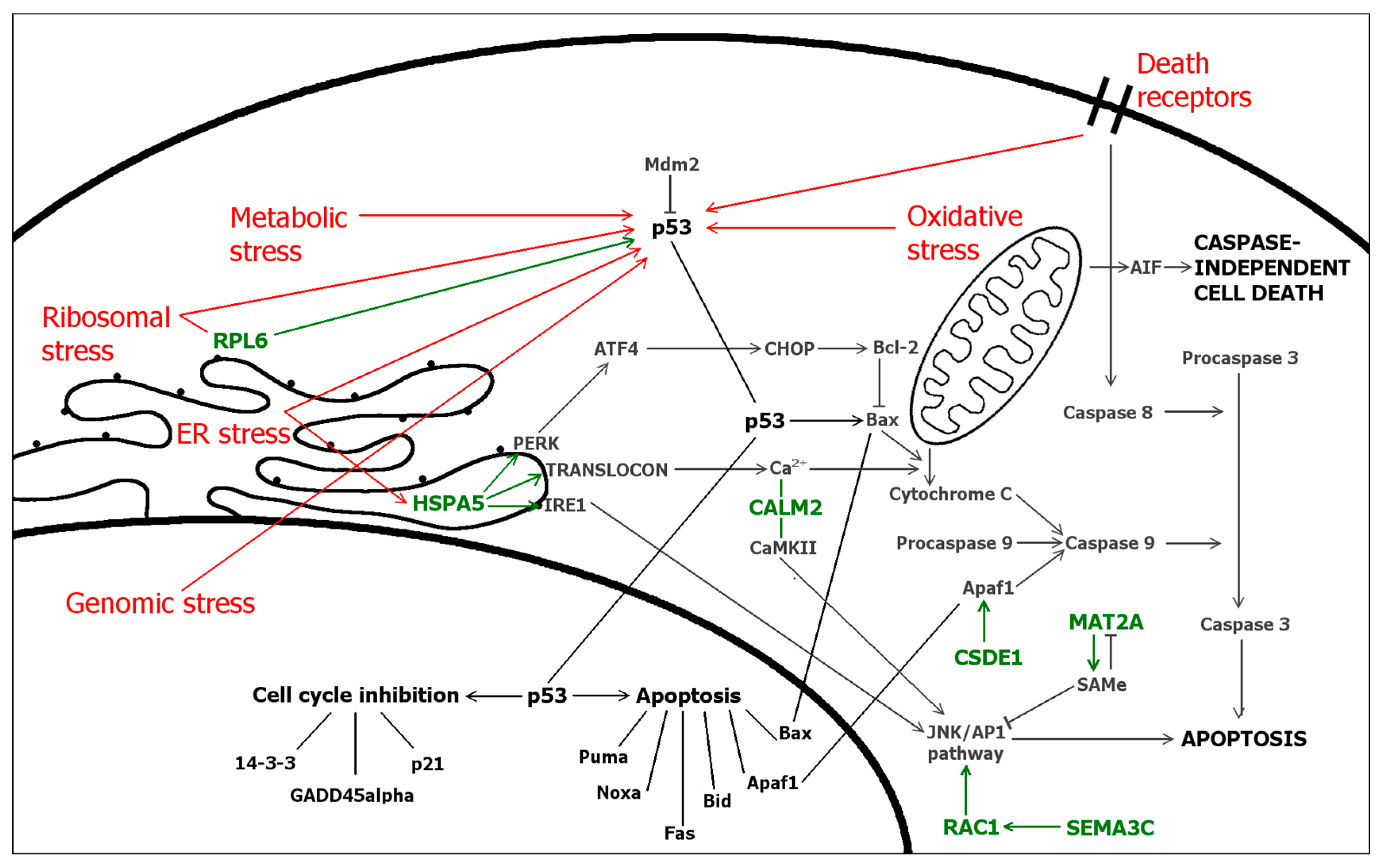
2.5. Differentially Expressed Genes
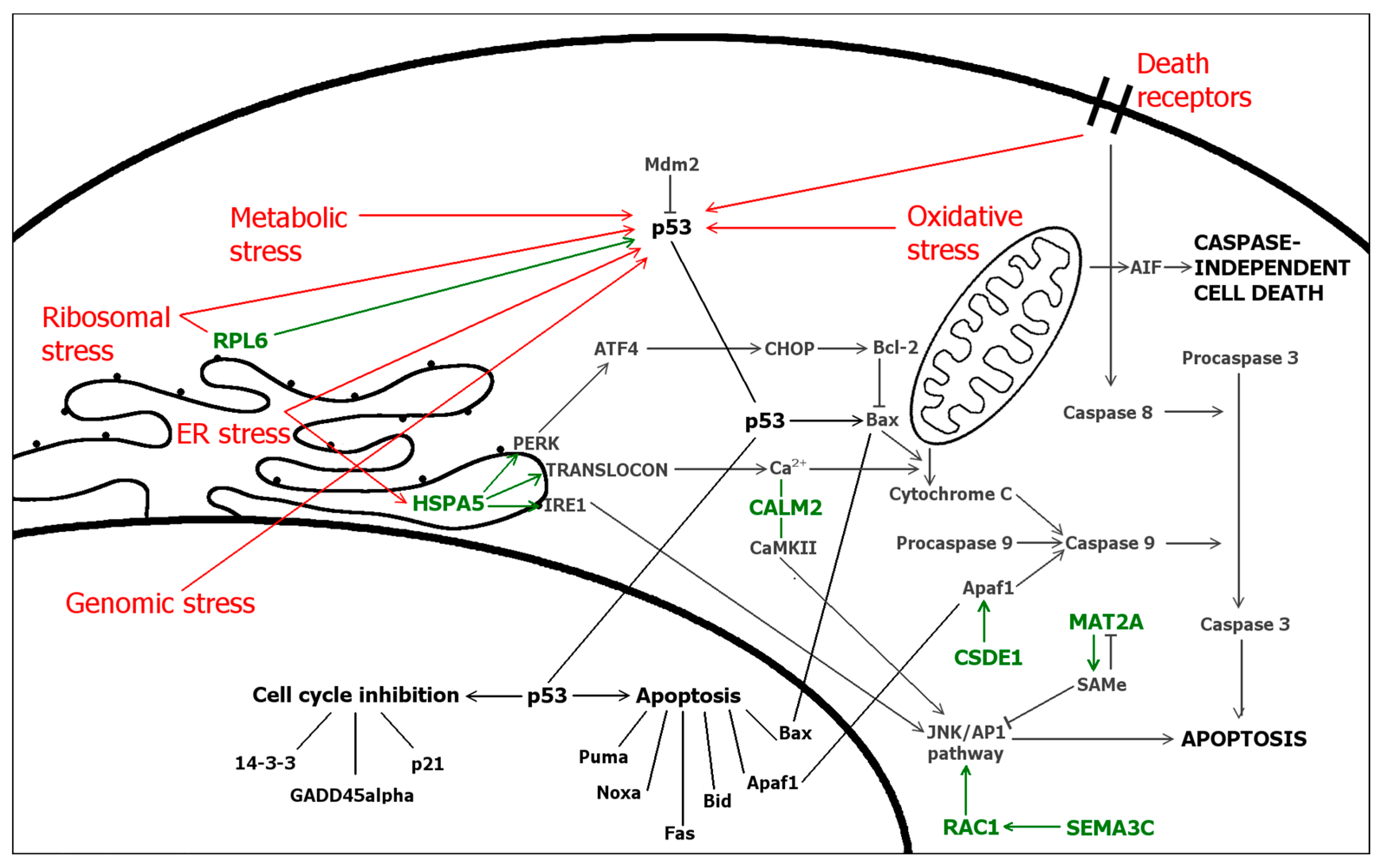
3. Discussion
4. Materials and Methods
4.1. Plant Material, Solvents and Reference Substances
4.2. Aqueous Extraction of Juniper Berries
4.3. Analysis of Volatile Oils by Gas Chromatography-Mass Spectrometry (GC–MS)
4.4. Analysis of Phenolic Composition by High Performance Liquid Chromatography (HPLC)
4.5. LC-MS-UV Systems and Analyses
4.6. Cell Culture and Treatments
4.7. Preparation of Protein Samples for Western Blot Analysis
4.8. Western Blot Analysis of Cytoplasmic and Nuclear p53
4.9. DNA Fragmentation
4.10. cDNA Representational Difference Analysis (RDA)
4.11. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CCD | charge coupled device |
| cDNA-RDA | cDNA representational difference analysis |
| QTOF | quadrupole time of flight |
References
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nature 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef]
- Hartwell, J.L. Plants used against cancer. A Surv. Lloydia 1970, 33, 288–392. [Google Scholar]
- Tunón, H.; Olavsdotter, C.; Bohlin, L. Evaluation of anti-inflammatory activity of some Swedish medicinal plants. Inhibition of prostaglandin biosynthesis and PAF-induced exocytosis. J. Ethnopharmacol. 1995, 48, 61–76. [Google Scholar] [CrossRef]
- Lim, T.K. Edible Medicinal and Non-Medicinal Plants; Springer: Dordrecht, The Netherlands, 2012; Volume 1. [Google Scholar]
- Innocenti, M.; Micheozzi, M.; Giaccherini, C.; Ieri, F.; Vincieri, F.F.; Mulinacci, N. Flavonoids and biflavonoids in Tuscan berries of Juniperus communis L.: Detection and quantitation by HPLC/DAD/ESI/MS. J. Agric. Food Chem. 2007, 55, 6596–6602. [Google Scholar] [CrossRef] [PubMed]
- Rajput, S.; Mandal, M. Antitumor promoting potential of selected phytochemicals derived from spices: A review. Eur. J. Cancer Prev. 2012, 21, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Lehár, J.; Krueger, A.S.; Avery, W.; Heilbut, A.M.; Johansen, L.M.; Price, E.R.; Rickles, R.J.; Short, G.F., III; Staunton, J.E.; Jin, X.; et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat. Biotechnol. 2009, 27, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Parhi, P.; Mohanty, C.; Sahoo, S.K. Nanotechnology-based combinational drug delivery: An emerging approach for cancer therapy. Drug Discov. Today 2012, 17, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Lantto, T.A.; Colucci, M.; Závadová, V.; Hiltunen, R.; Raasmaja, A. Cytotoxicity of curcumin, resveratrol and plant extracts from basil, juniper, laurel and parsley in SH-SY5Y and CV1-P cells. Food Chem. 2009, 117, 405–411. [Google Scholar] [CrossRef]
- Dorman, H.J.D.; Hiltunen, R. Antioxidant and pro-oxidant in vitro evaluation of water-soluble food-related botanical extracts. Food Chem. 2011, 129, 1612–1618. [Google Scholar] [CrossRef]
- Hinneburg, I.; Dorman, H.J.D.; Hiltunen, R. Antioxidant activities of extracts from selected culinary herbs and spices. Food Chem. 2006, 97, 122–129. [Google Scholar] [CrossRef]
- Sun, J.; Liang, F.; Bin, Y.; Li, P.; Duan, C. Screening non-colored phenolics in red wines using liquid chromatography/ultraviolet and mass spectrometry/mass spectrometry libraries. Molecules 2007, 12, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Aaby, K.; Ekeberg, D.; Skrede, G. Characterization of phenolic compounds in strawberry (Fragaria × ananassa) fruits by different HPLC detectors and contribution of individual compounds to total antioxidant capacity. J. Agric. Food Chem. 2007, 55, 4395–4406. [Google Scholar] [CrossRef] [PubMed]
- Miceli, N.; Trovato, A.; Dugo, P.; Cacciola, P.; Donato, P.; Marino, A.; Bellinghieri, V.; La Barbera, T.M.; Güvenç, A.; Taviano, M.F. Comparative analysis of flavonoid profile, antioxidant and antimicrobial activity of the berries of Juniperus communis L. var. communis and Juniperus communis L. var. saxatilis Pall. from Turkey. J. Agric. Food Chem. 2009, 57, 6570–6577. [Google Scholar] [PubMed]
- Nakanishi, T.; Iida, N.; Inatomi, Y.; Murata, H.; Inada, A.; Murata, J.; Lang, F.A.; Iinuma, M.; Tanaka, T. Neolignan and flavonoid glycosides in Juniperus communis var. depressa. Phytochemistry 2004, 65, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Almela, L.; Sánches-Muñoz, B.; Fernández-López, J.A.; Roca, M.J.; Rabe, V. Liquid chromatographic-mass spectrometric analysis of phenolics and free radical scavenging activity of rosemary extract from different raw material. J. Chromatogr. A 2006, 1120, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Eggers, C.M.; Kline, E.R.; Zhong, D.; Zhou, W.; Marcus, A.I. STE20-related kinase adaptor protein α (STRADα) regulates cell polarity and invasion through PAK1 signaling in LKB-null cells. J. Biol. Chem. 2012, 287, 18758–18768. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Pallas, D.C. STRIPAK complexes: Sructure, biological function, and involvement in human disease. Int. J. Biochem. Cell Biol. 2014, 47, 118–148. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yi, H.; Zhang, P.-F.; Li, M.-Y.; Li, C.; Li, F.; Peng, F.; Feng, X.-P.; Yang, Y.-X.; Yang, F.; et al. Identification of differential proteins in nasopharyngeal carcinoma cells with p53 silence by proteome analysis. FEBS Lett. 2007, 581, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Matassov, D.; Kagan, T.; Leblanc, J.; Sikorska, M.; Zakeri, Z. Measurement of apoptosis by DNA fragmentation. Methods Mol. Biol. 2004, 282, 1–17. [Google Scholar] [PubMed]
- Puttonen, K.A.; Lehtonen, S.; Lampela, P.; Männistö, P.T.; Raasmaja, A. Different viabilities and toxicity types after 6-OHDA and Ara-C exposure by four assays in five cell lines. Toxicol. In Vitro 2008, 22, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Kitazumi, I.; Tsukahara, M. Regulation of DNA fragmentation: The role of caspases and phophorylation. FEBS J. 2011, 278, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the nomenclature committee on cell death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, M.W.; Villalobo, A. The many faces of calmodulin in cell proliferation, programmed cell death, autophagy, and cancer. Biochim. Biophys. Acta 2014, 1843, 398–435. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.A.; Brown, E.C.; Coldwell, M.J.; Jackson, R.J.; Willis, A.E. Protein factor requirements of the Apaf-1 internal ribosome entry segment: Roles of polypyrimidine tract binding protein and upstream of N-ras. Mol. Cell. Biol. 2001, 21, 3364–3374. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanims. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Kim, Y.S. Phosphorylation of sIF2α attenuates statin-induced apoptosis by inhibiting the stabilization and translocation of p53 to the mitochondria. Int. J. Oncol. 2013, 42, 810–816. [Google Scholar] [PubMed]
