
Roles of d-Amino Acids on the Bioactivity of Host Defense Peptides



Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Natural d-Amino Acids (d-AA)-Containing HDPs
3. Synthetic d-AA-Containing HDPs
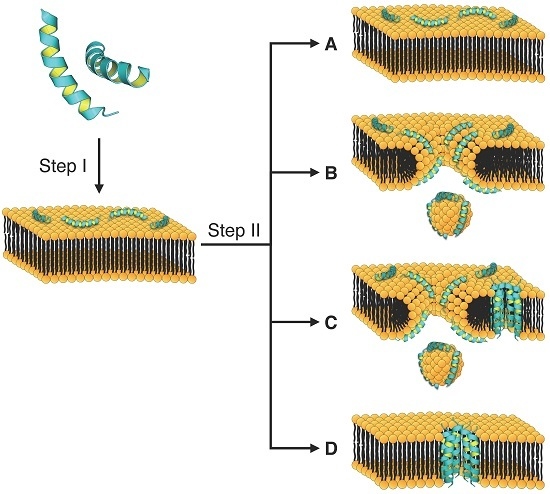
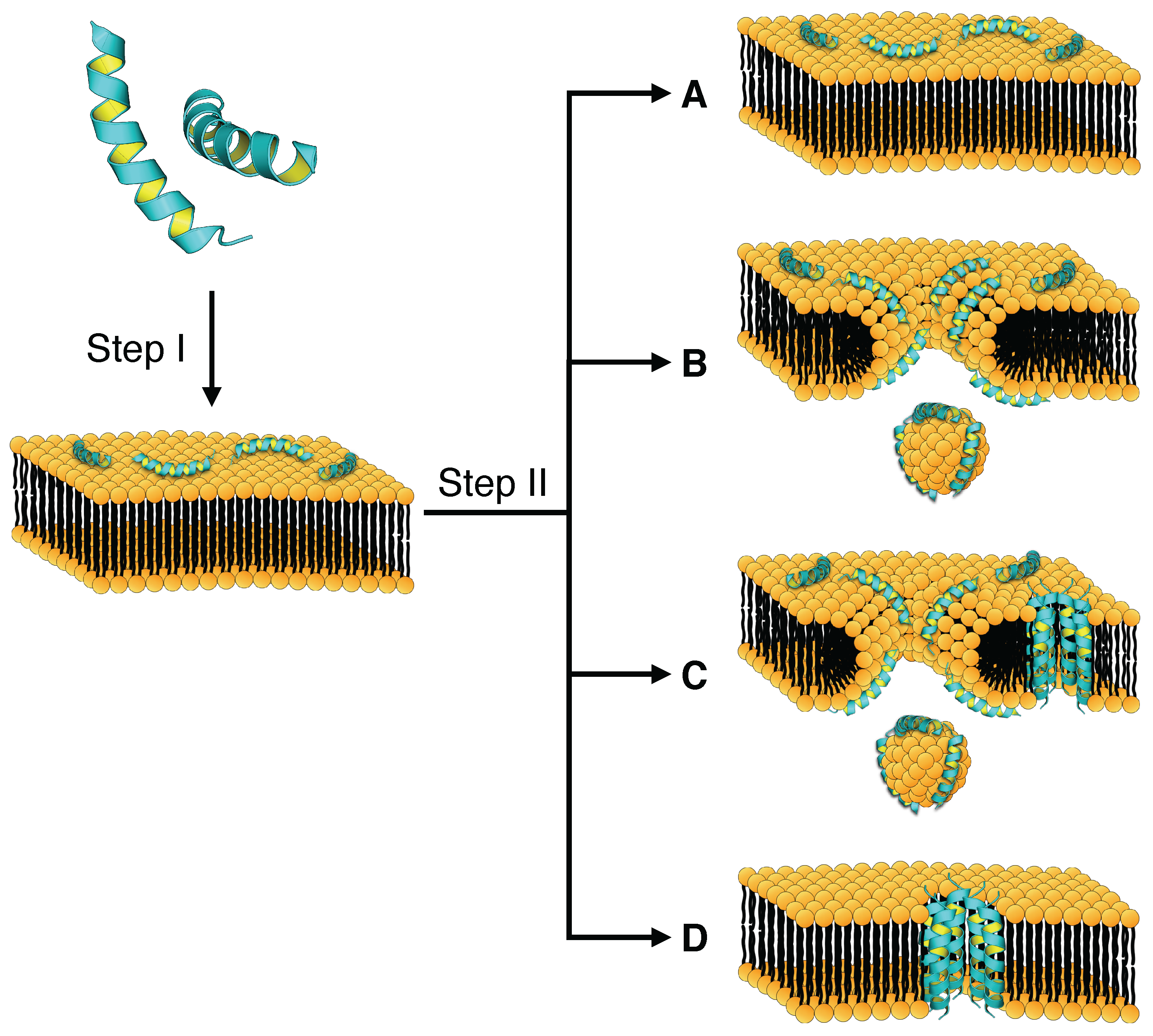
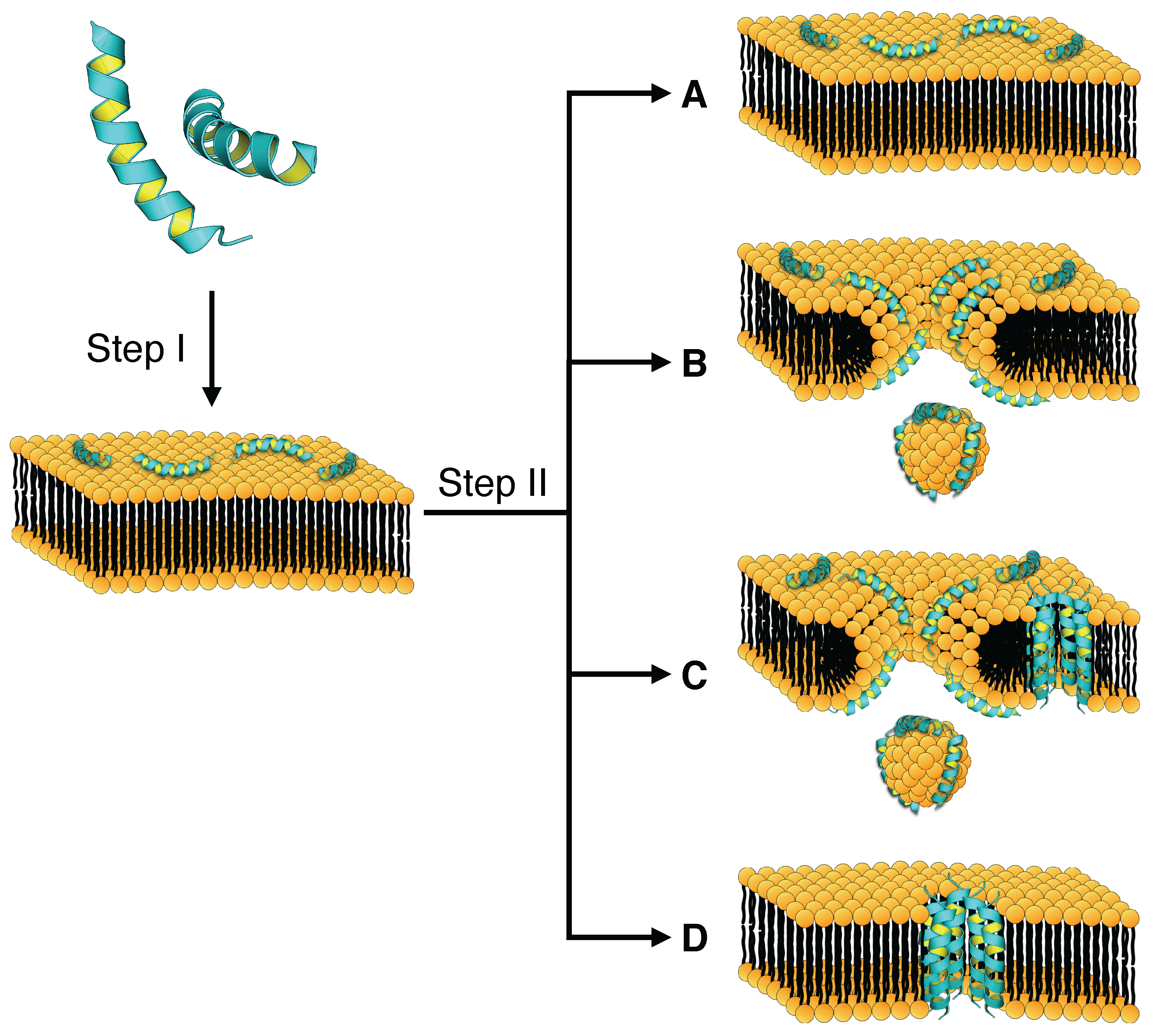
4. Mechanism of Membrane Destabilization
5. Mechanisms of Bioactivities
5.1. Antimicrobial Activity
5.2. Anticancer Activity
5.3. Antiviral Activity
5.4. Antibiofilm Activity
5.5. Immunological Effects of d-AA-Containing HDPs
5.5.1. Immune Modulation
5.5.2. Apoptotic Activity
5.6. Influence of Secondary Structure on Bioactivity
5.7. Diastereomeric vs. All-l and All-d Enantiomeric HDPs
6. In Vivo Testing of d-AA-Containing HDPs
6.1. Targeting Cancer Cells
- The benefit of long-term survival with nearly no side effects was demonstrated.
- The tumor was unable to develop resistance against the diastereomeric peptide tested by Papo et al. [13] even after prolonged exposure. This finding is in line with a previous observation by Hilchie et al. [2], who reported that no acquisition of resistance to HDPs by cancer has ever been documented. Furthermore, l-AA AMPs have been shown to induce resistance in bacteria when applied using graded doses for a prolonged period of time [8]. This finding may be due to the greater mutation potential of prokaryotes than that of eukaryotic cancer cells. However, a gradient exposure study performed with an all-d AMP in E. coli and S. aureus showed that resistance did not develop against the all-d AMP [9]. This result indicated that the inclusion of d-AAs not only stabilizes the peptide against serum proteases but also likely protects them against bacterial proteases. Thus, d-AA inclusion as a method of boosting HDP resistance against pathogen evasion is an avenue that cannot be overlooked.
- The diastereomeric peptide tested by Papo et al. [13] was suitable for systemic application and was not conjugated to any homing motifs, nor did it depend on any delivery vesicles. The development of anticancer HDPs suited for systemic administration is important because this application remains the only route for combating metastatic cancer.
6.2. Targeting Bacterial Cells
6.3. Targeting Other Pathogens
7. Toxicity
8. Synthesis of d-AA-Containing HDPs
9. Computational Modeling of d-AA-Containing HDPs
10. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, G.; Hanke, M.L.; Mishra, B.; Lushnikova, T.; Heim, C.E.; Thomas, V.C.; Bayles, K.W.; Kielian, T. Transformation of human cathelicidin LL-37 into selective, stable, and potent antimicrobial compounds. ACS Chem. Biol. 2014, 9, 1997–2002. [Google Scholar] [CrossRef] [PubMed]
- Hilchie, A.L.; Doucette, C.D.; Pinto, D.M.; Patrzykat, A.; Douglas, S.; Hoskin, D.W. Pleurocidin-family cationic antimicrobial peptides are cytolytic for breast carcinoma cells and prevent growth of tumor xenografts. Breast Cancer Res. 2011, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Nan, Y.H.; Shin, S.Y. Candidacidal mechanism of a Leu/Lys-rich α-helical amphipathic model antimicrobial peptide and its diastereomer composed of d,l-amino acids. J. Pept. Sci. 2010, 16, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Lynn, M.A.; Kindrachuk, J.; Marr, A.K.; Jenssen, H.; Pante, N.; Elliott, M.R.; Napper, S.; Hancock, R.E.; McMaster, W.R. Effect of BMAP-28 antimicrobial peptides on Leishmania major promastigote and amastigote growth: Role of leishmanolysin in parasite survival. PLoS Negl. Trop. Dis. 2011, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Li, T.; Song, Y.; Zhang, R.; Zeng, Z.; Han, S.; Zhang, X.; Wu, Y.; Li, W.; Cao, Z. Inhibitory activity and mechanism of two scorpion venom peptides against herpes simplex virus type 1. Antivir. Res. 2014, 102, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hoskin, D.W.; Ramamoorthy, A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2008, 1778, 357–375. [Google Scholar] [CrossRef] [PubMed]
- Dathe, M.; Wieprecht, T. Structural features of helical antimicrobial peptides: Their potential to modulate activity on model membranes and biological cells. Biochim. Biophys. Acta 1999, 1462, 71–87. [Google Scholar] [CrossRef]
- Perron, G.G.; Zasloff, M.; Bell, G. Experimental evolution of resistance to an antimicrobial peptide. Proc. R. Soc. B 2006, 273, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.O.; Cheng, J.; Huang, Y.; Xu, K.; Ji, Z.; Fan, W.; Yang, Y.Y. Effect of stereochemistry, chain length and sequence pattern on antimicrobial properties of short synthetic β-sheet forming peptide amphiphiles. Biomaterials 2014, 35, 1315–1325. [Google Scholar]
- Buri, M.V.; Domingues, T.M.; Paredes-Gamero, E.J.; Casaes-Rodrigues, R.L.; Rodrigues, E.G.; Miranda, A. Resistance to degradation and cellular distribution are important features for the antitumor activity of gomesin. PLoS ONE 2013, 8, e80924. [Google Scholar] [CrossRef] [PubMed]
- Mansour, S.C.; Pena, O.M.; Hancock, R.E.W. Host defense peptides: Front-line immunomodulators. Trends Immunol. 2014, 35, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Ma, Q.; Zhu, H. Distribution, industrial applications, and enzymatic synthesis of d-amino acids. Appl. Microbiol. Biotechnol. 2015, 99, 3341–3349. [Google Scholar] [CrossRef] [PubMed]
- Papo, N.; Seger, D.; Makovitzki, A.; Kalchenko, V.; Eshhar, Z.; Degani, H.; Shai, Y. Inhibition of tumor growth and elimination of multiple metastases in human prostate and breast xenografts by systemic inoculation of a host defense-like lytic peptide. Cancer Res. 2006, 66, 5371. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Brown, J.H.; Morell, J.L.; Huang, C.M. Synthetic magainin analogues with improved antimicrobial activity. FEBS Lett. 1988, 236, 462–466. [Google Scholar] [CrossRef]
- Bessalle, R.; Kapitkovsky, A.; Gorea, A.; Shalit, I.; Fridkin, M. All-d-magainin: Chirality, antimicrobial activity and proteolytic resistance. FEBS Lett. 1990, 274, 151–155. [Google Scholar] [PubMed]
- Papo, N.; Oren, Z.; Pag, U.; Sahl, H.G.; Shai, Y. The consequence of sequence alteration of an amphipathic α-helical antimicrobial peptide and its diastereomers. J. Biol. Chem. 2002, 277, 33913–33921. [Google Scholar] [CrossRef] [PubMed]
- Wakayama, M.; Yoshimune, K.; Hirose, Y.; Moriguchi, M. Production of d-amino acids by N-acyl-d-amino acid amidohydrolase and its structure and function. J. Mol. Catal. B Enzym. 2003, 23, 71–85. [Google Scholar] [CrossRef]
- Chen, Y.; Mant, C.T.; Farmer, S.W.; Hancock, R.E.W.; Vasil, M.L.; Hodges, R.S. Rational design of α-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J. Biol. Chem. 2005, 280, 12316–12329. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, X.; Xie, Z.; Xie, C.; Zhan, C.; Hu, X.; Shen, Q.; Wei, X.; Su, B.; Wang, J.; et al. D-peptides as recognition molecules and therapeutic agents. Chem. Rec. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Al-Benna, S.; Shai, Y.; Jacobsen, F.; Steinstraesser, L. Oncolytic activities of host defense peptides. Int. J. Mol. Sci. 2011, 12, 8027–8051. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, F. Cationic amphiphilic peptides with cancer-selective toxicity. Eur. J. Pharmacol. 2009, 625, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, D.; Veiga, S.A.; Miguel, A.R.B.C. From antimicrobial to anticancer peptides. A review. Front. Microbiol. 2013, 4, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ong, Z.Y.; Wiradharma, N.; Yang, Y.Y. Strategies employed in the design and optimization of synthetic antimicrobial peptide amphiphiles with enhanced therapeutic potentials. Adv. Drug Deliv. Rev. 2014, 78, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Morizawa, K. The extractive substances in Octopus octopodia. Acta Sch. Med. Univ. Imp. Kyoto 1927, 9, 285–298. [Google Scholar]
- Ollivaux, C.; Soyez, D.; Toullec, J.Y. Biogenesis of d-amino acid containing peptides/proteins: Where, when and how? J. Pept. Sci. 2014, 20, 595–612. [Google Scholar] [CrossRef] [PubMed]
- Noriko, F.; Yuichi, K.; Norihiko, F. d-Amino acids in aged proteins: Analysis and biological relevance. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 3141–3147. [Google Scholar]
- Fujii, N.; Kaji, Y.; Fujii, N. d-Amino acids in aged proteins: Analysis and biological relevance. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 3141–3147. [Google Scholar] [CrossRef] [PubMed]
- Radkov, A.D.; Moe, L.A. Bacterial synthesis of d-amino acids. Appl. Microbiol. Biotechnol. 2014, 98, 5363–5374. [Google Scholar] [CrossRef] [PubMed]
- Montecucchi, P.C.; de Castiglione, R.; Piani, S.; Gozzini, L.; Erspamer, V.R. Amino acid composition and sequence of dermorphin, a novel opiate-like peptide from the skin of Phyllomedusa sauvagei. Int. J. Pept. Protein Res. 1981, 3, 275–283. [Google Scholar] [CrossRef]
- Morishita, F.; Furukawa, Y.; Matsushima, O. Molecular cloning of two distinct precursor genes of NdWFamide, a d-tryptophan-containing neuropeptide of the sea hare, Aplysia kurodai. Peptides 2012, 38, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Allan, M.T.; Maria, T.; Chryssanthi, T.; Eleanor, C.K.; Katherine, B.; Dominic, P.G.; Paramjit, S.B.; Paul, F.A.; Philip, W.K. Mammalian L-to-d-amino-acid-residue isomerase from platypus venom. FEBS Lett. 2006, 580, 1587–1591. [Google Scholar]
- Heck, S.D.; Faraci, W.S.; Kelbaugh, P.R.; Saccomano, N.A.; Thadeio, P.F.; Volkmann, R.A. Posttranslational amino acid epimerization: Enzyme-catalyzed isomerization of amino acid residues in peptide chains. Proc. Natl. Acad. Sci. USA 1996, 93, 4036–4039. [Google Scholar] [CrossRef] [PubMed]
- Suda, S.; Lawton, E.M.; Wistuba, D.; Cotter, P.D.; Hill, C.; Ross, R.P. Homologues and bioengineered derivatives of LtnJ vary in ability to form D-alanine in the lantibiotic lacticin 3147. J. Bacteriol. 2012, 194, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Kreil, G. d-amino acids in animal peptides. Annu. Rev. Biochem. 1997, 66, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Amiche, M.; Delfour, A.; Nicolas, P. Structural requirements for dermorphin opioid receptor binding. Int. J. Pept. Protein Res. 1988, 32, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Moshe, T.; Chiara, M.; Alessandro, M.; Marco, G.; Gianluca, D.M.; Alberto, P.; Piero, G.G. Different transcription regulation routes are exerted by l- and d-amino acid enantiomers of peptide hormones. J. Exp. Biol. 2014, 217, 4337–4346. [Google Scholar]
- Bufe, B.; Schumann, T.; Zufall, F. Formyl peptide receptors from immune and vomeronasal system exhibit distinct agonist properties. J. Biol. Chem. 2012, 287, 33644–33655. [Google Scholar] [CrossRef] [PubMed]
- Kelkar, D.A.; Chattopadhyay, A. The gramicidin ion channel: A model membrane protein. Biochim. Biophys. Acta Biomembr. 2007, 1768, 2011–2025. [Google Scholar] [CrossRef] [PubMed]
- Hladky, S.B.; Haydon, D.A. Ion transfer across lipid membranes in the presence of gramicidin A: I. Studies of the unit conductance channel. Biochim. Biophys. Acta Biomembr. 1972, 274, 294–312. [Google Scholar] [CrossRef]
- Gall, Y.M.; Konashev, M.B. The discovery of Gramicidin S: The intellectual transformation of G.F. Gause from biologist to researcher of antibiotics and on its meaning for the fate of Russian genetic. Hist. Phil. Life Sci. 2001, 23, 137–150. [Google Scholar]
- Tamaki, M.; Imazeki, Y.; Shirane, A.; Fujinuma, K.; Shindo, M.; Kimura, M.; Uchida, Y. Novel gratisin derivatives with high antimicrobial activity and low hemolytic activity. Bioorg. Med. Chem. Lett. 2011, 21, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Loll, P.J.; Upton, E.C.; Nahoum, V.; Economou, N.J.; Cocklin, S. The high resolution structure of tyrocidine A reveals an amphipathic dimer. Biochim. Biophys. Acta Biomembr. 2014, 1838, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Maurizio, S.; Günther, K.; Barra, D. Bombinins, antimicrobial peptides from Bombina species. Biochim. Biophys. Acta Biomembr. 2009, 1788, 1551–1555. [Google Scholar]
- Papo, N.; Braunstein, A.; Eshhar, Z.; Shai, Y. Suppression of human prostate tumor growth in mice by a cytolytic D-, L-amino acid peptide: Membrane lysis, increased necrosis, and inhibition of prostate-specific antigen secretion. Cancer Res. 2004, 64, 5779–5786. [Google Scholar] [CrossRef] [PubMed]
- Hanae, U.; Tomohisa, H.; Oumi, N.; Koji, O.; Masayuki, K.; Koji, K. Semaphorin 3A lytic hybrid peptide binding to neuropilin-1 as a novel anti-cancer agent in pancreatic cancer. Biochem. Biophys. Res. Commun. 2011, 41, 60–66. [Google Scholar]
- Braunstein, A.; Papo, N.; Shai, Y. In vitro activity and potency of an intravenously injected antimicrobial peptide and its DL amino acid analog in mice infected with bacteria. Antimicrob. Agents Chemother. 2004, 48, 3127–3129. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Kim, J.K.; Shin, S.; Jeong, K.W.; Lee, J.; Lee, D.G.; Hwang, J.S.; Kim, Y. Enantiomeric 9-mer peptide analogs of protaetiamycine with bacterial cell selectivities and anti-inflammatory activities. J. Pept. Sci. 2011, 17, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Sinthuvanich, C.; Veiga, A.S.; Gupta, K.; Gaspar, D.; Blumenthal, R.; Schneider, J.P. Anticancer β-hairpin peptides: Membrane-induced folding triggers activity. J. Am. Chem. Soc. 2012, 134, 6210–6217. [Google Scholar] [CrossRef] [PubMed]
- Papo, N.; Shai, Y. Effect of drastic sequence alteration and d-amino acid incorporation on the membrane binding behavior of lytic peptides. Biochemistry 2004, 43, 6393–6403. [Google Scholar] [CrossRef] [PubMed]
- Oren, Z.; Shai, Y. Selective lysis of bacteria but not mammalian cells by diastereomers of melittin: Structure-function study. Biochemistry 1997, 36, 1826–1835. [Google Scholar] [CrossRef] [PubMed]
- Oren, Z.; Ramesh, J.; Avrahami, D.; Suryaprakash, N.; Shai, Y.; Jelinek, R. Structures and mode of membrane interaction of a short a helical lytic peptide and its diastereomer determined by NMR, FTIR, and fluorescence spectroscopy. Eur. J. Biochem. 2002, 269, 3869–3880. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Nan, Y.H.; Yang, S.T.; Kang, S.W.; Kim, Y.; Park, I.S.; Hahm, K.S.; Shin, S.Y. Cell selectivity and anti-inflammatory activity of a Leu/Lys-rich α-helical model antimicrobial peptide and its diastereomeric peptides. Peptides 2010, 31, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Almeida, P.F.; Pokorny, A. Interactions of antimicrobial peptides with lipid bilayers. Compr. Biophys. 2012, 5, 189–222. [Google Scholar]
- Katsumi, M. Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2009, 1788, 1687–1692. [Google Scholar]
- Bahar, A.A.; Ren, D. Antimicrobial peptides. Pharmaceuticals 2013, 6, 1543–1575. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. Human antimicrobial peptides and proteins. Pharmaceuticals 2014, 7, 545–594. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y.; Oren, Z. Diastereomers of cytolysins, a novel class of potent antibacterial peptides. J. Biol. Chem. 1996, 271, 7305–7308. [Google Scholar] [PubMed]
- Ojcius, D.M.; Young, J.D.E. Cytolytic pore-forming proteins and peptides is there a common structural motif? Trends Biol. Sci. 1991, 16, 225–229. [Google Scholar] [CrossRef]
- Ehrenstein, G.; Lecar, H. Electrically gated ionic channels in lipid bilayers. Quart. Rev. Biophys. 1977, 10, 1–34. [Google Scholar] [CrossRef]
- Jiang, H.; Ziv, O.; Yechiel, S. Structure and organization of hemolytic and nonhemolytic diastereomers of antimicrobial peptides in membranes. Biochemistry 1999, 38, 16963–16973. [Google Scholar]
- Sharon, M.; Oren, Z.; Shai, Y.; Anglister, J. 2D-NMR and ATR-FTIR study of the structure of a cell-selective diastereomer of melittin and its orientation in phospholipids. Biochemistry 1999, 38, 15305–15316. [Google Scholar] [CrossRef] [PubMed]
- Wieprecht, T.; Apostolov, O.; Beyermann, M.; Seelig, J. Membrane binding and pore formation of the antibacterial peptide PGLa: Thermodynamic and mechanistic aspects. Biochemistry 2000, 39, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Pouny, Y.; Shai, Y. Interaction of d-amino acid incorporated analogues of pardaxin with membranes. Biochemistry 1992, 31, 9482–9490. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.Z.; Yong, H.N.; Kyung, S.H.; Song, Y.S. Cell selectivity of an antimicrobial peptide melittin diastereomer with d-amino acid in the leucine zipper sequence. J. Biochem. Mol. Biol. 2007, 40, 1090–1094. [Google Scholar]
- Ziv, O.; Yechiel, S. Mode of action of linear amphipathic α-helical antimicrobial peptides. Biopolymers 1998, 47, 451–463. [Google Scholar]
- Papo, N.; Shai, Y. New lytic peptides based on the d,l-amphipathic helix motif preferentially kill tumor cells compared to normal cells. Biochemistry 2003, 42, 9346–9354. [Google Scholar] [CrossRef] [PubMed]
- Zwaal, R.F.A.; Schroit, A.J. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood 1997, 89, 1121–1132. [Google Scholar] [PubMed]
- Shai, Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta 1999, 1462, 55–70. [Google Scholar] [CrossRef]
- Bessalle, R.; Haas, H.; Goria, A.; Shalit, I.; Fridkin, M. Augmentation of the antibacterial activity of magainin by positive-charge chain extension. Antimicrob. Agents Chemother. 1992, 36, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, T.; Ishibashi, J.; Tanaka, H.; Sato, M.; Asaoka, A.; Taylor, D.; Yamakawa, M. Selective cancer cell cytotoxicity of enantiomeric 9-mer peptides derived from beetle defensins depends on negatively charged phosphatidylserine on the cell surface. Peptides 2009, 30, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Strøm, M.B.; Mekonnen, S.M.; Svendsen, J.S.; Rekdal, Ø. The effects of shortening lactoferrin derived peptides against tumour cells, bacteria and normal human cells. J. Pept. Sci. 2004, 10, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.E.; O’Brien, L.M.; Thwaite, J.E.; Fox, M.A.; Atkins, H.; Ulaeto, D.O. A carpet-based mechanism for direct antimicrobial peptide activity against vaccinia virus membranes. Peptides 2010, 31, 1966–1972. [Google Scholar] [CrossRef] [PubMed]
- Aldar, S.B.; Keith, K.; William, B. Anti-HIV effect of gramicidin in vitro: Potential for spermicide use. Life Sci. 1993, 54, 5–9. [Google Scholar]
- Barlow, P.G.; Svoboda, P.; Mackellar, A.; Nash, A.A.; York, I.A.; Pohl, J.; Davidson, D.J.; Donis, R.O. Antiviral activity and increased host defense against influenza infection elicited by the human cathelicidin LL-37. PLoS ONE 2011, 6, e25333. [Google Scholar] [CrossRef] [PubMed]
- Mariam, E.; Monta, C.; Dorothy, L.M.; Lee, W.M.; Milwood, M.; Caroline, A.G. Antiviral effects of synthetic membrane-active peptides on Herpes Simplex Virus, Type 1. Int. J. Antimicrob. Agents 1999, 13, 57–60. [Google Scholar]
- De la Fuente-Núñez, C.; Cardoso, M.H.; de Souza Cândido, E.; Franco, O.L.; Hancock, R.E.W. Synthetic antibiofilm peptides. Biochim. Biophys. Acta Biomembr. 2016, 1858, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Alice, E.B.; Katrin, M.; Carlos, J.S.; Miriam, L.B.; Joseph, C.W.; Clinton, K.M.; Kevin, S.A. Clinical infectious outcomes associated with biofilm-related bacterial infections: A retrospective chart review. BMC Infect. Dis. 2015, 15, 1–7. [Google Scholar]
- Giovanna, B.; Giuseppantonio, M.; Semih, E. Antimicrobial peptides and their interaction with biofilms of medically relevant bacteria. Biochim. Biophys. Acta Biomembr. 2016, 1858, 1044–1060. [Google Scholar]
- Segev-Zarko, L.A.; Saar-Dover, R.; Brumfeld, V.; Mangoni, M.L.; Shai, Y. Mechanisms of biofilm inhibition and degradation by antimicrobial peptides. Biochem. J. 2015, 468, 259–270. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente-Núñez, C.; Reffuveille, F.; Mansour, S.C.; Reckseidler-Zenteno, S.L.; Hernández, D.; Brackman, G.; Coenye, T.; Hancock, R.E.W. d-enantiomeric peptides that eradicate wild-type and multi-drug resistant biofilms and protect against lethal Pseudomonas aeruginosa infections. Chem. Biol. 2015, 22, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Dean, S.N.; Bishop, B.M.; Hoek, M.L.V. Natural and synthetic cathelicidin peptides with anti-microbial and anti-biofilm activity against Staphylococcus aureus. BMC Microbiol. 2011, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente-Núñez, C.; Korolikb, V.; Bainsa, M.; Nguyenc, U.; Breidensteina, E.B.M.; Horsmand, S.; Lewenzad, S.; Burrowsc, L.; Hancock, R.E.W. Inhibition of bacterial biofilm formation and swarming motility by a small synthetic cationic peptide. Antimicrob. Agents Chemother. 2012, 66, 2696–2704. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente-Núñez, C.; Reffuveille, F.; Haney, F.E.; Straus, K.S.; Hancock, R.E.W. Broad-spectrum anti-biofilm peptide that targets a cellular stress response. PLoS Pathog. 2014, 10, e1004152. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.B.; Wang, X.F.; Wang, H.Y.; Liu, Y.; Chen, Y. Studies on mechanism of action of anticancer peptides by modulation of hydrophobicity within a defined structural framework. Mol. Cancer Ther. 2011, 10, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Eliassen, L.T.; Berge, G.; Leknessund, A.; Wikman, M.; Lindin, I.; Løkke, C.; Ponthan, F.; Johnsen, J.I.; Sveinbjørnsson, B.; Kogner, P.; et al. The antimicrobial peptide, Lactoferricin B, is cytotoxic to neuroblastoma cells in vitro and inhibits xenograft growth in vivo. Int. J. Cancer 2006, 119, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Berge, G.; Eliassen, L.T.; Camilio, K.A.; Bartnes, K.; Sveinbjørnsson, B.; Rekdal, Ø. Therapeutic vaccination against a murine lymphoma by intratumoral injection of a cationic anticancer peptide. Cancer Immunol. Immunother. 2010, 59, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Hubert, P.; Herman, L.; Maillard, C.; Caberg, J.H.; Nikkels, A.; Pierard, G.; Foidart, J.M.; Noel, A.; Boniver, J.; Delvenne, P. Defensins induce the recruitment of dendritic cells in cervical human papillomavirus-associated (pre)neoplastic lesions formed in vitro and transplanted in vivo. FASEB J. 2007, 21, 2765–2775. [Google Scholar] [CrossRef] [PubMed]
- Tani, K.; Murphy, W.J.; Chertov, O.; Salcedo, R.; Koh, C.Y.; Utsunomiya, I.; Funakoshi, S.; Asai, O.; Herrmann, S.H.; Wang, J.M.; et al. Defensins act as potent adjuvant that promote cellular and humoral immune responses in mice to a lymphoma idiotype and carrier antigens. Int. Immunol. 2000, 12, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Chen, Q.; Oppenheim, J.J.; Kuusela, P.; Taylor, J.W.; Wade, D. Temporin/VesCP (T/V)-like antibiotic peptides, derived from frogs and wasps, induce migration of human monocytes and neutrophils. Lett. Pept. Sci. 2003, 10, 99–110. [Google Scholar] [CrossRef]
- Mader, J.S.; Salsman, J.; Conrad, D.M.; Hoskin, D.W. Bovine lactoferricin selectively induces apoptosis in human leukemia and carcinoma cell lines. Mol. Cancer Ther. 2012, 4, 612–624. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, I.W.; Kwon, Y.N.; Yun, E.Y.; Hwang, J.S. Synthetic coprisin analog peptide, D-CopA3 has antimicrobial activity and pro-apoptotic effects in human leukemia cells. J. Microbiol. Biotechnol. 2012, 22, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.W.; Burger, G.; Lang, B.F. Mitochondrial evolution. Science 1999, 283, 1476–1481. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Kolluri, S.K.; Gu, J.; Dawson, M.I.; Cao, X.; Hobbs, P.D.; Lin, B.; Chen, G.; Lu, J.; Lin, F.; et al. Cytochrome c release and apoptosis induced by mitochondrial targeting of nuclear orphan receptor TR3. Science 2000, 289, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, X.; Hong, S.; Chen, J.; Liu, N.; Underhill, C.B.; Creswell, K.; Zhang, L. RGD-Tachyplesin inhibits tumor growth. Cancer Res. 2001, 61, 2434–2438. [Google Scholar] [PubMed]
- Huang, Y.B.; He, L.Y.; Jiang, H.Y.; Chen, Y.X. Role of helicity on the anticancer mechanism of action of cationic-helical peptides. Int. J. Mol. Sci. 2012, 13, 6849–6862. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; He, L.; Li, G.; Zhai, N.; Jiang, H.; Chen, Y. Role of helicity of α-helical antimicrobial peptides to improve specificity. Protein Cell 2014, 5, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.; Tsubery, H.; Kolusheva, S.; Shames, A.; Fridkin, M.; Jelinek, R. Lipid binding and membrane penetration of polymyxin B derivatives studied in a biomimetic vesicle system. Biochem. J. 2003, 375, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Tsubery, H.; Ofek, I.; Cohen, S.; Fridkin, M. The functional association of polymyxin B with bacterial lipopolysaccharide is stereospecific: Studies on polymyxin B nonapeptide. Biochemistry 2000, 39, 11837–11844. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Wade, D.; Boman, A.; Wählin, B.; Drain, C.M.; Andreu, D.; Boman, H.G.; Merrifield, R.B. All-D amino acid-containing channel-forming antibiotic peptides. Proc. Natl. Acad. Sci. USA 1990, 87, 4761–4765. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, E.G.; Dobroff, A.S.; Cavarsan, C.F.; Paschoalin, T.; Nimrichter, L.; Mortara, R.A.; Santos, E.L.; Fázio, M.A.; Miranda, A.; Daffre, S.; et al. Effective topical treatment of subcutaneous murine B16F10-Nex2 melanoma by the antimicrobial peptide gomesin. Neoplasia 2008, 10, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Falciani, C.; Lozzi, L.; Pollini, S.; Luca, V.; Carnicelli, V.; Brunetti, J.; Lelli, B.; Bindi, S.; Scali, S.; Giulio, A.D.; et al. Isomerization of an antimicrobial peptide broadens antimicrobial spectrum to gram-positive bacterial pathogens. PLoS ONE 2012, 7, e46259. [Google Scholar] [CrossRef] [PubMed]
- Juba, M.; Porter, D.; Dean, S.; Gillmor, S.; Bishop, B. Characterization and performance of short cationic antimicrobial peptide isomers. Peptide Sci. 2013, 100, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.A.; Maloy, W.L.; Zasloff, M.; Jacob, L.S. Anticancer efficacy of magainin2 and analogue peptides. Cancer Res. 1993, 53, 3052–3057. [Google Scholar] [PubMed]
- Falagas, M.E.; Kasiakou, S.K. Colistin: The revival of polymyxins for the management of multidrug-resistant gram-negative bacterial infections. Clin. Infect. Dis. 2005, 40, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, L.O.; Merres, J.; Albrecht, L.J.; Varoga, D.; Pufe, T. Antimicrobial peptides: Multifunctional drugs for different applications. Polymers 2012, 4, 539–560. [Google Scholar] [CrossRef]
- Andrès, E. Cationic antimicrobial peptides in clinical development, with special focus on thanatin and heliomicin. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Curtis, K.K.; Sarantopoulos, J.; Northfelt, D.W.; Weiss, G.J.; Barnhart, K.M.; Whisnant, J.K.; Leuschner, C.; Alila, H.; Borad, M.J.; Ramanathan, R.K. Novel LHRH-receptor-targeted cytolytic peptide, EP-100: First-in-human phase I study in patients with advanced LHRH-receptor-expressing solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Van der Velden, W.J.F.M.; van Iersel, T.M.; Blijlevens, N.M.; Donnelly, J.P. Safety and tolerability of the antimicrobial peptide human lactoferrin 1-11 (hLF1-11). BMC Med. 2009, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nijnik, A. Immunomodulatory approaches for prevention and treatment of infectious diseases. Curr. Opin. Microbiol. 2013, 16, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Giulia, R.; Chiara, F.; Luisa, B.; Alessandro, P. The development of antimicrobial peptides as new antibacterial drugs. Curr. Protein Pept. Sci. 2013, 141, 641–649. [Google Scholar]
- Papo, N.; Shahar, M.; Eisenbach, L.; Shai, Y. A novel lytic peptide composed of dl-amino acids selectively kills cancer cells in culture and in mice. J. Biol. Chem. 2003, 278, 21018–21023. [Google Scholar] [CrossRef] [PubMed]
- Makovitzki, A.; Fink, A.; Shai, Y. Suppression of human solid tumor growth in mice by intratumor and systemic inoculation of histidine-rich and pH-dependent host defense-like lytic peptides. Cancer Res. 2009, 69, 3458–3463. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar] [PubMed]
- Ellerby, H.M.; Arap, W.; Ellerby, L.M.; Kain, R.; Andrusiak, R.; Rio, G.D.; Krajewski, S.; Lombardo, C.R.; Rao, R.; Rouslahti, E.; et al. Anti-cancer activity of targeted pro-apoptotic peptides. Nat. Med. 1999, 5, 1032–1038. [Google Scholar] [PubMed]
- Lan, Y.; Lam, J.T.; Siu, G.K.H.; Yam, W.C.; Mason, A.J.; Lam, J.K.W. Cationic amphipathic d-enantiomeric antimicrobial peptides with in vitro and ex vivo activity against drug-resistant Mycobacterium tuberculosis. Tuberculosis 2014, 94, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, J.; Sundar, S. Drug resistance in Leishmaniasis. J. Glob. Infect. Dis. 2010, 2, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Brotman, Y.; Makovitzki, A.; Shai, Y.; Chet, I.; Viterbo, A. Synthetic ultrashort cationic lipopeptides induce systemic plant defense responses against bacterial and fungal pathogens. Appl. Environ. Microbiol. 2009, 75, 5373–5379. [Google Scholar] [CrossRef] [PubMed]
- Makovitzki, A.; Viterbo, A.; Brotman, Y.; Chet, I.; Shai, Y. Inhibition of fungal and bacterial plant pathogens in vitro and in planta with ultrashort cationic lipopeptides. Appl. Environ. Microbiol. 2007, 73, 6629–6636. [Google Scholar] [CrossRef] [PubMed]
- López-García, B.; Pérez-Payá, E.; Marcos, J.F. Identification of novel hexapeptides bioactive against phytopathogenic fungi through screening of a synthetic peptide combinatorial library. Appl. Environ. Microbiol. 2002, 68, 2453–2460. [Google Scholar] [CrossRef] [PubMed]
- Slaninová, J.; Mlsová, V.; Kroupová, H.; Alán, L.; Tůmová, T.; Monincová, L.; Borovičková, L.; Fučík, V.; Čeřovský, V. Toxicity study of antimicrobial peptides from wild bee venom and their analogs toward mammalian normal and cancer cells. Peptides 2012, 33, 18–26. [Google Scholar]
- Reay, D.P.; Bastacky, S.I.; Wack, K.E.; Stolz, D.B.; Robbins, P.D.; Clemens, P.R. d-amino acid substitution of peptide-mediated NF-κB suppression in mdx mice preserves therapeutic benefit in skeletal muscle, but causes kidney toxicity. Mol. Med. 2015, 21, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.W.; Völker, K.; Dantzler, W.H.; Silbernagl, S. Why is d-serine nephrotoxic and α-aminoisobutyric acid protective? Am. J. Physiol. Ren. Physiol. 2007, 293, F382–F390. [Google Scholar] [CrossRef] [PubMed]
- Hils, M.; Münch, P.; Altenbuchner, J.; Syldatk, C.; Mattes, R. Cloning and characterization of genes from Agrobacterium sp. IP I-671 involved in hydantoin degradation. Appl. Microbiol. Biotechnol. 2001, 57, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Bommarius, A.S.; Schwarm, M.; Drauz, K. Biocatalysis to amino acid-based chiral pharmaceuticals— Examples and perspectives. J. Mol. Catal. B Enzym. 1998, 5, 1–11. [Google Scholar] [CrossRef]
- Komeda, H.; Asano, Y. Gene cloning, nucleotide sequencing, and purification and characterization of the d-stereospecific amino-acid amidase from Ochrobactrum anthropi SV3. Eur. J. Biochem. 2000, 267, 2028–2035. [Google Scholar] [CrossRef] [PubMed]
- Komeda, H.; Ishikawa, N.; Asano, Y. Enhancement of the thermostability and catalytic activity of d-stereospecific amino-acid amidase from Ochrobactrum anthropi SV3 by directed evolution. J. Mol. Catal. B Enzym. 2003, 21, 283–290. [Google Scholar] [CrossRef]
- Asano, Y.; Komeda, H. d-Aminopeptidase and Alkaline d-Peptidase. In Encyclopedia of Industrial Biotechnology; Flickinger, M.C., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2010. [Google Scholar]
- Asano, Y.; Ito, H.; Dairi, T.; Kato, Y. An alkaline d-stereospecific endopeptidase with β-lactamase activity from Bacillus cereus. J. Biol. Chem. 1996, 271, 30256–30262. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Shimizu, Y.; Mutaguchi, Y.; Doi, K.; Ohshima, T. Characterization of D-amino acid aminotransferase from Lactobacillus salivarius. J. Mol. Catal. B Enzym. 2013, 94, 15–22. [Google Scholar] [CrossRef]
- Bae, H.S.; Lee, S.G.; Hong, S.P.; Kwak, M.S.; Esaki, N.; Soda, K.; Sung, M.H. Production of aromatic D-amino acids from α-keto acids and ammonia by coupling of four enzyme reactions. J. Mol. Catal. B Enzym. 1999, 6, 241–247. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Mayr, L.M.; Minor, D.L.; Milhollen, M.A.; Burgess, M.W.; Kim, P.S. Identification of d-peptide ligands through mirror-image phage display. Science 1996, 271, 1854–1857. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.; Chorev, M. On the concept of linear modified retro-peptide structures. Acc. Chem. Res. 1979, 12, 1–7. [Google Scholar] [CrossRef]
- Nantasenamat, C.; Isarankura-Na-Ayudhya, C.; Naenna, T.; Prachayasittikul, V. A practical overview of quantitative structure-activity relationship. EXCLI J. 2009, 8, 74–88. [Google Scholar]
- Nantasenamat, C.; Isarankura-Na-Ayudhya, C.; Prachayasittikul, V. Advances in computational methods to predict the biological activity of compounds. Expert Opin. Drug Discov. 2010, 5, 633–654. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.; Eriksson, L.; Jonsson, J.; Sjöström, M.; Wold, S. New chemical descriptors relevant for the design of biologically active peptides. A multivariate characterization of 87 amino acids. J. Med. Chem. 1998, 41, 2481–2491. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.R.; Lin, H.H.; Han, L.Y.; Jiang, L.; Chen, X.; Chen, Y.Z. PROFEAT: A web server for computing structural and physicochemical features of proteins and peptides from amino acid sequence. Nucleic Acids Res. 2006, 34, W32–W37. [Google Scholar] [CrossRef] [PubMed]
- Xiao, N.; Cao, D.S.; Zhu, M.F.; Xu, Q.S. protr/ProtrWeb: R package and web server for generating various numerical representation schemes of protein sequences. Bioinformatics 2015, 31, 1857–1859. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, H.; Tuknait, A.; Chaudhary, K.; Singh, B.; Kumaran, S.; Raghava, G.P. PEPstrMOD: Structure prediction of peptides containing natural, non-natural and modified residues. Biol. Direct 2015, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Gfeller, D.; Michielin, O.; Zoete, V. SwissSidechain: A molecular and structural database of non-natural sidechains. Nucleic Acids Res. 2013, 41, D327–D332. [Google Scholar] [CrossRef] [PubMed]
- Yongye, A.B.; Li, Y.; Giulianotti, M.A.; Yu, Y.; Houghten, R.A.; Martinez-Mayorga, K. Modeling of peptides containing D-amino acids: Implications on cyclization. J. Comput. Aided Mol. Des. 2009, 23, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Moreau, V.; Fleury, C.; Piquer, D.; Nguyen, C.; Novali, N.; Villard, S.; Laune, D.; Granier, C.; Molina, F. PEPOP: Computational design of immunogenic peptides. BMC Bioinform. 2008, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Steinbeck, C.; Han, Y.; Kuhn, S.; Horlacher, O.; Luttmann, E.; Willighagen, E. The Chemistry Development Kit (CDK): An open-source Java library for chemo- and bioinformatics. J. Chem. Inf. Comput. Sci. 2003, 43, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.S.; Xu, Q.S.; Hu, Q.N.; Liang, Y.Z. ChemoPy: Freely available python package for computational biology and chemoinformatics. Bioinformatics 2013, 29, 1092–1094. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Cao, D.S.; Miao, H.Y.; Liu, S.; Deng, B.C.; Yun, Y.H.; Wang, N.N.; Lu, A.P.; Zeng, W.B.; Chen, A.F. ChemDes: An integrated web-based platform for molecular descriptor and fingerprint computation. J. Cheminform. 2015, 7, 60. [Google Scholar] [CrossRef] [PubMed]
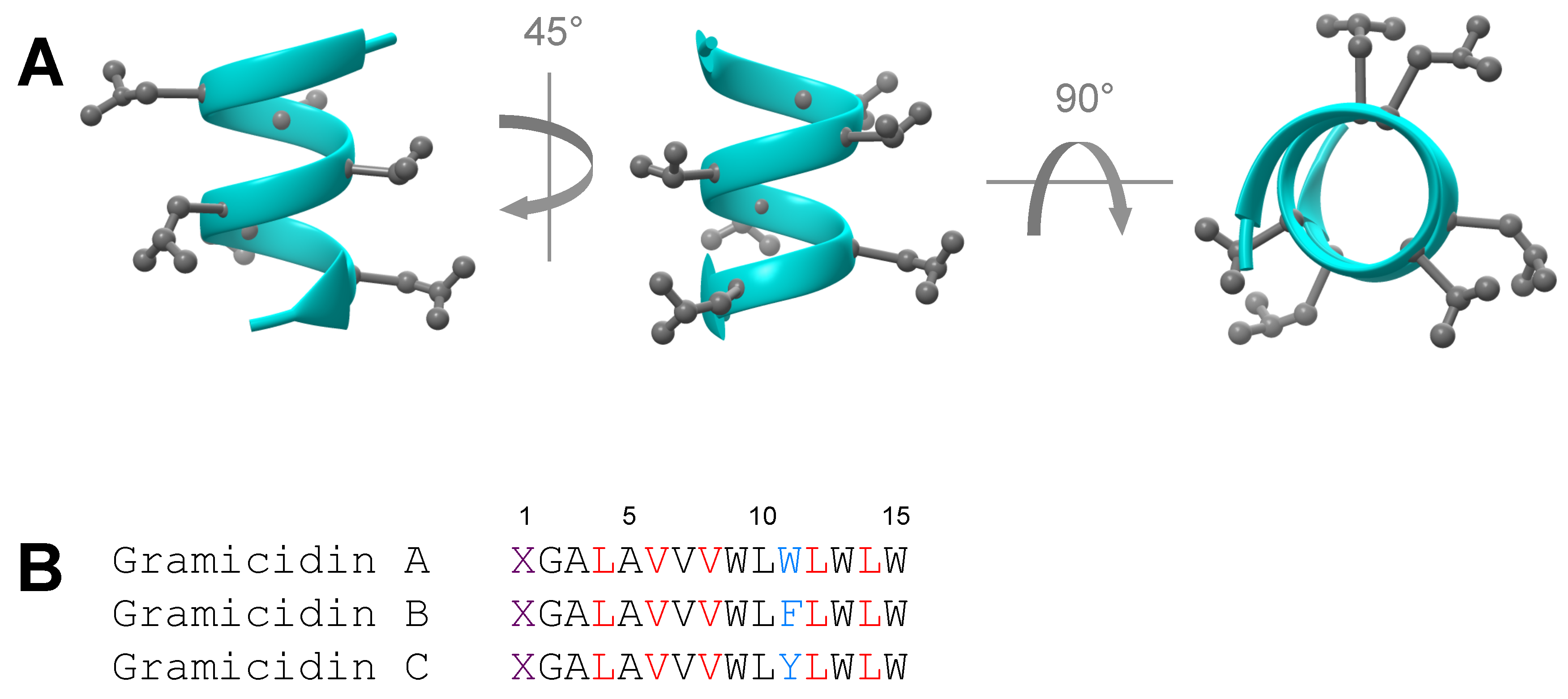
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Anuwongcharoen, N.; Malik, A.A.; Prachayasittikul, V.; Wikberg, J.E.S.; Nantasenamat, C. Roles of d-Amino Acids on the Bioactivity of Host Defense Peptides. Int. J. Mol. Sci. 2016, 17, 1023. https://doi.org/10.3390/ijms17071023
Li H, Anuwongcharoen N, Malik AA, Prachayasittikul V, Wikberg JES, Nantasenamat C. Roles of d-Amino Acids on the Bioactivity of Host Defense Peptides. International Journal of Molecular Sciences. 2016; 17(7):1023. https://doi.org/10.3390/ijms17071023
Chicago/Turabian StyleLi, Hao, Nuttapat Anuwongcharoen, Aijaz Ahmad Malik, Virapong Prachayasittikul, Jarl E. S. Wikberg, and Chanin Nantasenamat. 2016. "Roles of d-Amino Acids on the Bioactivity of Host Defense Peptides" International Journal of Molecular Sciences 17, no. 7: 1023. https://doi.org/10.3390/ijms17071023
APA StyleLi, H., Anuwongcharoen, N., Malik, A. A., Prachayasittikul, V., Wikberg, J. E. S., & Nantasenamat, C. (2016). Roles of d-Amino Acids on the Bioactivity of Host Defense Peptides. International Journal of Molecular Sciences, 17(7), 1023. https://doi.org/10.3390/ijms17071023