
A Guide to Non-Alcoholic Fatty Liver Disease in Childhood and Adolescence


Abstract
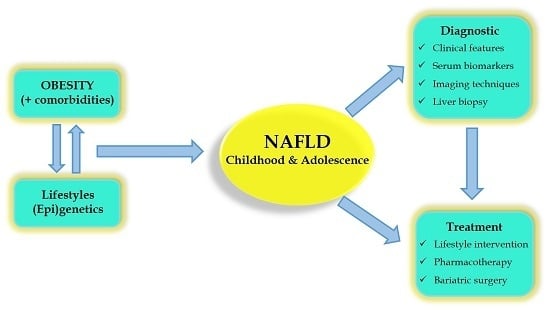
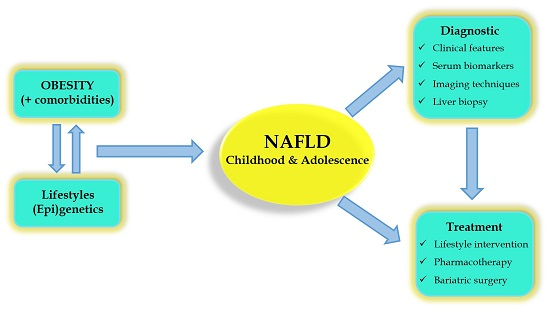
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Clinical Presentation of Paediatric Non-Alcoholic Fatty Liver Disease (NAFLD)
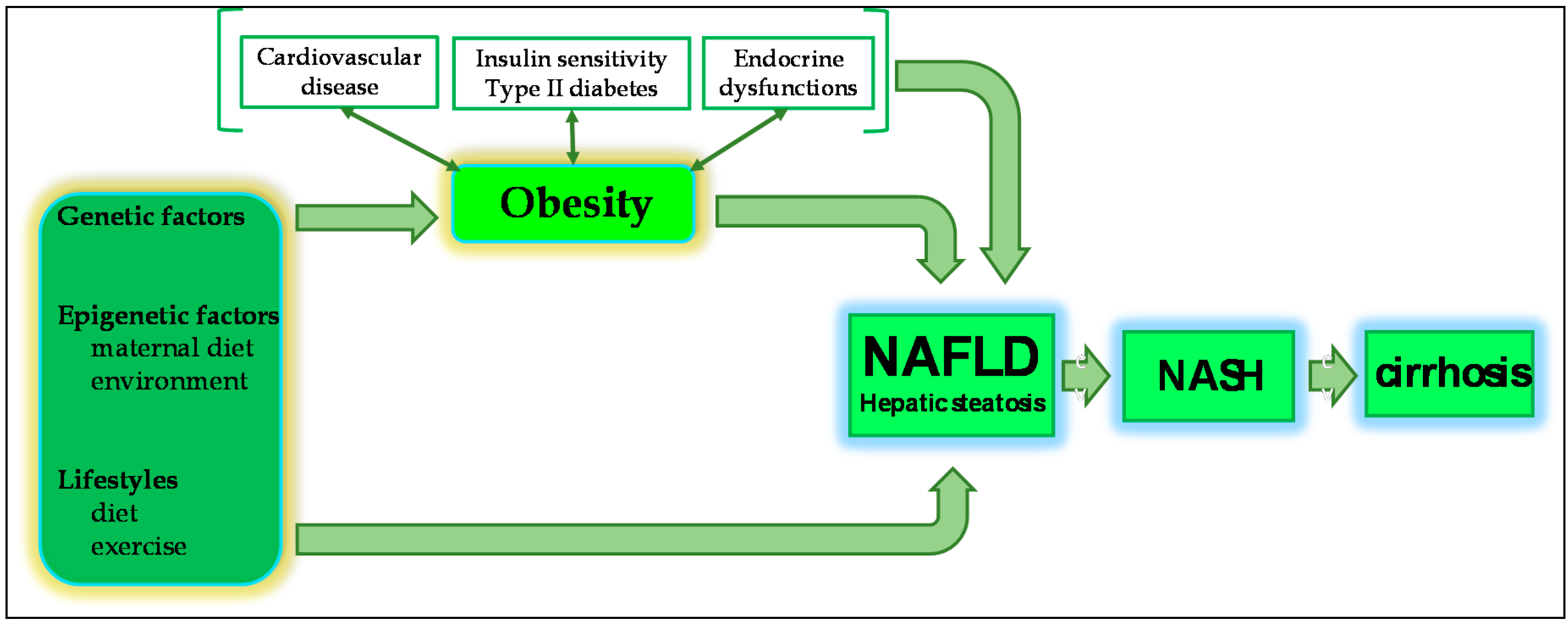
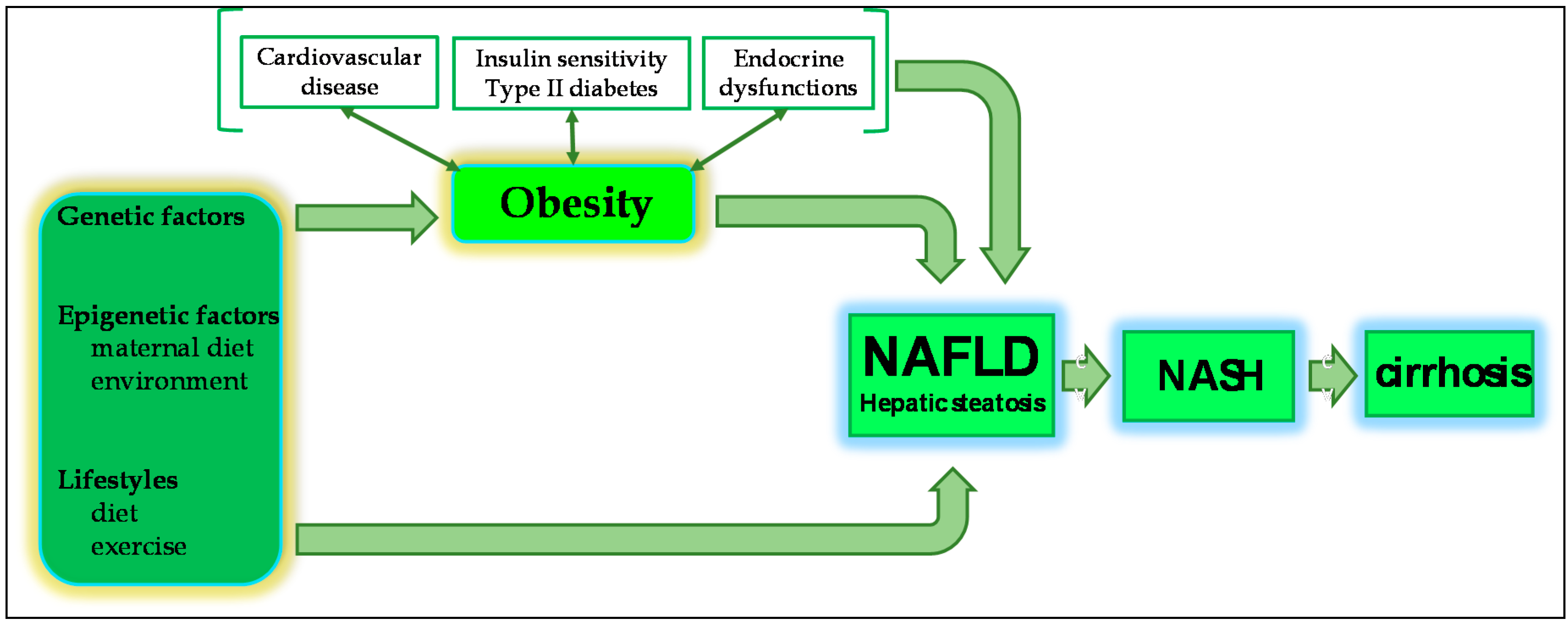
2.1. NAFLD and Obesity
2.2. Hepatic Complications of NAFLD
2.2.1. Fibrosis
2.2.2. Cirrhosis
2.2.3. Hepatocellular Carcinoma
2.3. Extra-Hepatic Complications of NAFLD
2.3.1. Cardiovascular Disease
2.3.2. Insulin Resistance and Type II Diabetes Mellitus
2.3.3. Other Endocrine Disorders
3. The Pathogenesis of NAFLD
3.1. Genetics of Paediatric NAFLD
3.2. Maternal Diet, Intrauterine Growth and Neonatal Diet
3.3. Gender Differences and Puberty
3.4. Dysregulation of Hedgehog Signalling Pathway in NAFLD
4. Making the Diagnosis
4.1. Alternative Classification System
4.2. Serum Biomarkers for Liver Damage
4.3. Abdominal Ultrasound
4.4. Magnetic Resonance Imaging
4.5. Other Imaging Techniques
4.6. Liver Biopsy and Histopathology
4.7. Non-Invasive Diagnostic Scoring Systems
5. Management of Paediatric NAFLD
5.1. Diet and Physical Exercise
5.1.1. Dietary Fructose
5.1.2. Vitamin D
5.1.3. ω-3 Fatty Acids
5.2. Alcohol
5.3. Bariatric Surgery
5.4. Pharmacological Intervention
5.4.1. Insulin Sensitizers
5.4.2. Weight Loss Drugs
5.4.3. Statins
5.5. Antioxidant Therapies
5.6. Vitamin E
5.7. Ursodeoxycholic Acid
5.8. Probiotic Therapy
6. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| LD | Linear dichroism |
| AASLD | American Association for the Study of Liver Diseases |
| ALT | Alanine Aminotransferase |
| AMPK | AMP-activated Protein Kinase |
| APOC3 | Adipoprotein C3 |
| AST | Aspartate Aminotransferase |
| BMI | Body Mass Index |
| CB2 | Cannabinoid Receptor 2 |
| CK-18 | Cytokeratin 18 |
| CT | Computerized Tomography |
| CYP2E1 | Cytochrome P450 family 2 subfamily E member 1 |
| DGAT2 | Diacylglycerol O-Acyltransferase 2 |
| DHA | Docosahexaenoic Acid |
| DPP-4 | Incretin Mimetics and Dipeptidyl Peptidase-4 |
| EASL | European Association for the Study of the Liver |
| ELF | Enhanced Liver Fibrosis |
| FTO | Fat Mass and Obesity associated |
| FXR | Farnesoid X Receptor |
| GCKR | Glucokinase Regulatory Protein |
| GGT | γ-glutamyl Transferase |
| GLP1 | Glucagon-Like Peptide-1 |
| GRP-120 | G Protein-coupled Receptor 120 |
| GWAS | Genome-Wide Association Study |
| HDL | High-Density Lipoprotein |
| HPC | Hepatic Progenitor Cells |
| HSC | Hepatic Stellate Cells |
| IL-6 | Interleukin 6 |
| IR | Insulin Resistance |
| IRS-1 | Insulin Receptor Substrate-1 |
| IUGR | Intrauterine Growth Restriction |
| KC | Kupffer Cells |
| KLF-6 | Kruppel-Like Factor 6 |
| LDL | Low Density Lipoprotein |
| LFTs | Liver Function Tests |