- Wek, R.C.; Jiang, H.-Y.; Anthony, T.G. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 2006, 34, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Guo, X.; Yu, W.; Li, C.; Gui, Y.; Cai, Z. Expression of methionine adenoslyltransferase 2A in renal cell carcinomas and potential mechanism for kidney carcinogenesis. BMC Cancer 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Lin, J.C.; Wang, K.; Chen, Y.J.; Liu, H.H.; Luan, R.; Jiang, S.; Che, T.; Zhao, Y.; Li, D.F. A nuclear ligand MRG15 involved in the proapoptotic activity of medicinal fungal galectin AAL (Agrocybe aegerita lectin). Biochim. Biophys. Acta 2010, 1800, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Kaziro, Y.; Satoh, T. Anti-apoptotic function of Rac in hematopoietic cells. Oncogene 1999, 18, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Al-Sammarraie, N.; DiPette, D.J.; Singh, U.S. Metformin impairs Rho GTPase signaling to induce apoptosis in neuroblastoma cells and inhibits growth of tumors in the xenograft mouse model of neuroblastoma. Oncotarget 2014, 5, 11709–11722. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.; McKay, M.J.; Christopherson, R.I.; Baxter, R.C. Biomarkers of breast cancer apoptosis induced by chemotherapy and TRAIL. J. Proteome Res. 2012, 11, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Nair, U.; Jotwani, A.; Geng, J.; Gammoh, N.; Richerson, D.; Yen, W.-L.; Griffith, J.; Nag, S.; Wang, K.; Moss, T. SNARE proteins are required for macroautophagy. Cell 2011, 146, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Notsuda, H.; Sakurada, A.; Endo, C.; Okada, Y.; Horii, A.; Shima, H.; Kondo, T. p190A RhoGAO is involved in EGFR pathways and promotes proliferation, invasion and migration in lung adenocarcinoma cells. Int. J. Oncol. 2013, 43, 1569–1577. [Google Scholar] [PubMed]
- Tinton, S.A.; Schepens, B.; Bruynooghe, Y.; Beyaert, R.; Cornelis, S. Regulation of the cell-cycle-dependent internal ribosome entry site of the PITSLRE protein kinase: Roles of Unr (upstream of N-ras) protein and phosphorylated translation initiation factor eIF-2α. Biochem. J. 2005, 385, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C.; Mato, J.M. S-adenosylmethionine in cell growth, apoptosis and liver cancer. J. Gastroenterol. Hepatol. 2008, 23, S73–S77. [Google Scholar] [CrossRef] [PubMed]
- Martrat, G.; Maxwell, C.A.; Tominaga, E.; Porta-de-la-Riva, M.; Bonifaci, N.; Gómez-Baldó, L.; Bogliolo, M.; Lázaro, C.; Blanco, I.; Brunet, J. Exploring the link between MORF4L1 and risk of breast cancer. Breast Cancer Res. 2011, 13. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Zhang, J.; Xiao, W.; Zheng, X. Regulation of the HDM2-p53 pathway by ribosomal protein L6 in response to ribosomal stress. Nucleic Acids Res. 2014, 42, 1799–1811. [Google Scholar] [CrossRef] [PubMed]
- Tiainen, M.; Ylikorkala, A.; Mäkelä, T.P. Growth supression by Lkb1 is mediated by a G(1) cell cycle arrest. Proc. Natl. Acad. Sci. USA 1996, 96, 9248–9251. [Google Scholar] [CrossRef]
- Hyodo, T.; Ito, S.; Hasegawa, H.; Asano, E.; Maeda, M.; Urano, T.; Takahashi, M.; Hamaguchi, M.; Senga, T. Misshapen-like kinase 1 (MINK1) is a novel component of striatin-interacting phosphatase and kinase (STRIPAK) and is required for the completion of cytokinesis. J. Biol. Chem. 2012, 287, 25019–25029. [Google Scholar] [CrossRef] [PubMed]
- Thayanidhi, N.; Liang, Y.; Hasegawa, H.; Nycz, D.C.; Oorschot, V.; Klumperman, J.; Hay, J.C. R-SNARE ykt6 resides in membrane-associated protease-resistant protein particles and modulates cell cycle progression when over-expressed. Biol. Cell 2012, 104, 397–417. [Google Scholar] [CrossRef] [PubMed]
- Hammadi, M.; Oulidi, A.; Gackière, F.; Katsogiannou, M.; Slomianny, C.; Roudbaraki, M.; Dewailly, E.; Delcourt, P.; Lepage, G.; Lotteau, S. Modulation of ER stress and apoptosis by endoplasmic reticulum calcium leak via translocon during unfolded protein response: Involvement of GRP78. FASEB J. 2013, 27, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Doyon, Y.; Selleck, W.; Lane, W.S.; Tan, S.; Côté, J. Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol. Cell. Biol. 2004, 24, 1884–1896. [Google Scholar] [CrossRef] [PubMed]
- Garcia, S.H.; Kirtane, B.M.; Podlutsky, A.J.; Pereira-Smith, O.M.; Tominaga, K. Mrg15 null heterozygous mouse embryonic fibroblasts exhibit DNA repair defects post exposure to gamma ionizing radiation. FEBS Lett. 2007, 13, 5275–5281. [Google Scholar] [CrossRef] [PubMed]
- Wilmet, J.-P.; Tastet, C.; Desruelles, E.; Ziental-Gelus, N.; Blanckaert, V.; Hondermarck, H.; Le Bourhis, X. Proteome changes induced by overexpression of the p75 neurotrophin receptor (p75NTR) in breast cancer cells. Int. J. Dev. Biol. 2011, 55, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Frozza, C.O.; da Silva Frozza, T.; Gambato, G.; Menti, C.; Moura, S.; Pinto, P.M.; Staats, C.C.; Padilha, F.F.; Begnini, K.R.; de Leon, P.M.M. Proteomic analysis identifies differentially expressed proteins after red propolis treatment in Hep-2 cells. Food Chem. Toxicol. 2014, 63, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Suga, K.; Saito, A.; Akagawa, K. ER stress response in NG108-15 cells involves upregulation of syntaxin 5 expression and reduced amyloid β peptide secretion. Exp. Cell Res. 2015, 332, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.-C.; Lin, J.-J.; Wen, H.-C.; Chen, L.-C.; Hsu, S.-P.; Lee, W.-S. Folic acid inhibits endothelial cell migration through inhibiting the RhoA activity mediated by activating the folic acid receptor/cSrc/p190RhoGAP-signaling pathway. Biochem. Pharmacol. 2013, 85, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Tse, E.Y.; Ching, Y.P. The role of p21-activated kinases in hepatocellular carcinoma metastasis. J. Mol. Signal. 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.-M.; Li, C.J.; Vagbo, C.B.; Shi, Y.; Wang, W.-L.; Song, S.-H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Salas, E.; Piñeiro, D.; Fernandez, N. Riboproteomic Approaches to Understand IRES Elements. In Biophysical Approaches to Translational Control of Gene Expression, Biophysics for the Life Sciences 1; Dinman, J.D., Ed.; Springer: New York, NY, USA, 2013; pp. 103–118. [Google Scholar]
- Zhu, B.; Mandal, S.S.; Pham, A.-D.; Zheng, Y.; Erdjument-Bromage, H.; Batra, S.K.; Tempst, P.; Reinberg, D. The human PAF complex coordinates transcription events downstream of RNA synthesis. Genes Dev. 2005, 19, 1668–1673. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; LaQuaglia, M.; Bénard, J.; Riou, G. Wild-type p53 protein undergoes cytoplasmic sequastration in undifferentiated neuroblastomas but not in differentiated tumors. Proc. Natl. Acad. Sci. USA 1995, 92, 4407–4411. [Google Scholar] [CrossRef] [PubMed]
- Tweddle, D.A.; Malcolm, A.J.; Cole, M.; Pearson, A.D.J.; Lunec, J. p53 Cellular localization and function in neuroblastoma: Evidence for defective G1 arrest despite WAF1 induction in MYCN-amplified cells. Am. J. Pathol. 2001, 158, 2067–2077. [Google Scholar] [CrossRef]
- Van Maerken, T.; Rihani, A.; Dreidax, D.; de Clercq, S.; Yigit, N.; Marine, J.-C.; Westermann, F.; de Paepe, A.; Vandesompele, J.; Speleman, F. Functional analysis of the p53 pathway in neuroblastoma cells using the small-molecule MDM2 antagonist Nutlin-3. Mol. Cancer Ther. 2011, 10, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Zhu, W.-G. Surf the post-translational modifications network of p53 regulation. Int. J. Biol. Sci. 2012, 8, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Green, D.R. Dissecting p53-dependent apoptosis. Cell Death Differ. 2006, 13, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Biagioli, M.; Pifferi, S.; Ragghianti, M.; Bucci, S.; Rizzuto, R.; Pinton, P. Endoplasmic reticulum stress and alteration in calcium homeostasis are involved in cadmium-induced apoptosis. Cell Calcium 2008, 43, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Ciechomska, I.A.; Gabrusiewicz, K.; Szczepankiewicz, A.A.; Kaminska, B. Endoplasmic reticulum stress triggers autophagy in malignant glioma cells undergoing cyclosporine A-induced cell death. Oncogene 2013, 32, 1518–1529. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Man, J.; Shoemake, J.; Zhou, W.; Fang, X.; Wu, Q.; Rizzo, A.; Prayson, R.; Bao, S.; Rich, J.N.; Yu, J.S. Sema3C promotes the survival and tumorigenecity of glioma stem cells through Rac1 activation. Cell Rep. 2014, 9, 1812–1826. [Google Scholar] [CrossRef] [PubMed]
- Benoist, M.; Gaillard, S.; Castets, F. The striatin family: A new signaling platform in dendritic spines. J. Physiol. Paris 2006, 99, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Gou, Y.; Wang, Q.; Jin, H.; Cui, L.; Zhang, Y.; He, L.; Wang, J.; Nie, Y.; Shi, Y. Downregulation of RPL6 by siRNA inhibits proliferation and cell cycle progression of human gastric cancer cell lines. PLoS ONE 2011, 6, e26401. [Google Scholar] [CrossRef] [PubMed]
- Korsse, S.E.; Peppelenbosch, M.P.; van Veelen, W. Targeting LKB1 signaling in cancer. Biochim. Biophys. Acta 2013, 1835, 194–210. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Boudeau, J.; Reid, J.L.; Mustard, K.J.; Udd, L.; Mäkelä, T.P.; Alessi, D.R.; Hardie, D.G. Complexes between the LKB1 tumor suppressor, STRADalpha/beta and MO25alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2003, 2. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.B.; Babcock, J.T.; Wells, C.D.; Quilliam, L.A. LKB1 tumor suppressor regulates AMP kinase/mTOR-independent cell growth and proliferation via the phosphorylation of Yap. Oncogene 2013, 32, 4100–4109. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, S.; Davis, K.A.; Choudhury, S.R.; Deeconda, A.; Banik, N.L.; Ray, S.K. Bcl-2 inhibitor and apigenin worked synergistically in human malignant neurblastoma cell lines and increased apoptosis with activation of extrinsic and intrinsic pathways. Biochem. Biophys. Res. Commun. 2009, 388, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Banik, N.L.; Ray, S.K. Mechanism of apoptosis with the involvement of calpain and caspase cascades in human malignant neuroblastoma SH-SY5Y cells exposed to flavonoids. Int. J. Cancer 2006, 119, 2575–2585. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Singh, A.; Mishra, A. Quercetin and taxifolin completely break MDM2-p53 association: Molecular dynamics simulation study. Med. Chem. Res. 2013, 22, 2778–2787. [Google Scholar] [CrossRef]
- Paulig Group Ltd. Available online: http://www.pauliggroup.com 2016 (accessed on 10 July 2016).
- Maisonneue, S.A. European Pharmacopeia Commission. In 5th European Pharmacopoeia; Maisonneue: Saint-Ruffine, France, 1975; Volume 3. [Google Scholar]
- Ossola, B.; Lantto, T.A.; Puttonen, K.A.; Tuominen, R.K.; Raasmaja, A.; Männistö, P.T. Minocycline protects SH-SY5Y cells from 6-hydroxydopamine by inhibiting both caspase-dependent and -independent programmed cell death. J. Neurosci. Res. 2012, 90, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Hubank, M.; Bryntesson, F.; Regan, J.; Schatz, D.G. Cloning of apoptosis-related genes by representational difference analysis of cDNA. Methods Mol. Biol. 2004, 282, 255–273. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak | Compounds | Rt min | a λmax nm | Mw | b ESI-MS− Ions m/z | MRM Ions | Amounts in Extract µg/g | Amounts in Treatments (10 µg/mL of Extract) nM (ng/mL) | References |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Protocatechuic acid | 11.11 | 260, 293 | 154 | 153, 109 | 153→109 | 412 d | 26.75 (4.12) | [16] |
| 2 | Catechin | 13.69 | 280 | 290 | 289, 245, 205 | - | 406 e | 14.00 (4.06) | [17] |
| 3 | Gossypetin-hexoside-pentoside | 15.07 | 271, 367 | 612 | 611, 479 | - | 155 e | 2.53 (1.55) | [18] |
| 4 | Rutin | 20.97 | 255, 354 | 610 | 609 | 609→301 | 1333 d | 21.85 (13.33) | [9] |
| 5 | Hyperoside | 21.14 | 255, 353 | 464 | 463, 301 | 463→300 | 561 d | 12.09 (5.61) | [9] |
| 6 | Quercetin pentoside | 24.09 | 275, 341 | 434 | 433, 301 | - | 174 e | 4.01 (1.74) | [18] |
| 7 | Isoscutellarein-8-O-hexoside | 24.72 | 276, 303, 327 | 448 | 447, 285, 895 c | - | 427 e | 9.53 (4.27) | [9,18,19] |
| 8 | Hypolaetin-7-O-pentoside | 25.00 | 275, 342 | 434 | 433, 301, 867 c | - | 887 e | 20.44 (8.87) | [9,18,19] |
| 9 | Apigenin-7-O-glucoside | 25.63 | 267, 338 | 432 | 431, 269 | 431→268 | 1646 d | 38.10 (16.46) | [9,18] |
| 10 | Luteolin pentoside | 26.16 | 273, 347 | 418 | 417, 285 | - | 109 e | 2.61 (1.09) | [9] |
| 11 | Hypolaetin hexoside | 26.79 | 257, 343 | 464 | 463, 301, 867 c | - | 305 e | 6.57 (3.15) | [9] |
| 12 | Isoscutellarein-7-O-pentoside | 27.25 | 275, 304, 326 | 418 | 417, 285, 835 c | - | 798 e | 19.09 (7.98) | [9,18] |
| 13 | Kaempferol-3-O-glucoside | 27.38 | 266, 346 | 448 | 447, 285 | 447→284 | 250 d | 5.58 (2.50) | [17] |
| 14 | Rosmarinic acid | 27.56 | 329 | 360 | 359, 161 | 359→161 | 103 d | 2.86 (1.03) | [20]] |
| 15 | Amentoflavone | 37.40 | 268, 340 | 538 | 537 | 537→375 | 167 d | 3.10 (1.67) | [9,18] |
| Gene | Accession Number | Score | E-Value | Length |
|---|---|---|---|---|
| ALKBH5 | NM_017758 | 706 | 0.0 | 988 |
| ARHGAP35 | NM_004491 | 771 | 0.0 | 948 |
| CALM2 | NM_001743 | 499 | 2 × 10−138 | 663 |
| CSDE1 | NM_001242893+ | 627 | 1 × 10−176 | 720 |
| EIF3A | NM_003750 | 508 | 4 × 10−141 | 706 |
| GNAS | NM_003259+ | 669 | 0.0 | 899 |
| HSPA5 | NM_005347 | 682 | 0.0 | 683 |
| ITFG3 | NM_032039 | 647 | 0.0 | 939 |
| MAT2A | NM_005911 | 363 | 4 × 10−97 | 363 |
| MORF4L1 | NM_006791 | 392 | 3 × 10−106 | 515 |
| PPAPDC1B | NM_001102559+ | 453 | 2 × 10−124 | 833 |
| PRSS12 | NM_003619 | 508 | 5 × 10−141 | 859 |
| RAC1 | NM_006908+ | 536 | 2 × 10−149 | 908 |
| RPL6 | NM_001024662 | 508 | 5 × 10−141 | 869 |
| RPLP0 | NM_001002+ | 623 | 2 × 10−175 | 874 |
| SEMA3C | NM_006379 | 616 | 3 × 10−173 | 907 |
| SF3B2 | NM_006842 | 544 | 1 × 10−151 | 932 |
| STRADA | NM_153335 | 468 | 9 × 10−129 | 998 |
| STRN4 | NM_013403 | 608 | 3 × 10−171 | 663 |
| TTC37 | NM_014639 | 573 | 1 × 10−160 | 702 |
| YKT6 | NM_006555 | 621 | 5 × 10−175 | 792 |
| Gene | Gene Full Name/Synonyms | Protein(s) Encoded/Synonyms |
|---|---|---|
| ALKBH5 | Alkylating repair homolog 5 (E. coli)/ABH5 | RNA demethylase ALKBH5 |
| ARHGAP35 | Rho GTPase activating protein 35/GRF1 | Rho GTPase activating protein 35/GRF1/p190-A |
| CALM2 | Calmodulin 2 (phosphorylase kinase, delta)/CAM2 | Calmodulin/CaM |
| CSDE1 | Cold shock domain containing E1/UNR | Cold shock domain-containing protein E1/UNR |
| EIF3A | Eukaryotic translation initiation factor 3, subunit A | Eukaryotic translation initiation factor 3, subunit A/eIF3a |
| GNAS | GNAS complex locus/NESP | ALEX |
| Guanine nucleotide-binding protein G(s), subunit alpha isoform Xlas/Xlalpha | ||
| Guanine nucleotide-binding protein G(s), subunit alpha isoform Short | ||
| Neuroendocrine secretory protein 55/NESP55 | ||
| HSPA5 | Heat shock 70 kDa protein 5/GRP78 | 78 kDa glucose-regulated protein/GRP78/BiP |
| ITFG3 | Integrin alpha FG-GAP repeat containing 3 | ITFG3 |
| MAT2A | Methionine adenosyltransferase II/MATA2 | S-adenosylmethionine synthase, isoform type-2/MAT2 |
| MORF4L1 | Mortality factor 4 like 1/Eaf3/MRG15 | Mortality factor 4-like protein 1/MRG15 |
| PPAPDC1B | Phosphatidic acid phosphatase type 2, domain containing 1B/HTPAP | Phosphatide phosphatase PPAPDC1B/HTPAP/DPPL1 |
| PRSS12 | Protease, serine, 12 (neurotrypsin, motopsin)/MRT1 | Neurotrypsin/Motopsin |
| RAC1 | Ras-related C3 botulinum toxin substrate 1/p21-Rac1 | p21-Rac1/TC25 |
| RPL6 | RPL6/TXREB1 | 60S ribosomal protein L6/TaxREB107 |
| RPLP0 | Ribosomal protein large P0 | 60S acidic ribosomal protein P0 |
| SEMA3C | Sema domain, immunoglobulin domain (Ig)/SemE | Semaphorin-3C/Semaphorin E/Sema E |
| SF3B2 | Splicing factor 3b, subunit 2, 145 kDa/Cus1 | Splicing factor 3B subunit 2/SAP145 |
| STRADA | STE20-related kinase adaptor alpha/LYK5 /STRAD | STRAD alpha |
| STRN4 | Striatin, calmodulin-binding protein 4/ZIN | Sriatin-4/Zinedin |
| TTC37 | Tetratricopeptide repeat domain 37/KIAA0372 | TPR repeat protein 37/Ski3/Thespin |
| YKT6 | YKT6 v-SNARE homolog (S. cerevisiae)/“R-SNARE” | Synaptobrevin homolog YKT6 |
| Functions | Genes | Specific Functions for Proteins Encoded by Differentially Expressed Genes |
|---|---|---|
| Cell death and cell survival | CALM2 | Positive or negative regulator of apoptosis [1], regulator of autophagy [28] |
| CSDE1 | Activates IRES-mediated translation of proapoptotic protein Apaf1 during apoptosis [29] | |
| HSPA5 | Activator of UPR-induced autophagy mediated by PERK/eIF2α/ATF4 pathway [30] | |
| Inhibits p53-dependent apoptosis via PERK/eIF2 activation [31] and apoptosis via PERK/eIF2/NF-κB [32] | ||
| Enhances apoptosis via PERK/eIF2/ATF4/CHOP pathway [32] | ||
| MAT2A | Downregulated in renal cancer cells/tissues increasing a cell survival via HO-1 [33] | |
| MORF4L1 | Activator of tumour suppressor-mediated apoptosisUP and nuclear ligand for proapoptotic agent [34] | |
| RAC1 | Mediates cell survival via NF-κB when activated by SemaC342 and via PAKs25 and PI3K/Akt and p38/MAPK pathways [35] | |
| Promotes drug-induced apopotosis via JNK pathway [36] | ||
| RPLP0 | Upregulated during drug-induced apoptosis [37] | |
| SEMA3C | Mediates cell survival by activating Rac1 and NF-κB [32] | |
| YKT6 | As a subunit of SNARE, mediates autophagy via comprising of phagophores [38] | |
| Cell cycle | ARHGAP35 | Positive regulator of cell cycle in lung carcinoma cells [39] |
| CALM2 | Controls cell cycle progression [28] | |
| CSDE1 | Activates IRES-mediated translation of cell cycle regulator PITSLRE during mitosis [40] | |
| MAT2A | Inhibits the growth of liver cancer cells via SAMe [41] | |
| MORF4L1 | Activator of tumour suppressor-mediated cell cycle arrestUP accompanied with DNA repair [42] | |
| RPL6 | Inhibition of cell cycle via stabilization of p53 by suppressing MDM2 activity [43] | |
| STRADA | Mediates G1 cell cycle arrest via activating tumor suppressor LKB1 [44] | |
| STRN4 | Required for the completion of cytokinesis (abscission) while interacting with Mink1 [45] | |
| YKT6 | Over-expression alters cell cycle by increasing mitotic index, DNA synthesis and decreasing cell size [46] | |
| Cellular stress | CALM2 | Regulates CaMKII, a significant mediator of ER stress induced apoptosis [28] |
| HSPA5 | Sensor and inducer of ER stress [30,47] and overexpressed itself during ER stress [47] | |
| Under ER stress, reduce translation and protein synthesis via PERK/eIF2 [5] | ||
| MORF4L1 | Responses to DNA damage repairing DNA double strand breaks [48,49] | |
| RAC1 | Under cellular stress, mediates cell survival via PAKs [9], PI3K/Akt- and p38/MAPK-signaling [35] | |
| RPL6 | Under ribosomal stress, stabilize p53 by suppressing MDM2 activity [43] | |
| RPLP0 | Upregulated under cellular stress, e.g., ribosomal stress or drug-induced stress [12,50,51] | |
| YKT6 | Under ER stress, mediates apoptosis as a subunit of SNARE [52] | |
| Cell shape, motility and polarity | ARHGAP35 | Inhibits cell motility and migration via folic acid receptor/cSrc/p190RhoGAP pathway [53] |
| RAC1 | Activator of cell motility via PAKs [54] | |
| STRADA | Regulation of cell polarity via activating LKB1 and via Rac1 and PAK1 [21] | |
| Protein synthesis | ALKBH5 | Involved in mRNA export and modifications [55] |
| CSDE1 | RNA binding proteinUP and IRES transacting factor (ITAF) [56] | |
| Under stress conditions, helps eIFs and other ITAFs to initiate alternative IRES-mediated protein synthesis [56] | ||
| EIF3A | Protein synthesis (translation)UP | |
| HSPA5 | Reduce translation and protein synthesis via PERK/eIF2 [32] | |
| MORF4L1 | Regulates chromatin remodeling during transcription via HAT and HDAC complexes [43] | |
| RPL6 | Ribosomal protein, protein synthesis (translation)UP | |
| RPLP0 | Ribosomal protein, protein synthesis (translation)UP | |
| SF3B2 | Splices mRNAUP | |
| TTC37 | As a subunit of SKI complex, mediates RNA surveillance with PAF complex [57] and assists exosome in mRNA degradationUP | |
| Ca2+-signaling | CALM2 | Ca2+-binding protein, the major regulator of calcium-mediated signaling [28] |
| HSPA5 | Ca2+-dependent chaperoneUP, involved in regulation of calcium leak during ER stress [47] | |
| STRN4 | Regulates Ca2+-signaling via calmodulin [57], possibly a sensor responding to the concentration of calcium [4] | |
| Enzymatic and protein-protein interactions | CALM2 | Interacts calcium-dependently with hundreds of proteins [28], e.g., striatin family proteins including zinedin [4,22] |
| EIF3A | Subunit of eukaryotic initiation factor eIF3 with eIF3bUP | |
| HSPA5 | Binds and inactivate UPR-related signaling molecules PERK and ATF4 [30], interacts with p53 [23] | |
| MAT2A | Catalyzes the synthesis of biological methyl donor SAMeUP | |
| MORF4L1 | Subunit of transcription regulators HAT and HDAC complexes mediating chromatin [24] | |
| RPL0 | Ribosomal protein—A component of a ribosome large 60S subunitHGNC | |
| RPLP0 | Ribosomal protein—A component of a ribosome large 60S subunitHGNC | |
| STRADA | Subunit of LKB1-STRADα-MO25 and STRADα-PAK1 complexes [21] | |
| STRN4 | Regulatory subunit of PP2A in a STRIPAK complex [22], binds with several proteins including Mink1 [45] and calmodulin [4] | |
| TTC37 | Subunit of a SKI complex which assists exosome in a mRNA degradationUP and associate with PAF in transcription [57] | |
| YKT6 | Subunit of SNARE complexes which function in tranportationUP |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lantto, T.A.; Laakso, I.; Dorman, H.J.D.; Mauriala, T.; Hiltunen, R.; Kõks, S.; Raasmaja, A. Cellular Stress and p53-Associated Apoptosis by Juniperus communis L. Berry Extract Treatment in the Human SH-SY5Y Neuroblastoma Cells. Int. J. Mol. Sci. 2016, 17, 1113. https://doi.org/10.3390/ijms17071113
Lantto TA, Laakso I, Dorman HJD, Mauriala T, Hiltunen R, Kõks S, Raasmaja A. Cellular Stress and p53-Associated Apoptosis by Juniperus communis L. Berry Extract Treatment in the Human SH-SY5Y Neuroblastoma Cells. International Journal of Molecular Sciences. 2016; 17(7):1113. https://doi.org/10.3390/ijms17071113
Chicago/Turabian StyleLantto, Tiina A., Into Laakso, H. J. Damien Dorman, Timo Mauriala, Raimo Hiltunen, Sulev Kõks, and Atso Raasmaja. 2016. "Cellular Stress and p53-Associated Apoptosis by Juniperus communis L. Berry Extract Treatment in the Human SH-SY5Y Neuroblastoma Cells" International Journal of Molecular Sciences 17, no. 7: 1113. https://doi.org/10.3390/ijms17071113
APA StyleLantto, T. A., Laakso, I., Dorman, H. J. D., Mauriala, T., Hiltunen, R., Kõks, S., & Raasmaja, A. (2016). Cellular Stress and p53-Associated Apoptosis by Juniperus communis L. Berry Extract Treatment in the Human SH-SY5Y Neuroblastoma Cells. International Journal of Molecular Sciences, 17(7), 1113. https://doi.org/10.3390/ijms17071113