
Cell Fusion in the War on Cancer: A Perspective on the Inception of Malignancy



Abstract
:1. Introduction
2. Cell Fusion in Health and Cancer
3. Our Interest in Cell Fusion
4. From Cell Fusion to Oncogenesis
5. Cell Fusion in Cancer Evolution and Progression
6. Reflections on the Role of Cell Fusion in Oncogenesis and Cancer Progression
7. Cell Fusion in the War on Cancer
Acknowledgments
Conflicts of Interest
References
- Price, D.E. The politics of the war on cancer. Science 1978, 199, 966–967. [Google Scholar] [CrossRef] [PubMed]
- Stebbing, G.F. Total war on cancer. Br. Med. J. 1946, 2, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Dukes, C.E. The origin and early history of the imperial cancer research fund. Ann. R. Coll. Surg. Engl. 1965, 36, 325–338. [Google Scholar] [PubMed]
- Epstein, S.S. Losing the war against cancer: Who’s to blame and what to do about it. Int. J. Health Serv. 1990, 20, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Hoyert, D.L. 75 years of mortality in the United States, 1935–2010. In NCHS Data Brief, No. 88; National Center for Health Statistics: Hyattsville, MD, USA, 2012; pp. 1–8. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Murphy, S.L.; Kochanek, K.D.; Bastian, B.A. Deaths: Final data for 2013. Natl. Vital Stat. Rep. 2016, 64, 1–119. [Google Scholar] [PubMed]
- Cornell, D.H. The war on cancer: From the benign to the malignant. N. Engl. J. Med. 1997, 94, 27–30. [Google Scholar]
- Topalian, S.L.; Wolchok, J.D.; Chan, T.A.; Mellman, I.; Palucka, K.; Banchereau, J.; Rosenberg, S.A.; Dane Wittrup, K. Immunotherapy: The path to win the war on cancer? Cell 2015, 161, 185–186. [Google Scholar] [PubMed]
- Hanahan, D. Rethinking the war on cancer. Lancet 2014, 383, 558–563. [Google Scholar] [CrossRef]
- Littman, D.R. Releasing the brakes on cancer immunotherapy. Cell 2015, 162, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Koch, M. Cancer immunotherapy booster. Cell 2016, 165, 253–255. [Google Scholar]
- Nordling, C.O. A new theory on cancer-inducing mechanism. Br. J. Cancer 1953, 7, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Armitage, P.; Doll, R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br. J. Cancer 1954, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Land, H.; Parada, L.F.; Weinberg, R.A. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 1983, 304, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. The path to cancer—Three strikes and you’re out. N. Engl. J. Med. 2015, 373, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Marchionni, L.; Nowak, M.A.; Parmigiani, G.; Vogelstein, B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Crossan, G.P.; Garaycoechea, J.I.; Patel, K.J. Do mutational dynamics in stem cells explain the origin of common cancers? Cell Stem Cell 2015, 16, 111–112. [Google Scholar] [CrossRef] [PubMed]
- Rozhok, A.I.; Wahl, G.M.; deGregori, J. A critical examination of the “bad luck” explanation of cancer risk. Cancer Prev. Res. 2015, 8, 762–764. [Google Scholar] [CrossRef] [PubMed]
- Belpomme, D.; Irigaray, P. Replicative random mutations as an unproven cause of cancer: A technical comment. Mol. Clin. Oncol. 2016, 4, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E.L. Are most cancers caused by specific risk factors acting on tissues with high underlying stem cell divisions? J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Little, M.P.; Hendry, J.H.; Puskin, J.S. Lack of correlation between stem-cell proliferation and radiation- or smoking-associated cancer risk. PLoS ONE 2016, 11, e0150335. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Powers, S.; Zhu, W.; Hannun, Y.A. Substantial contribution of extrinsic risk factors to cancer development. Nature 2016, 529, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Cavuto, S.; Apolone, G.; Noonan, D.M. Strategies to prevent “bad luck” in cancer. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Duelli, D.M.; Padilla-Nash, H.M.; Berman, D.; Murphy, K.M.; Ried, T.; Lazebnik, Y. A virus causes cancer by inducing massive chromosomal instability through cell fusion. Curr. Biol. 2007, 17, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Merchak, K.; Lee, W.; Grande, J.P.; Cascalho, M.; Platt, J.L. Cell fusion connects oncogenesis with tumor evolution. Am. J. Pathol. 2015, 185, 2049–2060. [Google Scholar] [CrossRef] [PubMed]
- Ogle, B.M.; Cascalho, M.; Platt, J.L. Biological implications of cell fusion. Nat. Rev. Mol. Cell Biol. 2005, 6, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.R. The cell theory: A restatement, history and critique. Part IV. The multiplication of cells. Q. J. Microsc. Sci. 1953, 94, 407–440. [Google Scholar]
- Wolpert, L. The evolution of ‘the cell theory’. Curr. Biol. CB 1996, 6, 225–228. [Google Scholar] [CrossRef]
- Oren-Suissa, M.; Podbilewicz, B. Cell fusion during development. Trends Cell Biol. 2007, 17, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Platt, J.L. Molecular and cellular mechanisms of mammalian cell fusion. Adv. Exp. Med. Biol. 2011, 713, 33–64. [Google Scholar] [PubMed]
- Helming, L.; Gordon, S. Molecular mediators of macrophage fusion. Trends Cell Biol. 2009, 19, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Barski, G.; Sorieul, S.; Cornefert, F. “Hybrid” type cells in combined cultures of two different mammalian cell strains. J. Natl. Cancer Inst. 1961, 26, 1269–1291. [Google Scholar] [PubMed]
- Sorieul, S.; Ephrussi, B. Karyological demonstration of hybridization of mammalian cells in vitro. Nature 1961, 190, 653–654. [Google Scholar] [CrossRef]
- Harris, H. Cell fusion and the analysis of malignancy. Proc. R. Soc. Lond. Ser. B Biol. Soc. 1971, 179, 1–20. [Google Scholar] [CrossRef]