| LPIN1 | Lipin 1 |
| LYPLAL1 | Lysophospholipase-Like 1 |
| MC4R | Melanocortin 4 Receptor |
| NAFLD | Non-Alcoholic Fatty Liver Disease |
| NASH | Non-Alcoholic Steatohepatitis |
| NCAN | Neurocan |
| NKT | Natural Killer T lymphocytes |
| PNFI | Pediatric NAFLD Fibrosis Index |
| PNPLA3 | Adiponutrin/Patatin-like Phospholipase Domain-containing 3 |
| PPARg | Peroxisome-Proliferator Activated Receptor-γ |
| PPP1R3B | Protein Phosphatase 1 Regulatory Subunit 3b |
| PYY | Peptide YY |
| ROS | Reactive Oxygen Species |
| SLD | Steatotic Liver Disease |
| SOD2 | Manganese-dependent Superoxide Dismutase |
| SREBP1c | Sterol Regulatory Binding Element |
| TNF-α | Tumor Necrosis Factor α |
| TONIC | Treatment of NAFLD in Children |
References
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, D.A.; Callaway, M.; Macdonald-Wallis, C.; Anderson, E.; Fraser, A.; Howe, L.D.; Day, C.; Sattar, N. Nonalcoholic fatty liver disease, liver fibrosis, and cardiometabolic risk factors in adolescence: A cross-sectional study of 1874 general population adolescents. J. Clin. Endocrinol. Metab. 2014, 99, E410–E417. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; Torbenson, M.; Wu, T.T.; Yeh, M.M. Non-alcoholic fatty liver disease contributes to hepatocarcinogenesis in non-cirrhotic liver: A clinical and pathological study. J. Gastroenterol. Hepatol. 2013, 28, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Cianfarani, S.; Manco, M.; Agostoni, C.; Nobili, V. Non-alcoholic fatty liver disease and metabolic syndrome in adolescents: Pathogenetic role of genetic background and intrauterine environment. Ann. Med. 2012, 44, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Perticone, M.; Cimellaro, A.; Maio, R.; Caroleo, B.; Sciacqua, A.; Sesti, G.; Perticone, F. Additive effect of non-alcoholic fatty liver disease on metabolic syndrome-related endothelial dysfunction in hypertensive patients. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Ayonrinde, O.T.; Olynyk, J.K.; Marsh, J.A.; Beilin, L.J.; Mori, T.A.; Oddy, W.H.; Adams, L.A. Childhood adiposity trajectories and risk of nonalcoholic fatty liver disease in adolescents. J. Gastroenterol. Hepatol. 2015, 30, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Moran, J.R.; Ghishan, F.K.; Halter, S.A.; Greene, H.L. Steatohepatitis in obese children: A cause of chronic liver dysfunction. Am. J. Gastroenterol. 1983, 78, 374–377. [Google Scholar] [PubMed]
- Marcason, W. What are the current guidelines for pediatric non-alcoholic fatty liver disease? J. Acad. Nutr. Diet. 2013, 113, 1772. [Google Scholar] [CrossRef] [PubMed]
- Mencin, A.A.; Lavine, J.E. Advances in pediatric nonalcoholic fatty liver disease. Pediatr. Clin. N. Am. 2011, 58, 1375–1392. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, P.A.; Antunes Bde, M.; Silveira, L.S.; Christofaro, D.G.; Fernandes, R.A.; Freitas Junior, I.F. Body composition variables as predictors of nafld by ultrasound in obese children and adolescents. BMC Pediatr. 2014, 14, 25. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Bellentani, S.; Cortez-Pinto, H.; Day, C.; Marchesini, G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J. Hepatol. 2010, 53, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Berardis, S.; Sokal, E. Pediatric non-alcoholic fatty liver disease: An increasing public health issue. Eur. J. Pediatr. 2014, 173, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Deutsch, R.; Kahen, T.; Lavine, J.E.; Stanley, C.; Behling, C. Prevalence of fatty liver in children and adolescents. Pediatrics 2006, 118, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Holterman, A.; Gurria, J.; Tanpure, S.; DiSomma, N. Nonalcoholic fatty liver disease and bariatric surgery in adolescents. Semin. Pediatr. Surg. 2014, 23, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Vajro, P.; Ferrante, L.; Lenta, S.; Mandato, C.; Persico, M. Management of adults with paediatric-onset chronic liver disease: Strategic issues for transition care. Dig. Liver Dis. 2014, 46, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Palmer, N.D.; Musani, S.K.; Yerges-Armstrong, L.M.; Feitosa, M.F.; Bielak, L.F.; Hernaez, R.; Kahali, B.; Carr, J.J.; Harris, T.B.; Jhun, M.A.; et al. Characterization of european ancestry nonalcoholic fatty liver disease-associated variants in individuals of african and hispanic descent. Hepatology 2013, 58, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Vajro, P.; Lenta, S.; Socha, P.; Dhawan, A.; McKiernan, P.; Baumann, U.; Durmaz, O.; Lacaille, F.; McLin, V.; Nobili, V. Diagnosis of nonalcoholic fatty liver disease in children and adolescents: Position paper of the espghan hepatology committee. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 700–713. [Google Scholar] [CrossRef] [PubMed]
- Boyraz, M.; Hatipoglu, N.; Sari, E.; Akcay, A.; Taskin, N.; Ulucan, K.; Akcay, T. Non-alcoholic fatty liver disease in obese children and the relationship between metabolic syndrome criteria. Obes. Res. Clin. Pract. 2014, 8, e356–e363. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American association for the study of liver diseases, American college of gastroenterology, and the American gastroenterological association. Am. J. Gastroenterol. 2012, 107, 811–826. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Fishbein, M.H.; Rigsby, C.K.; Zhang, G.; Schoeneman, S.E.; Donaldson, J.S. Quantitative MRI for hepatic fat fraction and T2* measurement in pediatric patients with non-alcoholic fatty liver disease. Pediatr. Radiol. 2014, 44, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Liccardo, D.; Alisi, A.; Porta, G.; Nobili, V. Is there any link between dietary pattern and development of nonalcoholic fatty liver disease in adolescence? An expert review. Expert Rev. Gastroenterol. Hepatol. 2013, 7, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Lerret, S.M.; Garcia-Rodriguez, L.; Skelton, J.; Biank, V.; Kilway, D.; Telega, G. Predictors of nonalcoholic steatohepatitis in obese children. Gastroenterol. Nurs. 2011, 34, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Jarabo, J.M.; Ubina-Aznar, E.; Tapia-Ceballos, L.; Ortiz-Cuevas, C.; Perez-Aisa, M.A.; Rivas-Ruiz, F.; Andrade, R.J.; Perea-Milla, E. Hepatic steatosis and severity-related factors in obese children. J. Gastroenterol. Hepatol. 2013, 28, 1532–1538. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Nobili, V. Non-alcoholic fatty liver disease in children now: Lifestyle changes and pharmacologic treatments. Nutrition 2012, 28, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Ozhan, B.; Ersoy, B.; Kiremitci, S.; Ozkol, M.; Taneli, F. Insulin sensitivity indices: Fasting versus glucose-stimulated indices in pediatric non-alcoholic fatty liver disease. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 3450–3458. [Google Scholar] [PubMed]
- Anderson, E.L.; Howe, L.D.; Jones, H.E.; Higgins, J.P.; Lawlor, D.A.; Fraser, A. The prevalence of non-alcoholic fatty liver disease in children and adolescents: A systematic review and meta-analysis. PLoS ONE 2015, 10, e0140908. [Google Scholar] [CrossRef] [PubMed]
- Kelsey, M.M.; Zaepfel, A.; Bjornstad, P.; Nadeau, K.J. Age-related consequences of childhood obesity. Gerontology 2014, 60, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Regnell, S.E.; Peterson, P.; Trinh, L.; Broberg, P.; Leander, P.; Lernmark, A.; Mansson, S.; Elding Larsson, H. Magnetic resonance imaging reveals altered distribution of hepatic fat in children with type 1 diabetes compared to controls. Metabolism 2015, 64, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Mansoor, S.; Yerian, L.; Kohli, R.; Xanthakos, S.; Angulo, P.; Ling, S.; Lopez, R.; Christine, C.K.; Feldstein, A.E.; Alkhouri, N. The evaluation of hepatic fibrosis scores in children with nonalcoholic fatty liver disease. Dig. Dis. Sci. 2015, 60, 1440–1447. [Google Scholar] [CrossRef] [PubMed]
- Trovato, F.M.; Martines, G.F.; Brischetto, D.; Catalano, D.; Musumeci, G.; Trovato, G.M. Fatty liver disease and lifestyle in youngsters: Diet, food intake frequency, exercise, sleep shortage and fashion. Liver Int. 2016, 36, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Stepanova, M.; Negro, F.; Hallaji, S.; Younossi, Y.; Lam, B.; Srishord, M. Nonalcoholic fatty liver disease in lean individuals in the United States. Medicine 2012, 91, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Silveira, L.S.; Monteiro, P.A.; Antunes Bde, M.; Seraphim, P.M.; Fernandes, R.A.; Christofaro, D.G.; Freitas Junior, I.F. Intra-abdominal fat is related to metabolic syndrome and non-alcoholic fat liver disease in obese youth. BMC Pediatr. 2013, 13, 115. [Google Scholar] [CrossRef] [PubMed]
- Kelishadi, R.; Cook, S.R.; Adibi, A.; Faghihimani, Z.; Ghatrehsamani, S.; Beihaghi, A.; Salehi, H.; Khavarian, N.; Poursafa, P. Association of the components of the metabolic syndrome with non-alcoholic fatty liver disease among normal-weight, overweight and obese children and adolescents. Diabetol. Metab. Syndr. 2009, 1, 29. [Google Scholar] [CrossRef] [PubMed]
- Manco, M.; Bedogni, G.; Marcellini, M.; Devito, R.; Ciampalini, P.; Sartorelli, M.R.; Comparcola, D.; Piemonte, F.; Nobili, V. Waist circumference correlates with liver fibrosis in children with non-alcoholic steatohepatitis. Gut 2008, 57, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Alterio, A.; Alisi, A.; Liccardo, D.; Nobili, V. Non-alcoholic fatty liver and metabolic syndrome in children: A vicious circle. Horm. Res. Paediatr. 2014, 82, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Mager, D.R.; Yap, J.; Rodriguez-Dimitrescu, C.; Mazurak, V.; Ball, G.; Gilmour, S. Anthropometric measures of visceral and subcutaneous fat are important in the determination of metabolic dysregulation in boys and girls at risk for nonalcoholic fatty liver disease. Nutr. Clin. Pract. 2013, 28, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Boyraz, M.; Cekmez, F.; Karaoglu, A.; Cinaz, P.; Durak, M.; Bideci, A. Relationship of adipokines (adiponectin, resistin and RBP4) with metabolic syndrome components in pubertal obese children. Biomark. Med. 2013, 7, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Sayin, O.; Tokgoz, Y.; Arslan, N. Investigation of adropin and leptin levels in pediatric obesity-related nonalcoholic fatty liver disease. J. Pediatr. Endocrinol. Metab. 2014, 27, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zheng, L.; Sheng, C.; Cheng, X.; Qing, L.; Qu, S. Systematic review on the treatment of pentoxifylline in patients with non-alcoholic fatty liver disease. Lipids Health Dis. 2011, 10, 49. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Lymp, J.F.; St Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Franzen, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006, 44, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Ong, J.P.; Pitts, A.; Younossi, Z.M. Increased overall mortality and liver-related mortality in non-alcoholic fatty liver disease. J. Hepatol. 2008, 49, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Newton, K.P.; Awai, H.I.; Choi, L.J.; Garcia, M.A.; Ellis, L.L.; Vanderwall, K.; Fontanesi, J. Paediatric gastroenterology evaluation of overweight and obese children referred from primary care for suspected non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2013, 38, 1267–1277. [Google Scholar] [CrossRef] [PubMed]
- Holterman, A.X.; Guzman, G.; Fantuzzi, G.; Wang, H.; Aigner, K.; Browne, A.; Holterman, M. Nonalcoholic fatty liver disease in severely obese adolescent and adult patients. Obesity 2013, 21, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Charlotte, F.; Le Naour, G.; Bernhardt, C.; Poynard, T.; Ratziu, V.; Group, L.S. A comparison of the fibrotic potential of nonalcoholic fatty liver disease and chronic hepatitis C. Hum. Pathol. 2010, 41, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.H.; Crespo, D.M.; Kang, H.S.; Al-Osaimi, A.M. Obesity and hepatocellular carcinoma. Gastroenterology 2004, 127, S97–S103. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Hampel, H.; Javadi, F. The association between diabetes and hepatocellular carcinoma: A systematic review of epidemiologic evidence. Clin. Gastroenterol. Hepatol. 2006, 4, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Berentzen, T.L.; Gamborg, M.; Holst, C.; Sorensen, T.I.; Baker, J.L. Body mass index in childhood and adult risk of primary liver cancer. J. Hepatol. 2014, 60, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Atabek, M.E.; Selver Eklioglu, B.; Akyurek, N. Which metabolic syndrome criteria best predict non-alcoholic fatty liver disease in children? Eat. Weight Disord. 2014, 19, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.K.; Vitola, B.E.; Holland, M.R.; Sekarski, T.; Patterson, B.W.; Magkos, F.; Klein, S. Alterations in ventricular structure and function in obese adolescents with nonalcoholic fatty liver disease. J. Pediatr. 2013, 162, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Fintini, D.; Chinali, M.; Cafiero, G.; Esposito, C.; Giordano, U.; Turchetta, A.; Pescosolido, S.; Pongiglione, G.; Nobili, V. Early left ventricular abnormality/dysfunction in obese children affected by NAFLD. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Pacifico, L.; Di Martino, M.; de Merulis, A.; Bezzi, M.; Osborn, J.F.; Catalano, C.; Chiesa, C. Left ventricular dysfunction in obese children and adolescents with nonalcoholic fatty liver disease. Hepatology 2014, 59, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Alkhouri, N.; Bartuli, A.; Manco, M.; Lopez, R.; Alisi, A.; Feldstein, A.E. Severity of liver injury and atherogenic lipid profile in children with nonalcoholic fatty liver disease. Pediatr. Res. 2010, 67, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Pacifico, L.; Bonci, E.; Andreoli, G.; Romaggioli, S.; Di Miscio, R.; Lombardo, C.V.; Chiesa, C. Association of serum triglyceride-to-HDL cholesterol ratio with carotid artery intima-media thickness, insulin resistance and nonalcoholic fatty liver disease in children and adolescents. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Donati, B.; Panera, N.; Vongsakulyanon, A.; Alisi, A.; Dallapiccola, B.; Valenti, L. A 4-polymorphism risk score predicts steatohepatitis in children with nonalcoholic fatty liver disease. J. Pediatr. Gastroenterol. Nutr. 2014, 58, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Baker, S.S.; Liu, W.; Alkhouri, R.; Baker, R.D.; Xie, J.; Ji, G.; Zhu, L. Endotoxemia unrequired in the pathogenesis of pediatric nonalcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2014, 29, 1292–1298. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Diehl, A.M. Hepatic triglyceride synthesis and nonalcoholic fatty liver disease. Curr. Opin. Lipidol. 2008, 19, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Wanless, I.R.; Shiota, K. The pathogenesis of nonalcoholic steatohepatitis and other fatty liver diseases: A four-step model including the role of lipid release and hepatic venular obstruction in the progression to cirrhosis. Semin. Liver Dis. 2004, 24, 99–106. [Google Scholar] [PubMed]
- Morandi, A.; Maffeis, C. Predictors of metabolic risk in childhood obesity. Horm. Res. Paediatr. 2014, 82, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Torun, E.; Ozgen, I.T.; Gokce, S.; Aydin, S.; Cesur, Y. Thyroid hormone levels in obese children and adolescents with non-alcoholic fatty liver disease. J. Clin. Res. Pediatr. Endocrinol. 2014, 6, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Pacifico, L.; Bonci, E.; Ferraro, F.; Andreoli, G.; Bascetta, S.; Chiesa, C. Hepatic steatosis and thyroid function tests in overweight and obese children. Int. J. Endocrinol. 2013, 2013, 381014. [Google Scholar] [CrossRef] [PubMed]
- De Vito, R.; Alisi, A.; Masotti, A.; Ceccarelli, S.; Panera, N.; Citti, A.; Salata, M.; Valenti, L.; Feldstein, A.E.; Nobili, V. Markers of activated inflammatory cells correlate with severity of liver damage in children with nonalcoholic fatty liver disease. Int. J. Mol. Med. 2012, 30, 49–56. [Google Scholar] [PubMed]
- Ferreyra Solari, N.E.; Inzaugarat, M.E.; Baz, P.; de Matteo, E.; Lezama, C.; Galoppo, M.; Galoppo, C.; Chernavsky, A.C. The role of innate cells is coupled to a Th1-polarized immune response in pediatric nonalcoholic steatohepatitis. J. Clin. Immunol. 2012, 32, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Barshop, N.J.; Sirlin, C.B.; Schwimmer, J.B.; Lavine, J.E. Review article: Epidemiology, pathogenesis and potential treatments of paediatric non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2008, 28, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Carpino, G.; Alisi, A.; Franchitto, A.; Alpini, G.; de Vito, R.; Onori, P.; Alvaro, D.; Gaudio, E. Hepatic progenitor cells activation, fibrosis, and adipokines production in pediatric nonalcoholic fatty liver disease. Hepatology 2012, 56, 2142–2153. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.J.; Cha, B.S. Pathogenesis and therapeutic approaches for non-alcoholic fatty liver disease. World J. Hepatol. 2014, 6, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Swiderska-Syn, M.; Suzuki, A.; Guy, C.D.; Schwimmer, J.B.; Abdelmalek, M.F.; Lavine, J.E.; Diehl, A.M. Hedgehog pathway and pediatric nonalcoholic fatty liver disease. Hepatology 2013, 57, 1814–1825. [Google Scholar] [CrossRef] [PubMed]
- Mouralidarane, A.; Soeda, J.; Visconti-Pugmire, C.; Samuelsson, A.M.; Pombo, J.; Maragkoudaki, X.; Butt, A.; Saraswati, R.; Novelli, M.; Fusai, G.; et al. Maternal obesity programs offspring nonalcoholic fatty liver disease by innate immune dysfunction in mice. Hepatology 2013, 58, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Stienstra, R.; Saudale, F.; Duval, C.; Keshtkar, S.; Groener, J.E.; van Rooijen, N.; Staels, B.; Kersten, S.; Muller, M. Kupffer cells promote hepatic steatosis via interleukin-1β-dependent suppression of peroxisome proliferator-activated receptor α activity. Hepatology 2010, 51, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.L.; Sigala, B.; Soeda, J.; Cordero, P.; Nguyen, V.; McKee, C.; Mouraliderane, A.; Vinciguerra, M.; Oben, J.A. Acetylcholine induces fibrogenic effects via M2/M3 acetylcholine receptors in non-alcoholic steatohepatitis and in primary human hepatic stellate cells. J. Gastroenterol. Hepatol. 2016, 31, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Sigala, B.; McKee, C.; Soeda, J.; Pazienza, V.; Morgan, M.; Lin, C.I.; Selden, C.; Vander Borght, S.; Mazzoccoli, G.; Roskams, T.; et al. Sympathetic nervous system catecholamines and neuropeptide y neurotransmitters are upregulated in human nafld and modulate the fibrogenic function of hepatic stellate cells. PLoS ONE 2013, 8, e72928. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.R.; Song, J.Y.; Liu, F.H.; Ma, J.; Wang, H.J. Gwas-identified common variants with nonalcoholic fatty liver disease in Chinese children. J. Pediatr. Gastroenterol. Nutr. 2015, 60, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Celedon, M.A.; Lavine, J.E.; Salem, R.; Campbell, N.; Schork, N.J.; Shiehmorteza, M.; Yokoo, T.; Chavez, A.; Middleton, M.S.; et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology 2009, 136, 1585–1592. [Google Scholar] [CrossRef] [PubMed]
- Deboer, M.D.; Wiener, R.C.; Barnes, B.H.; Gurka, M.J. Ethnic differences in the link between insulin resistance and elevated ALT. Pediatrics 2013, 132, e718–e726. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Chang, P.F.; Chang, M.H.; Ni, Y.H. Genetic variants in GCKR and PNPLA3 confer susceptibility to nonalcoholic fatty liver disease in obese individuals. Am. J. Clin. Nutr. 2014, 99, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Hernaez, R.; McLean, J.; Lazo, M.; Brancati, F.L.; Hirschhorn, J.N.; Borecki, I.B.; Harris, T.B.; