
Analysis of the Mitochondrial Genome in Hypomyces aurantius Reveals a Novel Twintron Complex in Fungi

Abstract
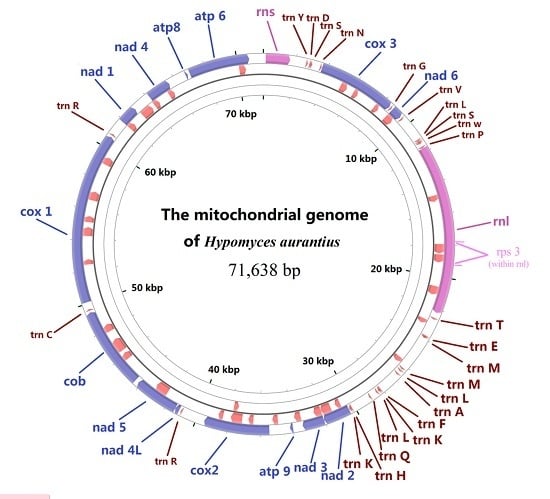
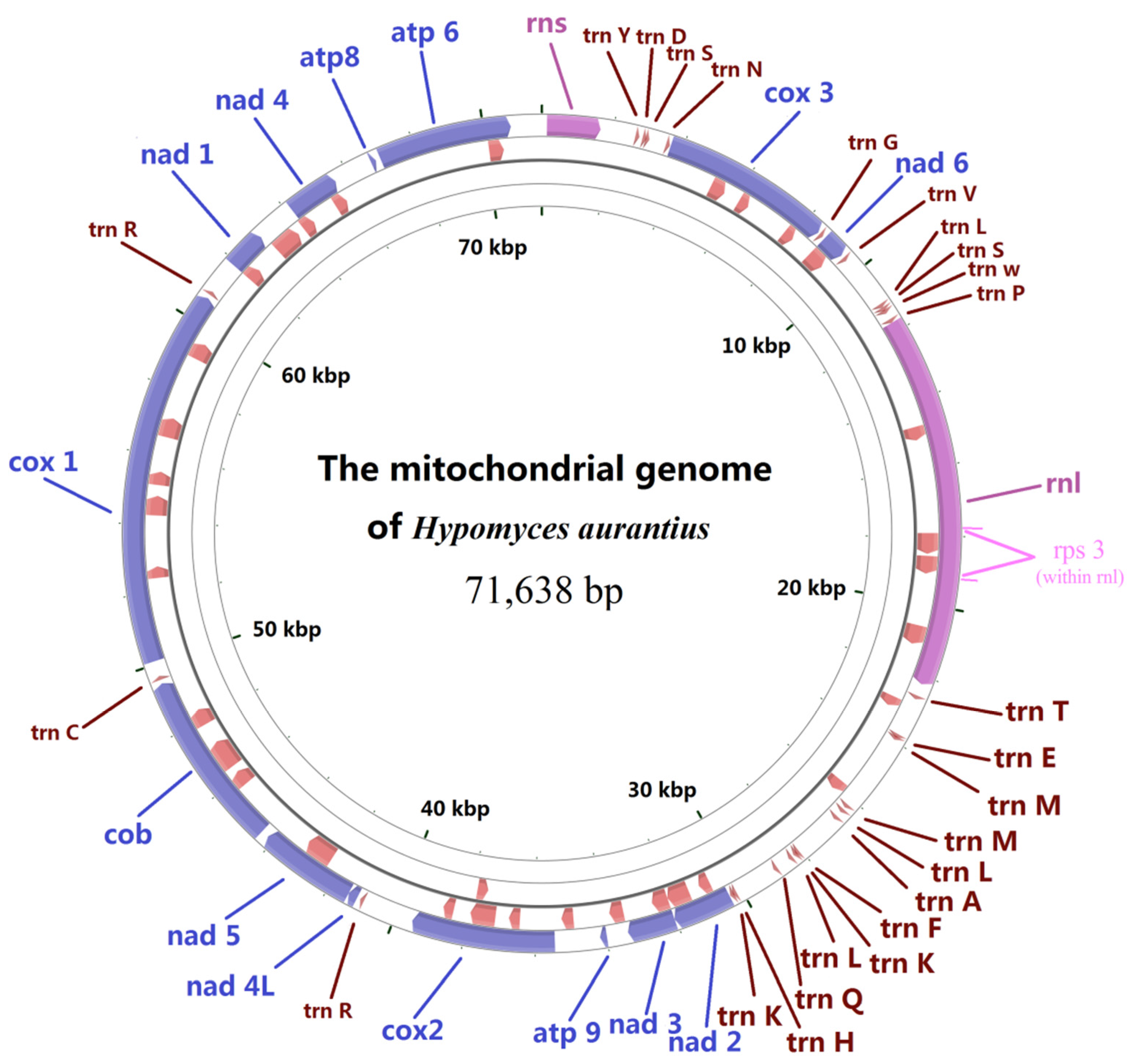
:
1. Introduction
2. Results
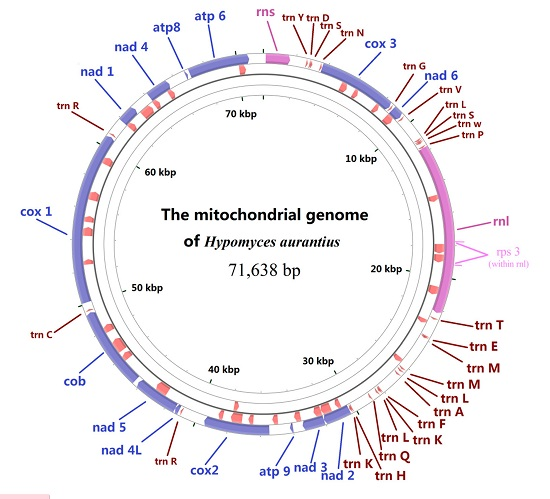
2.1. Gene Content of the Mitogenome in H. aurantius
2.2. Genetic Code
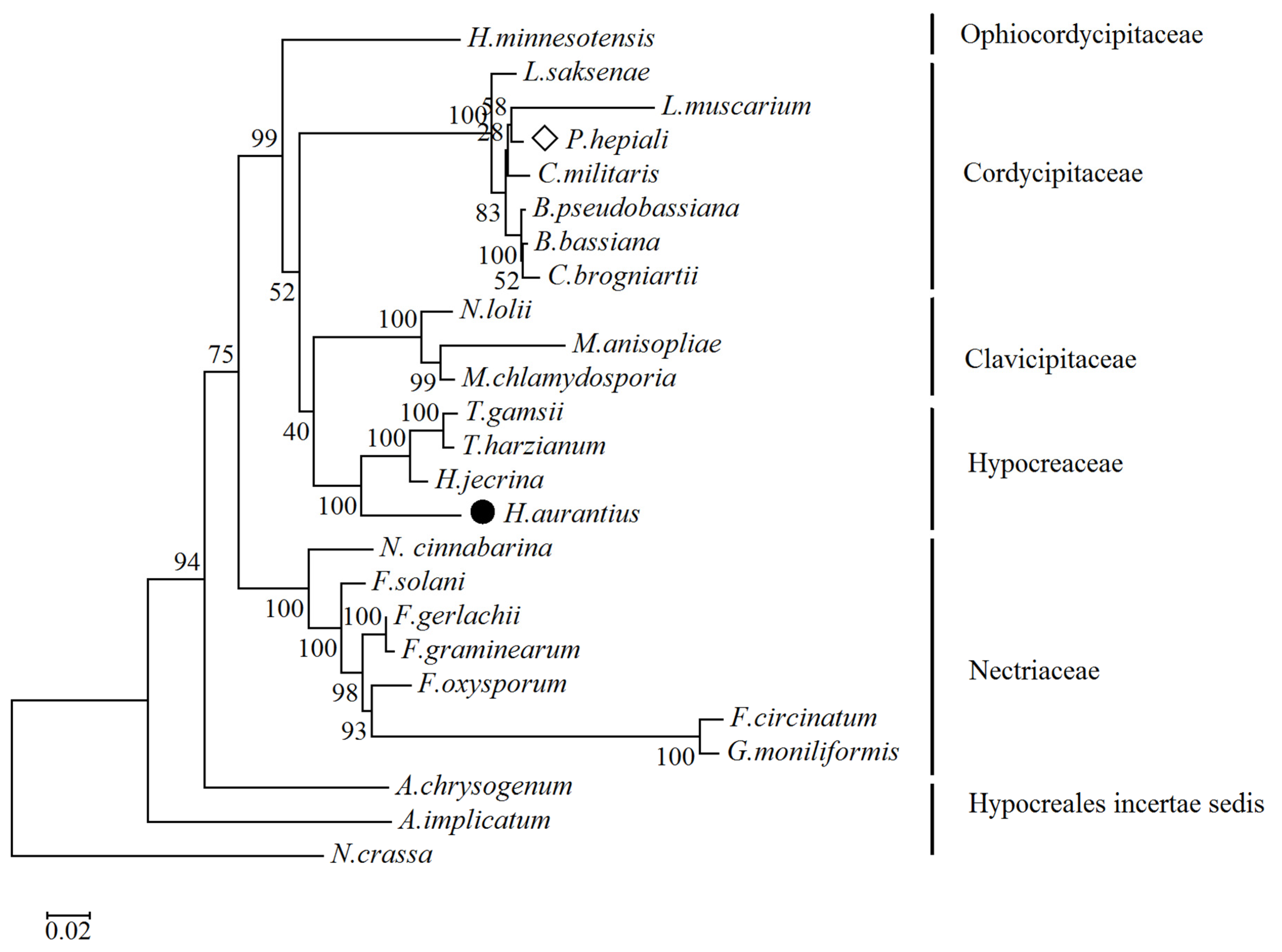
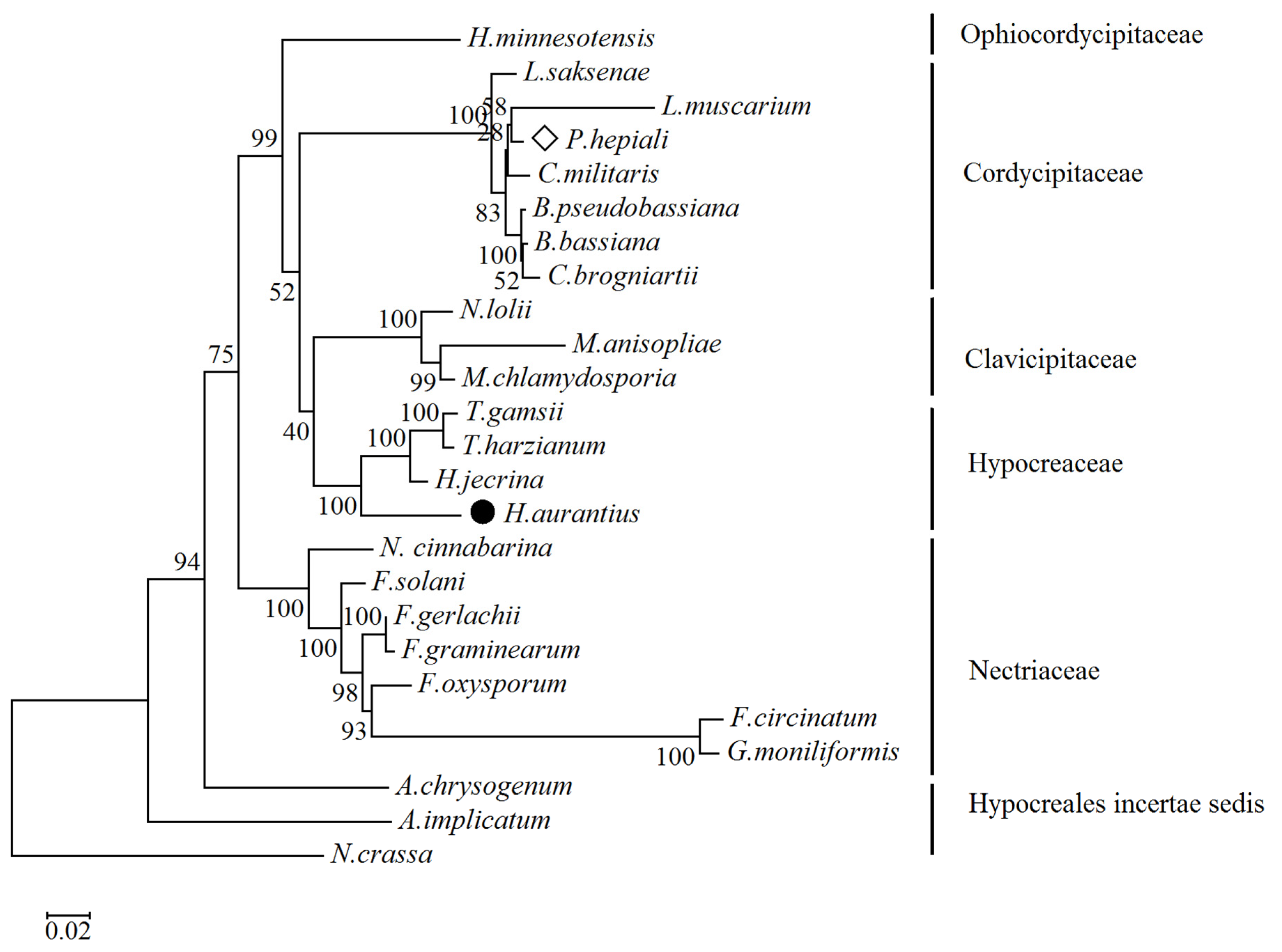
2.3. Phylogenetic Relationship Analysis
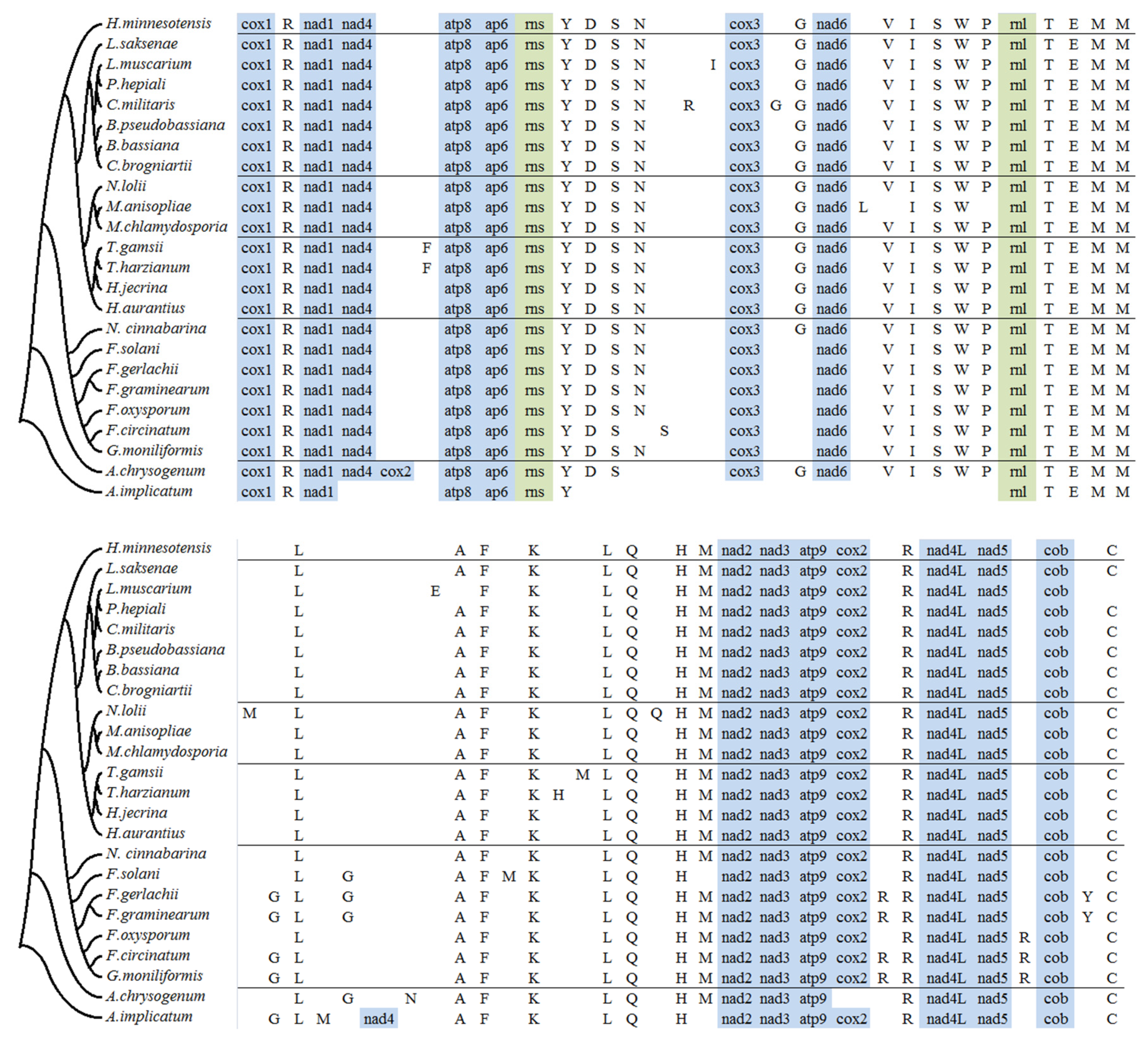
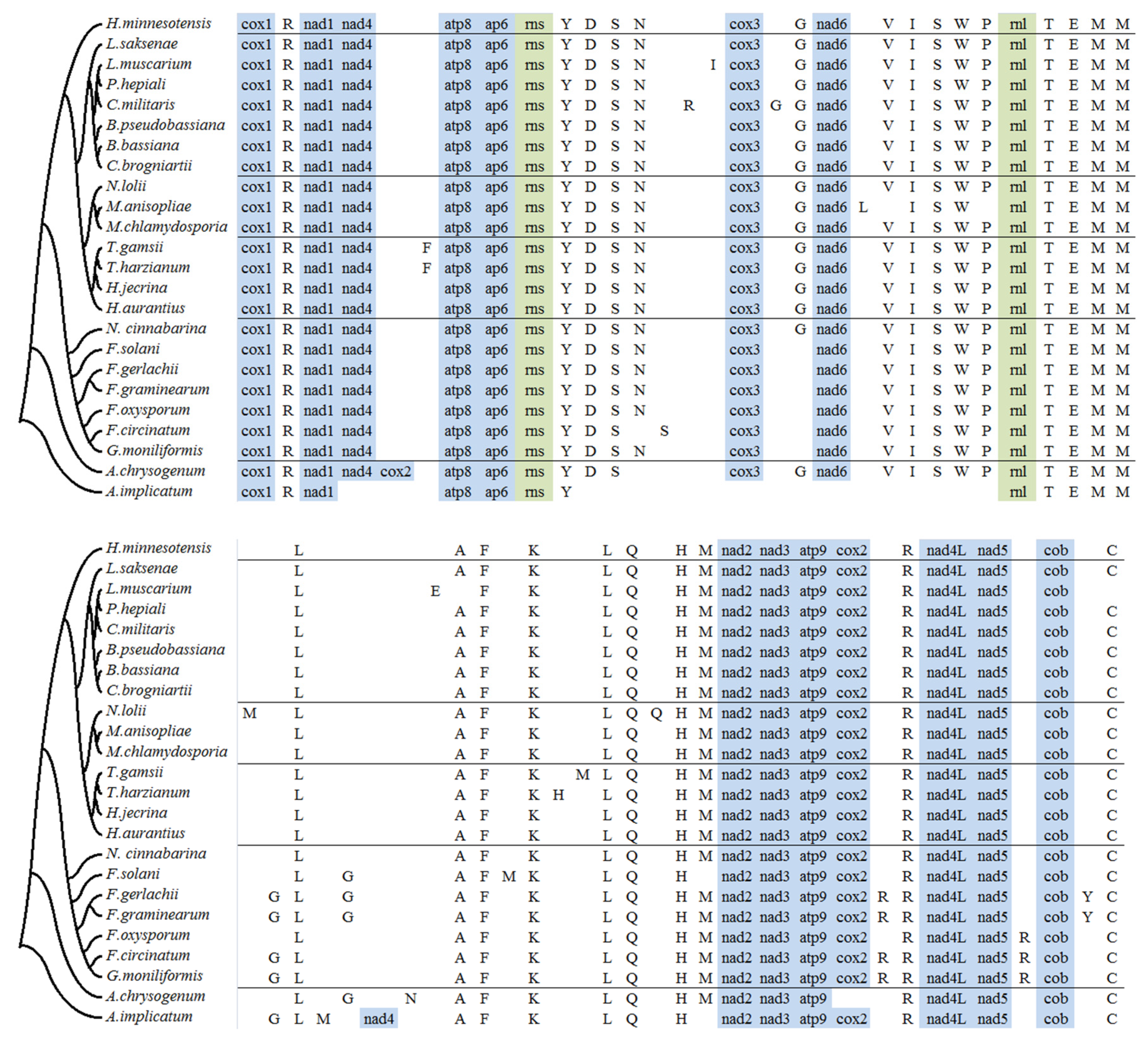
2.4. tRNA Gene Distribution and Gene Order Comparison in Hypocreales
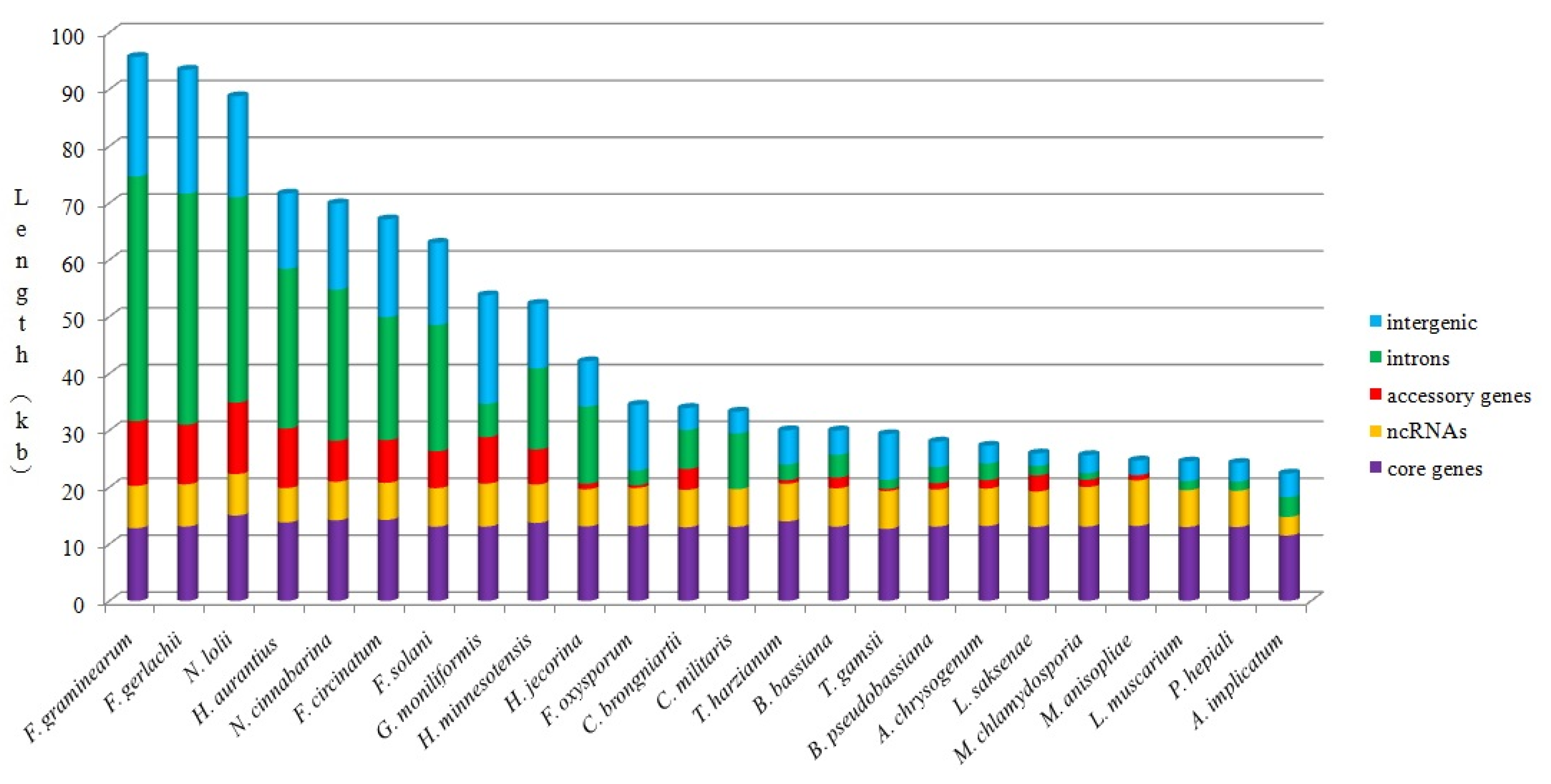
2.5. Analysis of Mitogenome Size Variation
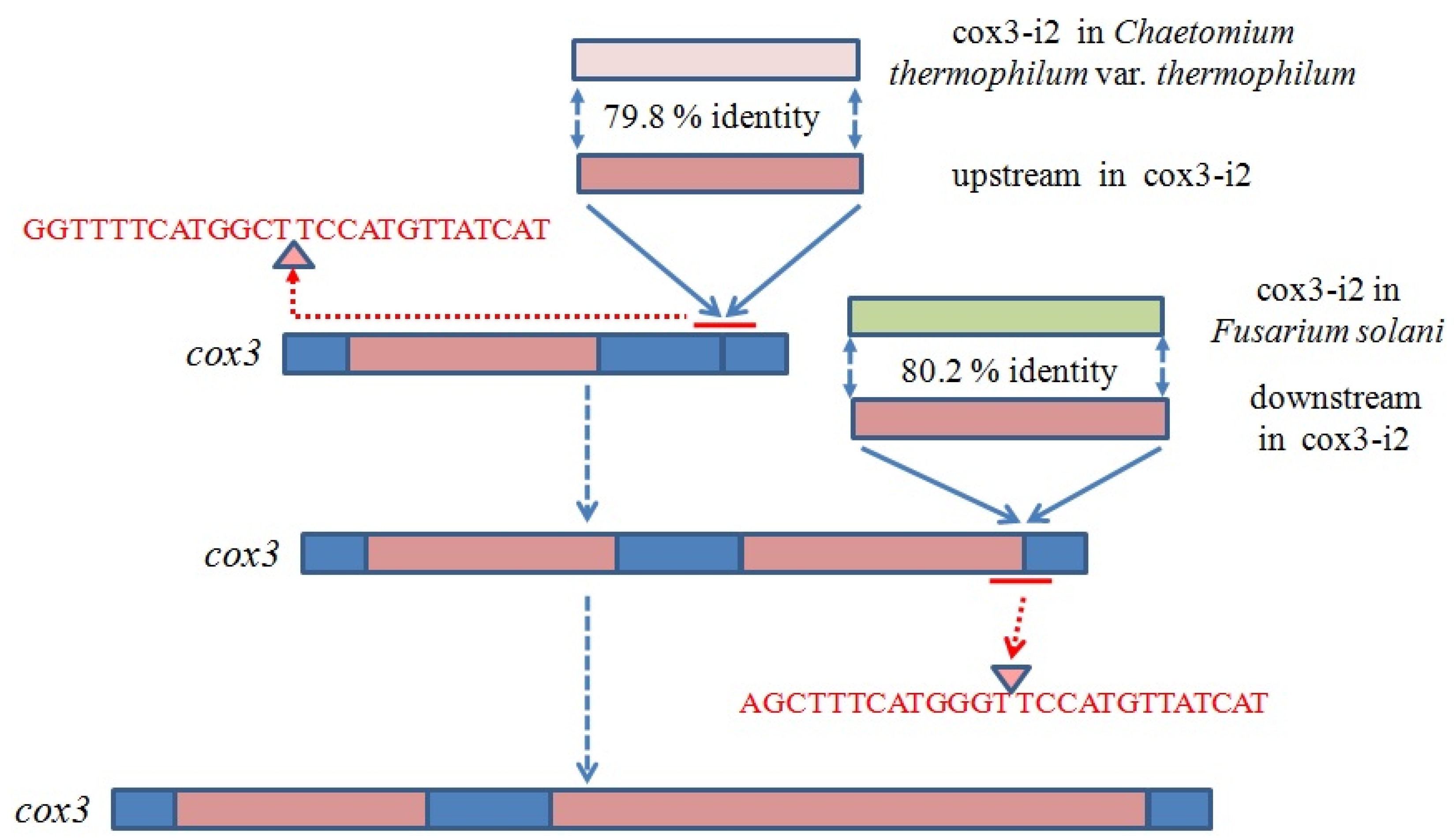
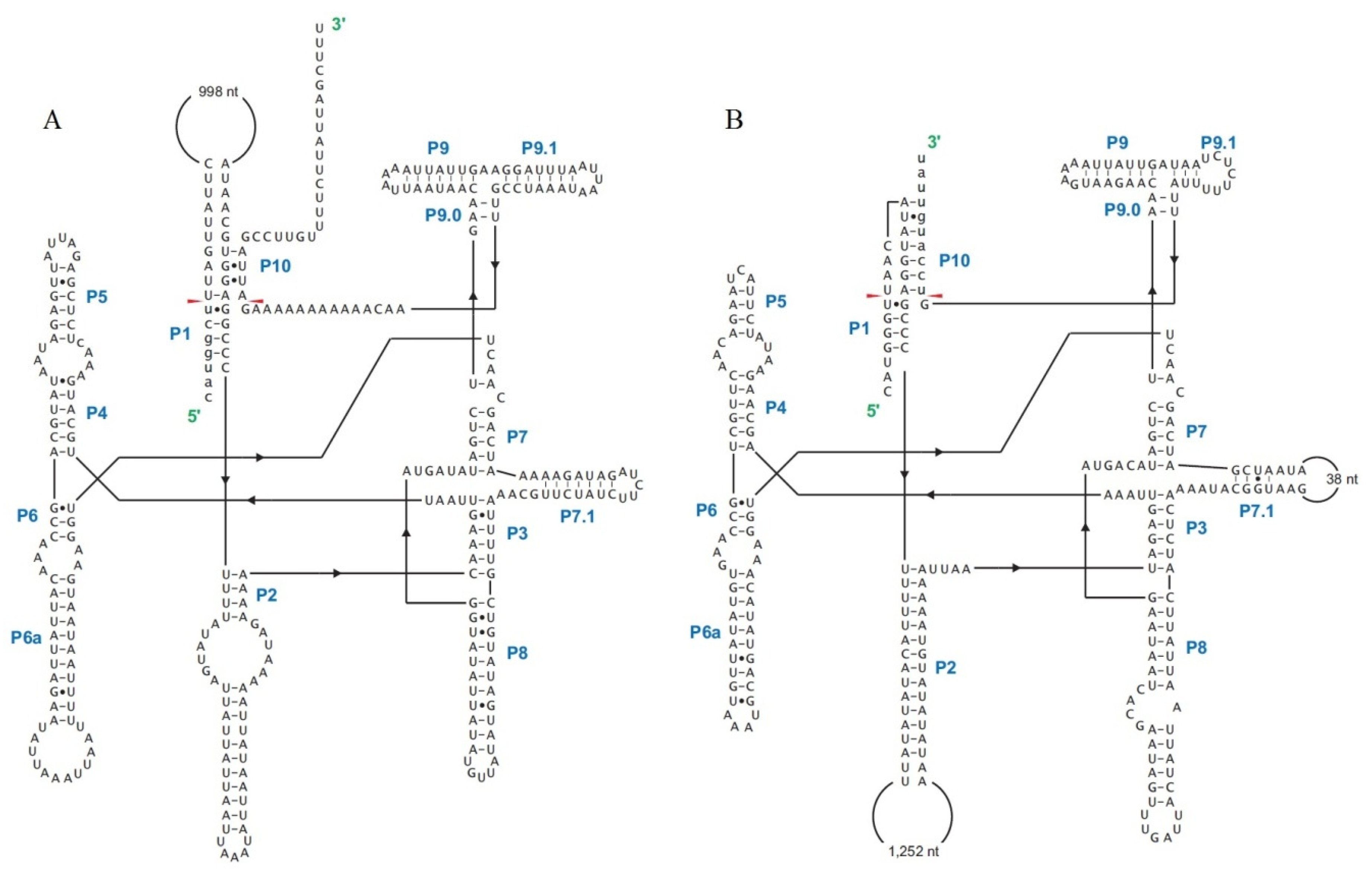
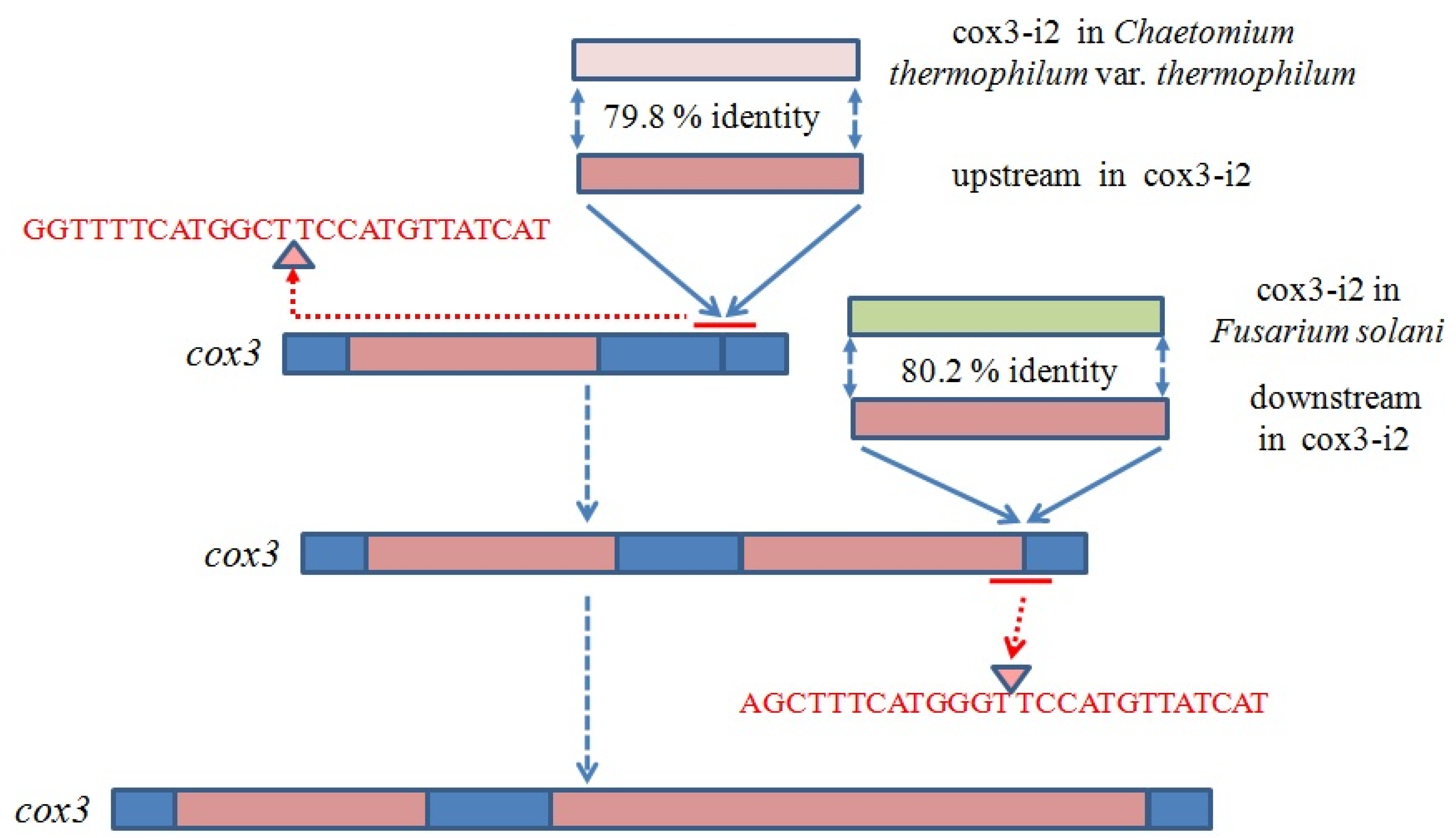
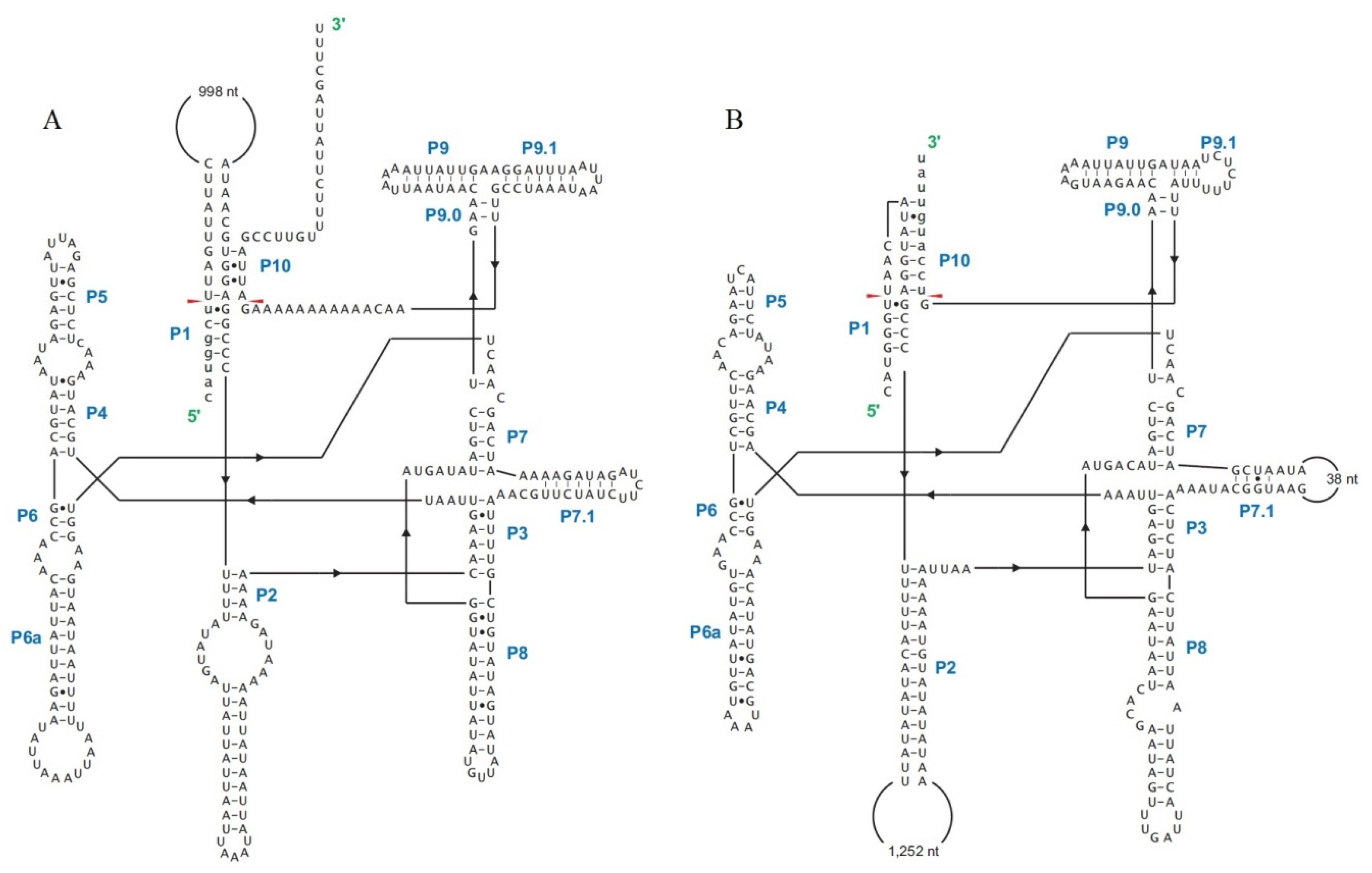
2.6. Introns in Conserved Genes
3. Discussion
4. Materials and Methods
4.1. Fungal Strains and DNA Preparation
4.2. Genome Sequencing and Mitochondrial DNA Assembly
4.3. Gene Annotation and Bioinformatic Analysis
4.4. Gene Order and Phylogenetic Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Potočnik, I.; Milijašević, S.; Rekanović, E.; Todorović, B.; Stepanović, M. Sensitivity of Cladobotryum spp., a pathogen of the button mushroom (Agaricus bisporus), to some fungicides. Pestic. I Fitomedicina 2007, 22, 233–240. [Google Scholar]
- Back, C.-G.; Kim, Y.-H.; Jo, W.-S.; Chung, H.; Jung, H.-Y. Cobweb disease on Agaricus bisporus caused by Cladobotryum mycophilum in Korea. J. Gen. Plant Pathol. 2010, 76, 232–235. [Google Scholar] [CrossRef]
- Grogan, H.M.; Gaze, R.H. Fungicide resistance among Cladobotryum spp.—Causal agents of cobweb disease of the edible mushroom Agaricus bisporus. Mycol. Res. 2000, 104, 357–364. [Google Scholar]
- Kim, H.-K.; Seok, S.-J.; Kim, G.-P.; Moon, B.-J.; Terashita, T. Occurrence of disease caused by Cladobotryum varium on Flammulina velutipes in Korea. Korean J. Mycol. 1999, 27, 415–419. [Google Scholar]
- Kim, T.; Lee, H.; Song, G.; Shin, W. King oyster mushroom (Pleurotus eryngii) white mold disease caused by Cladobotryum varium. KSM News Lett. 1998, 11, 46. [Google Scholar]
- Back, C.-G.; Lee, C.-Y.; Seo, G.-S.; Jung, H.-Y. Characterization of species of Cladobotryum which cause cobweb disease in edible mushrooms grown in Korea. Mycobiology 2012, 40, 189–194. [Google Scholar] [CrossRef] [PubMed]