- Bjerkvig, R.; Tysnes, B.B.; Aboody, K.S.; Najbauer, J.; Terzis, A.J. Opinion: The origin of the cancer stem cell: Current controversies and new insights. Nat. Rev. Cancer 2005, 5, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, B.M.; Harrell, J.C.; Jedlicka, P.; Borges, V.F.; Varella-Garcia, M.; Horwitz, K.B. Spontaneous fusion with, and transformation of mouse stroma by, malignant human breast cancer epithelium. Cancer Res. 2006, 66, 8274–8279. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Kang, Y. Cell fusion as a hidden force in tumor progression. Cancer Res. 2009, 69, 8536–8539. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.E.; Anderson, E.C.; Davies, P.S.; Silk, A.D.; Pelz, C.; Impey, S.; Wong, M.H. Fusion between intestinal epithelial cells and macrophages in a cancer context results in nuclear reprogramming. Cancer Res. 2011, 71, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Berndt, B.; Zanker, K.S.; Dittmar, T. Cell fusion is a potent inducer of aneuploidy and drug resistance in tumor cell/normal cell hybrids. Crit. Rev. Oncog. 2013, 18, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, T.; Zanker, K.S. Tissue regeneration in the chronically inflamed tumor environment: Implications for cell fusion driven tumor progression and therapy resistant tumor hybrid cells. Int. J. Mol. Sci. 2015, 16, 30362–30381. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Lazova, R.; Davies, S.; Backvall, H.; Ponten, F.; Brash, D.; Pawelek, J. Donor DNA in a renal cell carcinoma metastasis from a bone marrow transplant recipient. Bone Marrow Transplant. 2004, 34, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Y.; Lazova, R.; Qumsiyeh, M.; Cooper, D.; Pawelek, J. Donor y chromosome in renal carcinoma cells of a female bmt recipient: Visualization of putative bmt-tumor hybrids by fish. Bone Marrow Transplant. 2005, 35, 1021–1024. [Google Scholar] [CrossRef] [PubMed]
- Lazova, R.; Laberge, G.S.; Duvall, E.; Spoelstra, N.; Klump, V.; Sznol, M.; Cooper, D.; Spritz, R.A.; Chang, J.T.; Pawelek, J.M. A melanoma brain metastasis with a donor-patient hybrid genome following bone marrow transplantation: First evidence for fusion in human cancer. PLoS ONE 2013, 8, e66731. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, D.M.; Gotz, H. On the ‘human’ nature of highly malignant heterotransplantable tumors of human origin. Eur. J. Cancer 1968, 4, 547–548. [Google Scholar] [CrossRef]
- Miller, F.R.; McInerney, D.; Rogers, C.; Miller, B.E. Spontaneous fusion between metastatic mammary tumor subpopulations. J. Cell. Biochem. 1988, 36, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Rachkovsky, M.; Sodi, S.; Chakraborty, A.; Avissar, Y.; Bolognia, J.; McNiff, J.M.; Platt, J.; Bermudes, D.; Pawelek, J. Melanoma × macrophage hybrids with enhanced metastatic potential. Clin. Exp. Metastasis 1998, 16, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Ogle, B.M.; Knudsen, B.E.; Nishitai, R.; Ogata, K.; Platt, J.L. Toward the development of human T cells in swine for potential use in adoptive T cell immunotherapy. Tissue Eng. 2009, 15, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Zanjani, E.D.; Pallavicini, M.G.; Ascensao, J.L.; Flake, A.W.; Langlois, R.G.; Reitsma, M.; MacKintosh, F.R.; Stutes, D.; Harrison, M.R.; Tavassoli, M. Engraftment and long-term expression of human fetal hemopoietic stem cells in sheep following transplantation in utero. J. Clin. Investig. 1992, 89, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Zanjani, E.D.; Flake, A.W.; Rice, H.; Hedrick, M.; Tavassoli, M. Long-term repopulating ability of xenogeneic transplanted human fetal liver hematopoietic stem cells in sheep. J. Clin. Investig. 1994, 93, 1051–1055. [Google Scholar] [CrossRef] [PubMed]
- Liechty, K.W.; MacKenzie, T.C.; Shaaban, A.F.; Radu, A.; Moseley, A.M.; Deans, R.; Marshak, D.R.; Flake, A.W. Human mesenchymal stem cells engraft and demonstrate site-specific differentiation after in utero transplantation in sheep. Nat. Med. 2000, 6, 1282–1286. [Google Scholar] [PubMed]
- Ogle, B.M.; Butters, K.A.; Plummer, T.B.; Ring, K.R.; Knudsen, B.E.; Litzow, M.R.; Cascalho, M.; Platt, J.L. Spontaneous fusion of cells between species yields transdifferentiation and retroviral transfer in vivo. FASEB J. 2004, 18, 548–550. [Google Scholar] [CrossRef] [PubMed]
- Quaroni, A.; Wands, J.; Trelstad, R.L.; Isselbacher, K.J. Epithelioid cell cultures from rat small intestine. Characterization by morphologic and immunologic criteria. J. Cell Biol. 1979, 80, 248–265. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Oates, P.S. IEC-6 cells are an appropriate model of intestinal iron absorption in rats. J. Nutr. 2002, 132, 680–687. [Google Scholar] [PubMed]
- Ouko, L.; Ziegler, T.R.; Gu, L.H.; Eisenberg, L.M.; Yang, V.W. Wnt11 signaling promotes proliferation, transformation, and migration of IEC6 intestinal epithelial cells. J. Biol. Chem. 2004, 279, 26707–26715. [Google Scholar] [CrossRef] [PubMed]
- Boucher, M.J.; Jean, D.; Vezina, A.; Rivard, N. Dual role of MEK/ERK signaling in senescence and transformation of intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G736–G746. [Google Scholar] [CrossRef] [PubMed]
- Nandan, M.O.; McConnell, B.B.; Ghaleb, A.M.; Bialkowska, A.B.; Sheng, H.; Shao, J.; Babbin, B.A.; Robine, S.; Yang, V.W. Kruppel-like factor 5 mediates cellular transformation during oncogenic kras-induced intestinal tumorigenesis. Gastroenterology 2008, 134, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Voisin, L.; Julien, C.; Duhamel, S.; Gopalbhai, K.; Claveau, I.; Saba-El-Leil, M.K.; Rodrigue-Gervais, I.G.; Gaboury, L.; Lamarre, D.; Basik, M.; et al. Activation of MEK1 or MEK2 isoform is sufficient to fully transform intestinal epithelial cells and induce the formation of metastatic tumors. BMC Cancer 2008, 8, 337. [Google Scholar] [CrossRef] [PubMed]
- Duhamel, S.; Hebert, J.; Gaboury, L.; Bouchard, A.; Simon, R.; Sauter, G.; Basik, M.; Meloche, S. Sef downregulation by ras causes MEK1/2 to become aberrantly nuclear localized leading to polyploidy and neoplastic transformation. Cancer Res. 2012, 72, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Swanton, C. Cancer evolution constrained by mutation order. N. Engl. J. Med. 2015, 372, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; de Lange, T. The causes and consequences of polyploidy in normal development and cancer. Annu. Rev. Cell Dev. Biol. 2011, 27, 585–610. [Google Scholar] [CrossRef] [PubMed]
- Dewhurst, S.M.; McGranahan, N.; Burrell, R.A.; Rowan, A.J.; Gronroos, E.; Endesfelder, D.; Joshi, T.; Mouradov, D.; Gibbs, P.; Ward, R.L.; et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 2014, 4, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Aktipis, C.A.; Boddy, A.M.; Gatenby, R.A.; Brown, J.S.; Maley, C.C. Life history trade-offs in cancer evolution. Nat. Rev. Cancer 2013, 13, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Sage, J.C.; Miller, M.R.; Verhaak, R.G.; Hippenmeyer, S.; Vogel, H.; Foreman, O.; Bronson, R.T.; Nishiyama, A.; Luo, L.; et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 2011, 146, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Blyth, K.; Morton, J.P.; Sansom, O.J. The right time, the right place: Will targeting human cancer-associated mutations to the mouse provide the perfect preclinical model? Curr. Opin. Genet. Dev. 2012, 22, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Goldstein, A.S.; Witte, O.N. Tissue recombination models for the study of epithelial cancer. Cold Spring Harb. Protoc. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Cairns, J. Mutation selection and the natural history of cancer. Nature 1975, 255, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Doll, R. An epidemiological perspective of the biology of cancer. Cancer Res. 1978, 38, 3573–3583. [Google Scholar] [PubMed]
- Armitage, P. Multistage models of carcinogenesis. Environ. Health Perspect. 1985, 63, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Peto, J. Cancer epidemiology in the last century and the next decade. Nature 2001, 411, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Meza, R.; Jeon, J.; Moolgavkar, S.H.; Luebeck, E.G. Age-specific incidence of cancer: Phases, transitions, and biological implications. Proc. Natl. Acad. Sci. USA 2008, 105, 16284–16289. [Google Scholar] [CrossRef] [PubMed]
- Noble, R.; Kaltz, O.; Hochberg, M.E. Peto’s paradox and human cancers. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Rosen, J.M.; Jordan, C.T. The increasing complexity of the cancer stem cell paradigm. Science 2009, 324, 1670–1673. [Google Scholar] [CrossRef] [PubMed]
- Tomasson, M.H. Cancer stem cells: A guide for skeptics. J. Cell Biochem. 2009, 106, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Stem cells and cancer—The promise and puzzles. Mol. Oncol. 2010, 4, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Coquelle, A.; Vivet, S.; Vitale, I.; Kauffmann, A.; Dessen, P.; Pequignot, M.O.; Casares, N.; Valent, A.; Mouhamad, S.; et al. Apoptosis regulation in tetraploid cancer cells. EMBO J. 2006, 25, 2584–2595. [Google Scholar] [CrossRef] [PubMed]
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009, 460, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Ganem, N.J.; Cornils, H.; Chiu, S.Y.; O’Rourke, K.P.; Arnaud, J.; Yimlamai, D.; Thery, M.; Camargo, F.D.; Pellman, D. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell 2014, 158, 833–848. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Ganem, N.J. Tetraploidy and tumor development. Oncotarget 2014, 5, 10959–10960. [Google Scholar] [CrossRef] [PubMed]
- Lissa, D.; Senovilla, L.; Rello-Varona, S.; Vitale, I.; Michaud, M.; Pietrocola, F.; Boileve, A.; Obrist, F.; Bordenave, C.; Garcia, P.; et al. Resveratrol and aspirin eliminate tetraploid cells for anticancer chemoprevention. Proc. Natl. Acad. Sci. USA 2014, 111, 3020–3025. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Guan, K.L. Hippo pathway key to ploidy checkpoint. Cell 2014, 158, 695–696. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Huch, M.; van Boxtel, R.; Karthaus, W.; Wedge, D.C.; Tamuri, A.U.; Martincorena, I.; Petljak, M.; Alexandrov, L.B.; Gundem, G.; et al. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature 2014, 513, 422–425. [Google Scholar] [CrossRef] [PubMed]
- Vijg, J.; Busuttil, R.A.; Bahar, R.; Dolle, M.E. Aging and genome maintenance. Ann. N. Y. Acad. Sci. 2005, 1055, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Busuttil, R.A.; Garcia, A.M.; Reddick, R.L.; Dolle, M.E.; Calder, R.B.; Nelson, J.F.; Vijg, J. Intra-organ variation in age-related mutation accumulation in the mouse. PLoS ONE 2007, 2, e876. [Google Scholar] [CrossRef] [PubMed]
- Cairns, J. Somatic stem cells and the kinetics of mutagenesis and carcinogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 10567–10570. [Google Scholar] [CrossRef] [PubMed]
- Potten, C.S.; Owen, G.; Booth, D. Intestinal stem cells protect their genome by selective segregation of template DNA strands. J. Cell Sci. 2002, 115, 2381–2388. [Google Scholar] [PubMed]
- Gandara, R.M.; Mahida, Y.R.; Potten, C.S. Regional differences in stem and transit cell proliferation and apoptosis in the terminal ileum and colon of mice after 12 Gy. Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, e521–e528. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.W.; Taylor, M.H.; Hickey, R.D.; Hanlon Newell, A.E.; Lenzi, M.L.; Olson, S.B.; Finegold, M.J.; Grompe, M. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 2010, 467, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.W.; Hickey, R.D.; Paulk, N.K.; Culberson, A.J.; Olson, S.B.; Finegold, M.J.; Grompe, M. Ploidy reductions in murine fusion-derived hepatocytes. PLoS Genet. 2009, 5, e1000385. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Munger, K. Viruses associated with human cancer. Biochim. Biophys. Acta 2008, 1782, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Plafker, K.; Vorozhko, V.; Zuna, R.E.; Hanigan, M.H.; Gorbsky, G.J.; Plafker, S.M.; Angeletti, P.C.; Ceresa, B.P. Human papillomavirus 16 E5 induces bi-nucleated cell formation by cell-cell fusion. Virology 2009, 384, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Zheng, J. Oncogenic virus-mediated cell fusion: New insights into initiation and progression of oncogenic viruses-related cancers. Cancer Lett. 2011, 303, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.H.; Chan, J.; Lambele, M.; Thali, M. Clustering and mobility of HIV-1 Env at viral assembly sites predict its propensity to induce cell-cell fusion. J. Virol. 2013, 87, 7516–7525. [Google Scholar] [CrossRef] [PubMed]
- Pertel, P.E. Human herpesvirus 8 glycoprotein B (gB), gH, and gL can mediate cell fusion. J. Virol. 2002, 76, 4390–4400. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, H.; Shimoyama, M.; Miwa, M.; Sugimura, T. Detection of lymphocytes producing a human retrovirus associated with adult T-cell leukemia by syncytia induction assay. Proc. Natl. Acad. Sci. USA 1983, 80, 7337–7341. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, G.J.; Wolf, H. An Epstein—Barr virus early protein induces cell fusion. Proc. Natl. Acad. Sci. USA 1981, 78, 7162–7165. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.; Miller, O.J.; Klein, G.; Worst, P.; Tachibana, T. Suppression of malignancy by cell fusion. Nature 1969, 223, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Harris, H. The Biology of Tumour Suppression. In Genetic Analysis of Tumor Suppression; Bock, G., Marsh, J., Eds.; CIBA Foundation Symposium: Chichester, NY, USA, 1989; Volume 142, pp. 199–208. [Google Scholar]
- Winton, D.J.; Blount, M.A.; Ponder, B.A. Polyclonal origin of mouse skin papillomas. Br. J. Cancer 1989, 60, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, D.M.; Rooney, R.J.; Loo, M.; Liu, D.; Chang, C.H. In-vivo fusion of human cancer and hamster stromal cells permanently transduces and transcribes human DNA. PLoS ONE 2014, 9, e107927. [Google Scholar] [CrossRef] [PubMed]
- Lazebnik, Y. The shock of being united: Another lesson from plants? Cell Cycle 2014, 13. [Google Scholar] [CrossRef] [PubMed]
- Platt, J.L.; Cascalho, M. IgM in the kidney: A multiple personality disorder. Kidney Int. 2015, 88, 439–441. [Google Scholar] [CrossRef] [PubMed]
- Saadi, S.; Wrenshall, L.E.; Platt, J.L. Regional manifestations and control of the immune system. FASEB J. 2002, 16, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Cascalho, M.; Platt, J.L. The immunologic barriers to replacing damaged organs. Curr. Immunol. Rev. 2006, 2, 65–72. [Google Scholar] [CrossRef]
- Platt, J.L.; Cascalho, M. Transplantation immunology. In Greenfield’s Surgery: Scientific Principles and Practice, 5th ed.; Mulholland, M.W., Lillemoe, K.D., Doherty, G.M., Maier, R.V., Simeone, D.M., Upchurch, G.R.J., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2011; pp. 497–514. [Google Scholar]
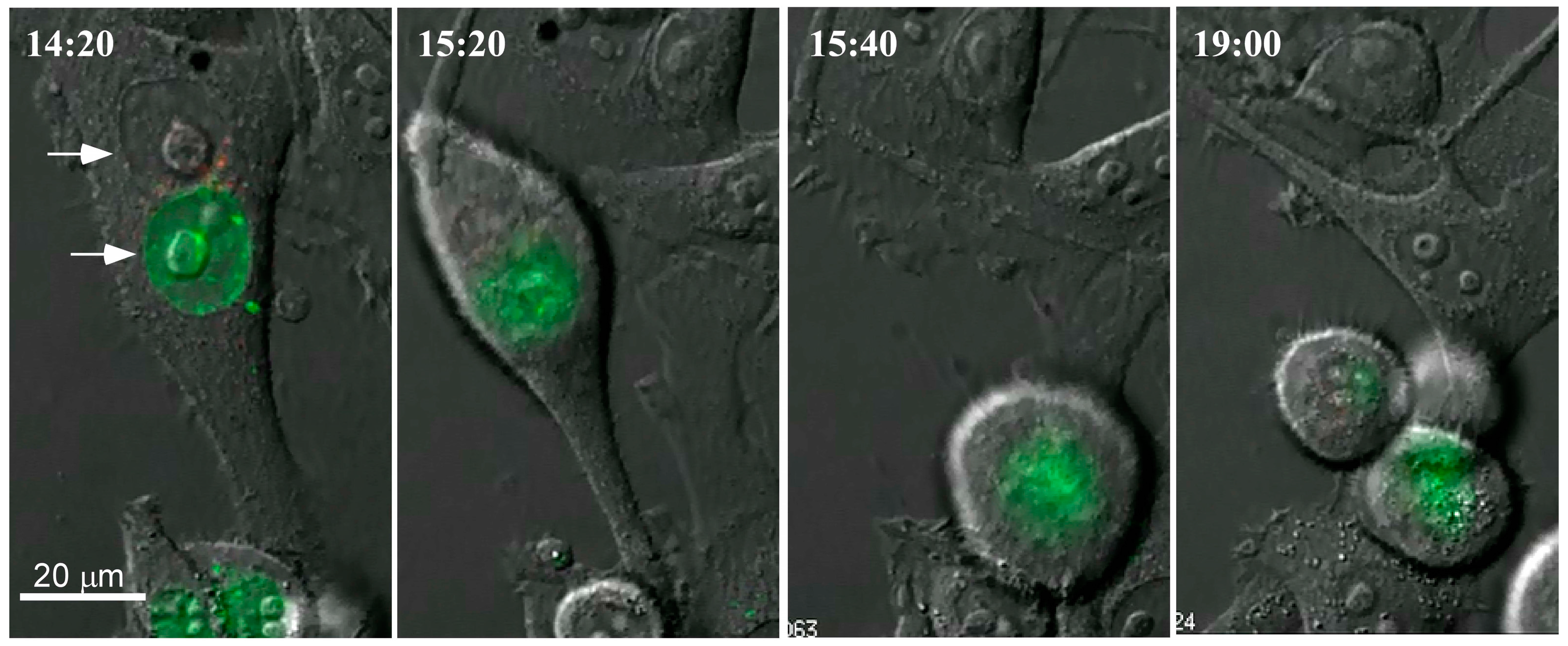

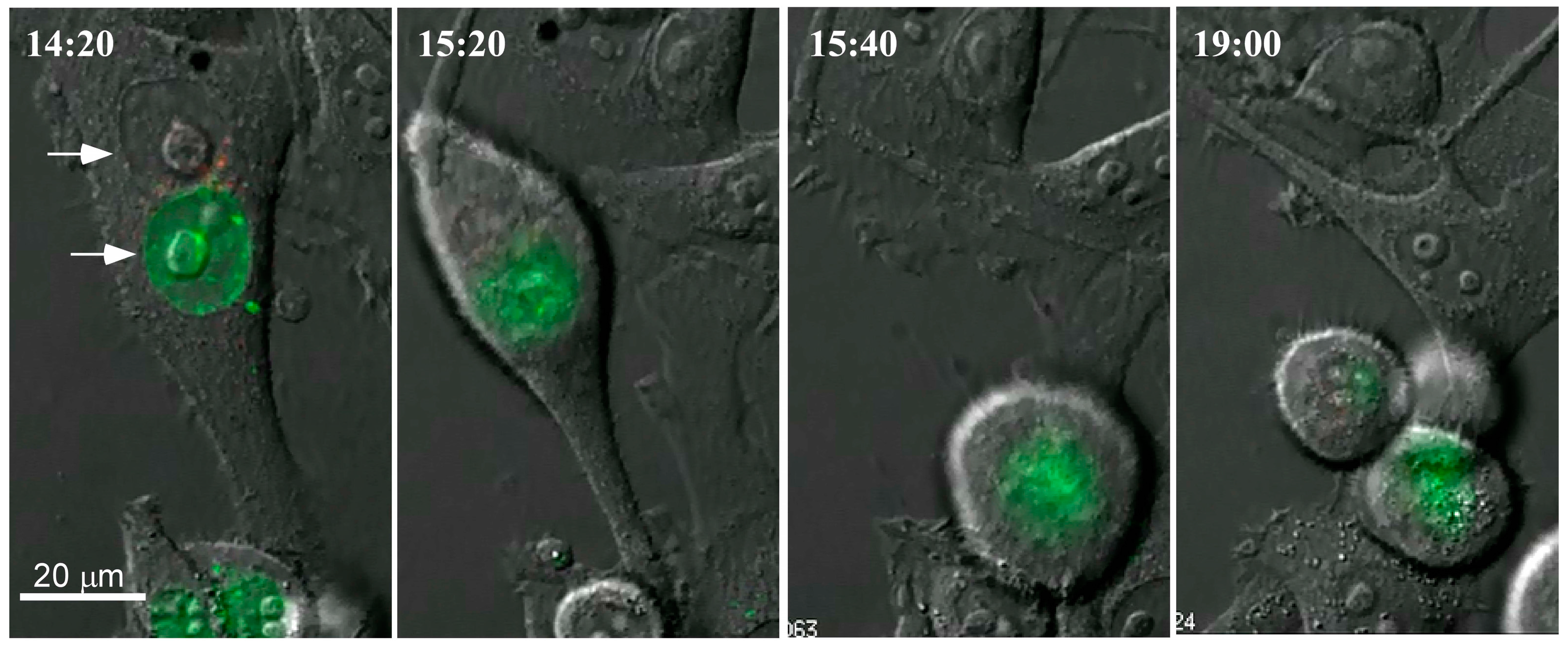{kind=link}
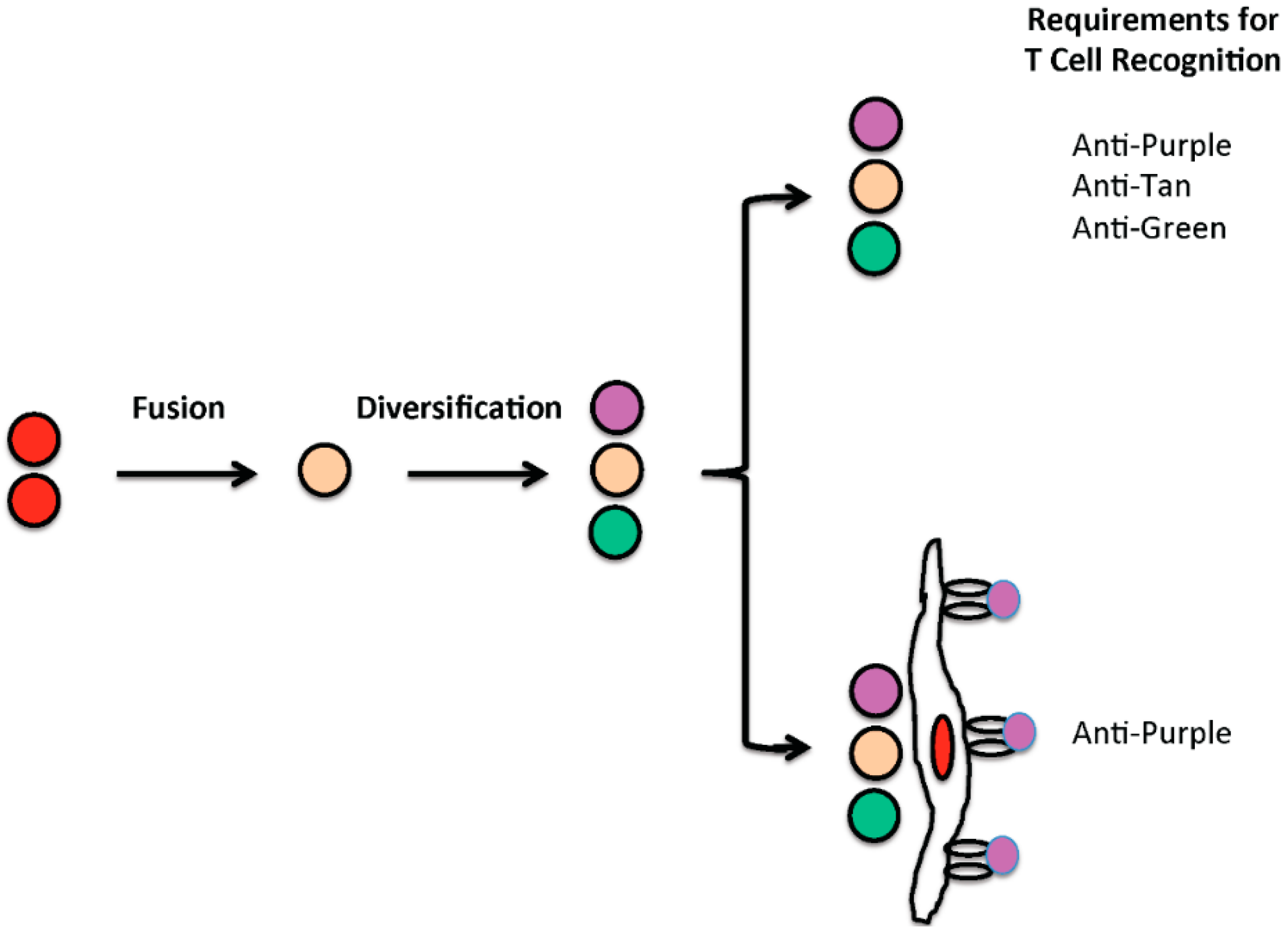
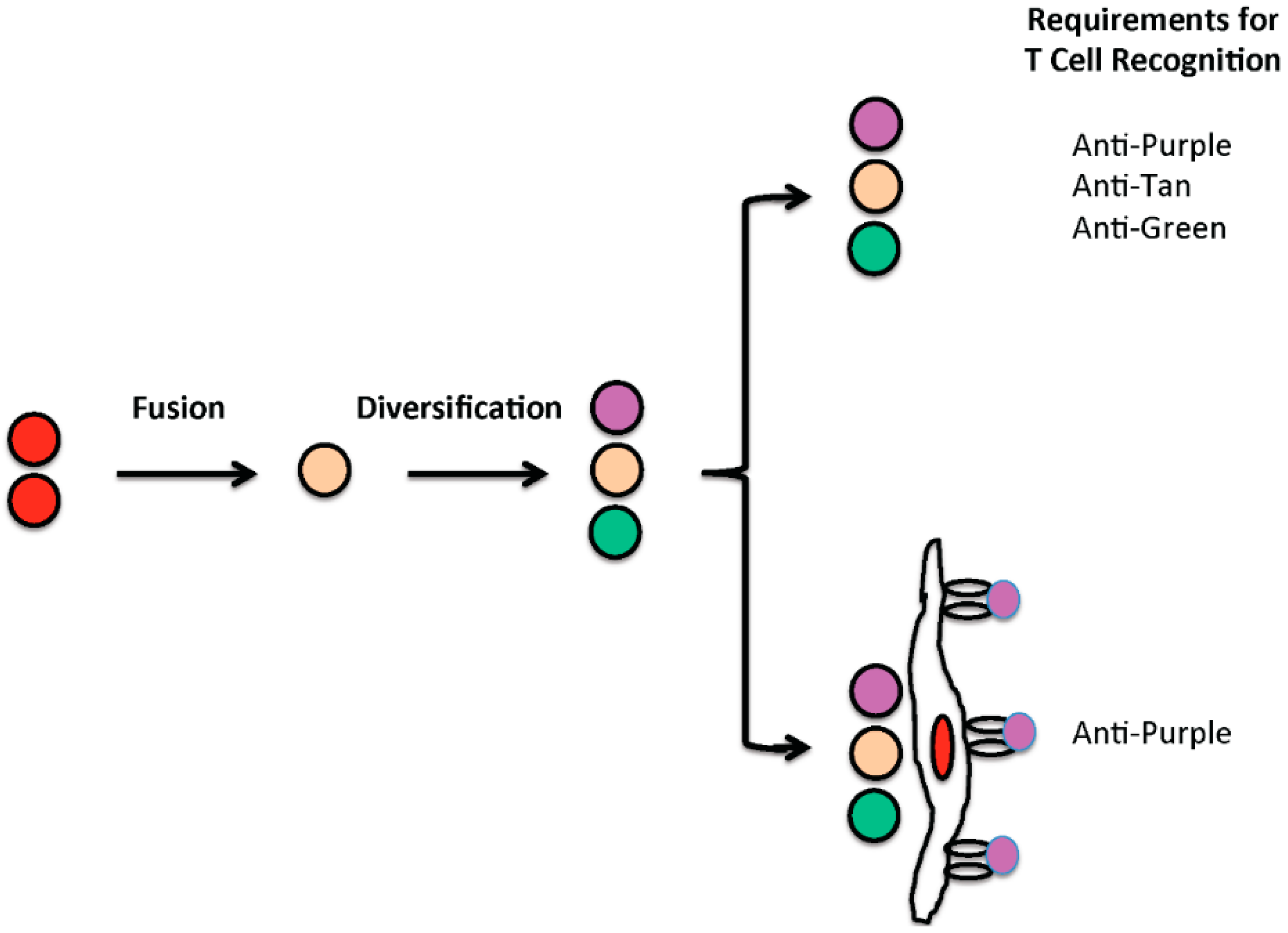{kind=link}
| Year | US Population × 106 | Number of Cancer Deaths | Crude Death Rate |
|---|---|---|---|
| 2013 | 315 | 611,105 | 193 |
| 1971 | 208 | 330,730 | 163 |
| 1946 | 141 | 182,005 | 130 |
| 1923 | 112 | 85,575 | 88 |
| Cancer | New Cases (2013) | Percent of Total |
|---|---|---|
| Prostate | 238,590 | 14.37 |
| Breast | 234,580 | 14.13 |
| Lung | 228,190 | 13.74 |
| Lymphoid ** | 123,130 | 7.42 |
| Colon | 102,480 | 6.17 |
| Kidney | 65,150 | 3.92 |
| Thyroid | 60,220 | 3.63 |
| Pancreas | 45,220 | 2.72 |
| Liver | 30,640 | 1.85 |
| Myeloid | 20,510 | 1.24 |
| Muscle # | 11,410 | 0.07 |
| Small intestine | 8810 | 0.05 |
| Cardiac + | 102 | 0.00006 |
| Total new cancers ^ | 1,660,290 |
| Virus | Affected Cells | Cancer | Frequency (Estimated) | Reference |
|---|---|---|---|---|
| Human papillomavirus | epithelial cells | cervical | 12,900 | [100] |
| anal | 7270 | |||
| oropharyngeal | 15,520 | |||
| Hepatitis C, hepatitis B | hepatocytes | hepatoma | 30,640 # | [101] |
| HIV * | T cells | [102] | ||
| endothelial cells | Kaposi (soft tissue) sarcoma | 1943 | ||
| HHV-8 | endothelial cells | Kaposi sarcoma | 1943 | [103] |
| HTLV-1 | T cells | T cell leukemia | rare | [104] |
| EBV | B cells | Burkitt’s | 1652 | [105] |
| epithelial cells | Lymphoma Nasopharyngeal | 3200 | ||
| Total virus associated | 73,125 | |||
| Total new cancers * | 1,660,290 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Platt, J.L.; Zhou, X.; Lefferts, A.R.; Cascalho, M. Cell Fusion in the War on Cancer: A Perspective on the Inception of Malignancy. Int. J. Mol. Sci. 2016, 17, 1118. https://doi.org/10.3390/ijms17071118
Platt JL, Zhou X, Lefferts AR, Cascalho M. Cell Fusion in the War on Cancer: A Perspective on the Inception of Malignancy. International Journal of Molecular Sciences. 2016; 17(7):1118. https://doi.org/10.3390/ijms17071118
Chicago/Turabian StylePlatt, Jeffrey L., Xiaofeng Zhou, Adam R. Lefferts, and Marilia Cascalho. 2016. "Cell Fusion in the War on Cancer: A Perspective on the Inception of Malignancy" International Journal of Molecular Sciences 17, no. 7: 1118. https://doi.org/10.3390/ijms17071118
APA StylePlatt, J. L., Zhou, X., Lefferts, A. R., & Cascalho, M. (2016). Cell Fusion in the War on Cancer: A Perspective on the Inception of Malignancy. International Journal of Molecular Sciences, 17(7), 1118. https://doi.org/10.3390/ijms17071118