- McKay, G.J.; Egan, D.; Morris, E.; Scott, C.; Brown, A.E. Genetic and morphological characterization of Cladobotryum species causing cobweb disease of mushrooms. Appl. Environ. Microbiol. 1999, 65, 606–610. [Google Scholar] [PubMed]
- Adie, B.; Grogan, H.; Archer, S.; Mills, P. Temporal and spatial dispersal of Cladobotryum conidia in the controlled environment of a mushroom growing room. Appl. Environ. Microbiol. 2006, 72, 7212–7217. [Google Scholar] [CrossRef] [PubMed]
- Joardar, V.; Abrams, N.F.; Hostetler, J.; Paukstelis, P.J.; Pakala, S.; Pakala, S.B.; Zafar, N.; Abolude, O.O.; Payne, G.; Andrianopoulos, A. Sequencing of mitochondrial genomes of nine Aspergillus and Penicillium species identifies mobile introns and accessory genes as main sources of genome size variability. BMC Genom. 2012, 13, 698. [Google Scholar] [CrossRef] [PubMed]
- Kouvelis, V.N.; Ghikas, D.V.; Typas, M.A. The analysis of the complete mitochondrial genome of Lecanicillium muscarium (synonym Verticillium lecanii) suggests a minimum common gene organization in mtDNAs of Sordariomycetes: Phylogenetic implications. Fungal Genet. Biol. 2004, 41, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Paquin, B.; Laforest, M.-J.; Forget, L.; Roewer, I.; Wang, Z.; Longcore, J.; Lang, B.F. The fungal mitochondrial genome project: Evolution of fungal mitochondrial genomes and their gene expression. Curr. Genet. 1997, 31, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.W.; Burger, G.; Lang, B.F. Mitochondrial evolution. Science 1999, 283, 1476–1481. [Google Scholar] [CrossRef] [PubMed]
- Seifert, K.A. Progress towards DNA barcoding of fungi. Mol. Ecol. Resour. 2009, 9, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Min, X.J.; Hickey, D.A. Assessing the effect of varying sequence length on DNA barcoding of fungi. Mol. Ecol. Notes 2007, 7, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Zhang, H.; Hou, Y.; Zhuang, Y.; Miranda, L. High-level diversity of dinoflagellates in the natural environment, revealed by assessment of mitochondrial cox1 and cob genes for dinoflagellate DNA barcoding. Appl. Environ. Microbiol. 2009, 75, 1279–1290. [Google Scholar] [CrossRef] [PubMed]
- Beaudet, D.; Nadimi, M.; Iffis, B.; Hijri, M. Rapid mitochondrial genome evolution through invasion of mobile elements in two closely related species of arbuscular mycorrhizal fungi. PLoS ONE 2013, 8, e60768. [Google Scholar] [CrossRef] [PubMed]
- Beaudet, D.; Terrat, Y.; Halary, S.; de la Providencia, I.E.; Hijri, M. Mitochondrial genome rearrangements in glomus species triggered by homologous recombination between distinct mtDNA haplotypes. Genome Biol. Evol. 2013, 5, 1628–1643. [Google Scholar] [CrossRef] [PubMed]
- Losada, L.; Pakala, S.B.; Fedorova, N.D.; Joardar, V.; Shabalina, S.A.; Hostetler, J.; Pakala, S.M.; Zafar, N.; Thomas, E.; Rodriguez-Carres, M. Mobile elements and mitochondrial genome expansion in the soil fungus and potato pathogen Rhizoctonia solani AG-3. FEMS Microbiol. Lett. 2014, 352, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Pramateftaki, P.V.; Kouvelis, V.N.; Lanaridis, P.; Typas, M.A. The mitochondrial genome of the wine yeast Hanseniaspora uvarum: A unique genome organization among yeast/fungal counterparts. FEMS Yeast Res. 2006, 6, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Férandon, C.; Xu, J.; Barroso, G. The 135 kbp mitochondrial genome of Agaricus bisporus is the largest known eukaryotic reservoir of group I introns and plasmid-related sequences. Fungal Genet. Biol. 2013, 55, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, R.; Mohr, G.; Belfort, M.; Lambowitz, A.M. Group I and group II introns. FASEB J. 1993, 7, 15–24. [Google Scholar] [PubMed]
- Pellenz, S.; Harington, A.; Dujon, B.; Wolf, K.; Schäfer, B. Characterization of the I-Spom I endonuclease from fission yeast: Insights into the evolution of a group I intron-encoded homing endonuclease. J. Mol. Evol. 2002, 55, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, P.; Barroso, G.; Labarère, J. Molecular analysis of the split cox1 gene from the Basidiomycota Agrocybe aegerita: Relationship of its introns with homologous Ascomycota introns and divergence levels from common ancestral copies. Gene 1998, 220, 45–53. [Google Scholar] [CrossRef]
- Copertino, D.; Hallick, R.B. Group II twintron: An intron within an intron in a chloroplast cytochrome B-559 gene. EMBO J. 1991, 10, 433. [Google Scholar] [PubMed]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Liu, C.; Shen, B.; Bai, M.; Ling, J.; Chen, G.; Mao, Z.; Cheng, X.; Xie, B. Analysis of the complete mitochondrial genome of Pochonia chlamydosporia suggests a close relationship to the invertebrate-pathogenic fungi in Hypocreales. BMC Microbiol. 2015, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.F.; Laforest, M.J.; Burger, G. Mitochondrial introns: A critical view. Trends Genet. 2007, 23, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, S.; Zhang, G.; Liu, X.; Wang, C.; Xu, J. Comparison of mitochondrial genomes provides insights into intron dynamics and evolution in the caterpillar fungus Cordyceps militaris. Fungal Genet. Biol. 2015, 77, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Jung, P.P.; Friedrich, A.; Reisser, C.; Hou, J.; Schacherer, J. Mitochondrial genome evolution in a single protoploid yeast species. G3 Genes Genomes Genet. 2012, 2, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Jalalzadeh, B.; Saré, I.C.; Férandon, C.; Callac, P.; Farsi, M.; Savoie, J.-M.; Barroso, G. The intraspecific variability of mitochondrial genes of Agaricus bisporus reveals an extensive group I intron mobility combined with low nucleotide substitution rates. Curr. Genet. 2015, 61, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Torriani, S.F.; Penselin, D.; Knogge, W.; Felder, M.; Taudien, S.; Platzer, M.; McDonald, B.A.; Brunner, P.C. Comparative analysis of mitochondrial genomes from closely related Rhynchosporium species reveals extensive intron invasion. Fungal Genet. Biol. 2014, 62, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Mardanov, A.V.; Beletsky, A.V.; Kadnikov, V.V.; Ignatov, A.N.; Ravin, N.V. The 203 kbp mitochondrial genome of the phytopathogenic fungus sclerotinia borealis reveals multiple invasions of introns and genomic duplications. PLoS ONE 2014, 9, e107536. [Google Scholar] [CrossRef] [PubMed]
- Salavirta, H.; Oksanen, I.; Kuuskeri, J.; Mäkelä, M.; Laine, P.; Paulin, L.; Lundell, T. Mitochondrial genome of Phlebia radiata is the second largest (156 kbp) among fungi and features signs of genome flexibility and recent recombination events. PLoS ONE 2014, 9, e97141. [Google Scholar]
- Duò, A.; Bruggmann, R.; Zoller, S.; Bernt, M.; Grünig, C.R. Mitochondrial genome evolution in species belonging to the Phialocephala fortinii sl-Acephala applanata species complex. BMC Genom. 2012, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Sethuraman, J.; Majer, A.; Friedrich, N.C.; Edgell, D.R.; Hausner, G. Genes within Genes: Multiple LAGLIDADG homing endonucleases target the ribosomal protein S3 gene encoded within an rnl group I intron of ophiostoma and related taxa. Mol. Biol. Evol. 2009, 26, 2299–2315. [Google Scholar] [CrossRef] [PubMed]
- Pantou, M.P.; Kouvelis, V.N.; Typas, M.A. The complete mitochondrial genome of Fusarium oxysporum: Insights into fungal mitochondrial evolution. Gene 2008, 419, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Aguileta, G.; de Vienne, D.M.; Ross, O.N.; Hood, M.E.; Giraud, T.; Petit, E.; Gabaldon, T. High variability of mitochondrial gene order among fungi. Genome Biol. Evol. 2014, 6, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Einvik, C.; Elde, M.; Johansen, S. Group I twintrons: Genetic elements in myxomycete and schizopyrenid amoeboflagellate ribosomal DNAs. J. Biotechnol. 1998, 64, 63–74. [Google Scholar] [CrossRef]
- Copertino, D.W.; Hall, E.T.; van Hook, F.W.; Jenkins, K.P.; Hallick, R.B. A group III twintron encoding a maturase-like gene excises through lariat intermediates. Nucleic Acids Res. 1994, 22, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Hafez, M.; Majer, A.; Sethuraman, J.; Rudski, S.M.; Michel, F.; Hausner, G. The mtDNA rns gene landscape in the Ophiostomatales and other fungal taxa: Twintrons, introns, and intron-encoded proteins. Fungal Genet. Biol. 2013, 53, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.; Archibald, J.M. Lateral transfer of introns in the cryptophyte plastid genome. Nucleic Acids Res. 2008, 36, 3043–3053. [Google Scholar] [CrossRef] [PubMed]
- Copertino, D.; Shigeoka, S.; Hallick, R. Chloroplast group III twintron excision utilizing multiple 5′- and 3′-splice sites. EMBO J. 1992, 11, 5041. [Google Scholar] [PubMed]
- Li, H.; Ulge, U.Y.; Hovde, B.T.; Doyle, L.A.; Monnat, R.J. Comprehensive homing endonuclease target site specificity profiling reveals evolutionary constraints and enables genome engineering applications. Nucleic Acids Res. 2012, 40, 2587–2598. [Google Scholar] [CrossRef] [PubMed]
- Barzel, A.; Privman, E.; Peeri, M.; Naor, A.; Shachar, E.; Burstein, D.; Lazary, R.; Gophna, U.; Pupko, T.; Kupiec, M. Native homing endonucleases can target conserved genes in humans and in animal models. Nucleic Acids Res. 2011, 39, 6646–6659. [Google Scholar] [CrossRef] [PubMed]
- Scalley-Kim, M.; McConnell-Smith, A.; Stoddard, B.L. Coevolution of a homing endonuclease and its host target sequence. J. Mol. Biol. 2007, 372, 1305–1319. [Google Scholar] [CrossRef] [PubMed]
- Sellem, C.H.; d'Aubenton-Carafa, Y.; Rossignol, M.; Belcour, L. Mitochondrial intronic open reading frames in Podospora: Mobility and consecutive exonic sequence variations. Genetics 1996, 143, 777–788. [Google Scholar] [PubMed]
- Sellem, C.H.; Belcour, L. Intron open reading frames as mobile elements and evolution of a group I intron. Mol. Biol. Evol. 1997, 14, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Manicon, B.; Bar-Joseph, M.; Rosner, A.; Vigodsky-Haas, H.; Kotze, J. Potential applications of random DNA probes and restriction fragment length polymorphisms in the taxonomy of the Fusaria. Phytopathology 1987, 77, 669–672. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Beck, N.; Lang, B. RNAweasel: A Webserver for Identification of Mitochondrial, Structured RNAs; University of Montreal: Montreal, QC, Canada, 2009. [Google Scholar]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.M.; Belfort, M.; Cech, T.R.; Davies, R.W.; Schweyen, R.J.; Shub, D.A.; Szostak, J.W.; Tabak, H.F. Structural conventions for group I introns. Nucleic Acids Res. 1987, 15, 7217–7221. [Google Scholar] [CrossRef] [PubMed]
- Cech, T.R.; Damberger, S.H.; Gutell, R.R. Representation of the secondary and tertiary structure of group I introns. Nat. Struct. Mol. Biol. 1994, 1, 273–280. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y. Predicting the secondary structures and tertiary interactions of 211 group I introns in IE subgroup. Nucleic Acids Res. 2005, 33, 2118–2128. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lu, C.; Wu, Q.-J.; Wang, Y.; Sun, Z.-T.; Deng, J.-C.; Zhang, Y. GISSD: Group I intron sequence and structure database. Nucleic Acids Res. 2008, 36, D31–D37. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Intron | Position | Intron Size, bp | Intron Type | ORF | Conserved Domain | E-Value | Id | Similarity | Accession |
|---|---|---|---|---|---|---|---|---|---|---|
| cox3 | Intron 1 | 73 aa | 1128 | IB | ORF309 | LAGLIDADG_1 | 1.00 × 10−172 | 79% | Ceratocystis cacaofunesta | YP_007507087 |
| Intron 2 | 213 aa | 2877 | IA(5’) | degraded ORF | LAGLIDADG_1 | 9 × 10−119 | 59% | Annulohypoxylon stygium | YP_008964946 | |
| IA | degraded ORF | LAGLIDADG_1 | 0 | 80% | Fusarium solani | YP_005088126 | ||||
| cox2 | Intron 1 | 76 aa | 3295 | IB | * | GIY-YIG | 1.00 × 10−92 | 73% | Ganoderma lucidum | CCQ18569 |
| ORF591 | LAGLIDADG_1 | 6.00 × 10−78 | 46% | Podospora anserina | CAA38805 | |||||
| cob | Intron 1 | 67 aa | 1586 | IB(3’) | ORF453 | LAGLIDADG_1 | 0 | 90% | Sordaria macrospora k-hell | XP_003342387 |
| Intron 2 | 131 aa | 1202 | ID | ORF287 | GIY-YIG | 7.00 × 10−144 | 79% | Fusarium acuminatum | CDL73465 | |
| Intron 3 | 169 aa | 1069 | IB(3’) | ORF322 | LAGLIDADG_1 | 0 | 83% | Aspergillus nidulans | P03880 | |
| rnl | Intron 1 | 622 bp | 1790 | IC1 | ORF308 | GIY-YIG | 8.00 × 10−77 | 74% | Sclerotinia borealis | YP_009072316 |
| Intron 2 | 811 bp | 1666 | IC1 | ORF342 | GIY-YIG | 3.00 × 10−112 | 70% | Cordyceps brongniartii | YP_002213592 | |
| Intron 3 | 2494 bp | 2090 | IA | ORF474 | rps3 | 0 | 82% | Trichoderma harzianum | AKK32420 | |
| Intron 4 | 2599 bp | 1852 | IB(5’) | ORF294 | GIY-YIG | 5.00 × 10−70 | 49% | Sclerotinia borealis | YP_009072319 | |
| cox1 | Intron 1 | 94 aa | 1260 | IB | ORF301 | LAGLIDADG_1 | 5 × 10−162 | 80% | Ganoderma meredithae | YP_009129958 |
| Intron 2 | 180 aa | 1385 | IC2 | ORF245 | LAGLIDADG_1 | ‒ | ‒ | ‒ | ‒ | |
| Intron 3 | 352 aa | 2310 | IB | ORF422 | GIY-YIG | 0 | 74% | Madurella mycetomatis | YP_006576207 | |
| ORF308 | GIY-YIG | 2 × 10−167 | 83% | Fusarium culmorum | CDL73521 | |||||
| Intron 4 | 375 aa | 2334 | IB | ORF357 | LAGLIDADG_1 | 0 | 79% | Ceratocystis cacaofunesta | YP_007507075 | |
| ORF392 | ‒ | ‒ | ‒ | ‒ | ‒ | |||||
| Intron 5 | 427 aa | 1759 | IB | ORF447 | GIY-YIG | 0 | 75% | Fusarium graminearum | AKB93468 | |
| atp6 | Intron 1 | 116 aa | 1404 | IB | ORF365 | LAGLIDADG_1 | 1.00 × 10−145 | 71% | Botrytis cinerea | AGN49025 |
| Intron 2 | 189 aa | 1584 | IC2 | ORF302 | GIY-YIG | 7.00 × 10−133 | 72% | Podospora anserina | NP_074919 |
| Genome a | Mitogenome Size | GC % | Protein-Coding Genes | Protein-Coding Genes with Introns | Length (and Number) of Introns in Protein Coding Genes (bp) | Length (and Number) of Intron in Large Subunit rRNA (bp) | Length (and Number) of Intron in Small Subunit rRNA (bp) | tRNAs b | Intronic ORFS | Length (and Number) of Non-Intronic Accessory Genes (bp) | Accession |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Acremonium chrysogenum | 27,266 | 26.54 | 19 | 1 | 1331 (2) | 1602 (1) | 0 | 26 | 2 | 1482 (3) | NC_023268 |
| Acremonium implicatum | 22,376 | 26.12 | 15 | 0 | 0 | 0 | 0 | 17 | 0 | 2556 (3) | NC_026534 |
| Beauveria bassiana | 29,961 | 27.25 | 20 | 2 | 2272 (2) | 1745 (1) | 0 | 25 | 2 | 1872 (3) | NC_010652 |
| Beauveria pseudobassiana | 28,006 | 27.54 | 17 | 1 | 1078 (1) | 1755 (1) | 0 | 25 | 2 | 1116 (1) | NC_022708 |
| Cordyceps brongniartii | 33,926 | 27.34 | 22 | 2 | 3776 (3) | 3095 (2) | 0 | 25 | 4 | 3663 (4) | NC_011194 |
| Cordyceps militaris | 33,277 | 26.79 | 22 | 4 | 4707 (4) | 5116 (4) | 0 | 27 | 7 | 0 | NC_022834 |
| Fusarium circinatum | 67,109 | 31.45 | 33 | 5 | 19,269 (15) | 2379 (1) | 0 | 27 | 17 | 7506 (2) | NC_022681 |
| Fusarium gerlachii | 93,428 | 31.91 | 53 | 8 | 38,673 (27) | 1999 (1) | 0 | 28 | 31 | 10,479 (8) | NC_025928 |
| Fusarium graminearum | 95,676 | 31.84 | 53 | 8 | 41,057 (28) | 1999 (1) | 0 | 28 | 30 | 11,388 (9) | NC_009493 |
| Fusarium oxysporum | 34,477 | 30.98 | 17 | 1 | 1010 (1) | 1602 (1) | 0 | 25 | 2 | 456 (1) | AY945289 |
| Fusarium solani | 62,978 | 28.88 | 34 | 6 | 20,427 (14) | 1796 (1) | 0 | 25 | 14 | 6495 (2) | NC_016680 |
| Gibberella moniliformis | 53,753 | 32.61 | 24 | 2 | 3909 (3) | 1953 (1) | 0 | 27 | 4 | 8175 (2) | NC_016687 |
| Hirsutella minnesotensis | 52,245 | 28.42 | 30 | 4 | 12,428 (10) | 1818 (1) | 0 | 25 | 11 | 6145 (5) | NC_027660 |
| Hypocrea jecorina | 42,130 | 27.24 | 25 | 3 | 11,934 (9) | 1655 (1) | 0 | 26 b | 9 | 960 (2) | NC_003388 |
| Lecanicillium muscarium | 24,499 | 27.15 | 15 | 0 | 0 | 1617 (1) | 0 | 25 | 1 | 0 | NC_004514 |
| Lecanicillium saksenae | 25,919 | 26.53 | 17 | 0 | 0 | 1668 (1) | 0 | 25 | 1 | 2829 (2) | NC_028330 |
| Metarhizium anisopliae | 24,673 | 28.40 | 15 | 0 | 0 | 0 | 0 | 24 | 0 | 924 (1) | NC_008068 |
| Metacordyceps chlamydosporia | 25,615 | 28.28 | 18 | 0 | 0 | 1147 (1) | 0 | 25 | 1 | 1293 (2) | NC_022835 |
| Neotyphodium lolii | 88,756 | 27.53 | 60 | 7 | 30,040 (19) | 4809 (4) | 1288 (1) | 28 b | 21 | 12,576 (22) | KF906135 |
| Nectria cinnabarina | 69,895 | 28.71 | 33 | 6 | 23,840 (15) | 2762 (2) | 0 | 25 | 11 | 7197 (8) | KT731105 |
| Paecilomyces hepiali | 24,245 | 26.64 | 15 | 0 | 0 | 1610 (1) | 0 | 25 | 1 | 0 | KJ764671 |
| Trichoderma harzianum | 29,999 | 27.78 | 17 | 1 | 1118 (1) | 1631 (1) | 0 | 27 | 2 | 639 (1) | KR952346 |
| Trichoderma gamsii | 29,303 | 28.25 | 16 | 0 | 0 | 1609 (1) | 0 | 28 b | 1 | 348 (1) | KU687109 |
| Hypomyces aurantius | 71,638 | 28.31 | 42 | 5 | 20,136 (13) | 7972 (4) | 0 | 27 b | 18 | 10,482 (12) | KU666552 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, Y.; Zhang, Q.; Ming, R.; Lin, L.; Lin, X.; Lin, Y.; Li, X.; Xie, B.; Wen, Z. Analysis of the Mitochondrial Genome in Hypomyces aurantius Reveals a Novel Twintron Complex in Fungi. Int. J. Mol. Sci. 2016, 17, 1049. https://doi.org/10.3390/ijms17071049
Deng Y, Zhang Q, Ming R, Lin L, Lin X, Lin Y, Li X, Xie B, Wen Z. Analysis of the Mitochondrial Genome in Hypomyces aurantius Reveals a Novel Twintron Complex in Fungi. International Journal of Molecular Sciences. 2016; 17(7):1049. https://doi.org/10.3390/ijms17071049
Chicago/Turabian StyleDeng, Youjin, Qihui Zhang, Ray Ming, Longji Lin, Xiangzhi Lin, Yiying Lin, Xiao Li, Baogui Xie, and Zhiqiang Wen. 2016. "Analysis of the Mitochondrial Genome in Hypomyces aurantius Reveals a Novel Twintron Complex in Fungi" International Journal of Molecular Sciences 17, no. 7: 1049. https://doi.org/10.3390/ijms17071049
APA StyleDeng, Y., Zhang, Q., Ming, R., Lin, L., Lin, X., Lin, Y., Li, X., Xie, B., & Wen, Z. (2016). Analysis of the Mitochondrial Genome in Hypomyces aurantius Reveals a Novel Twintron Complex in Fungi. International Journal of Molecular Sciences, 17(7), 1049. https://doi.org/10.3390/ijms17071049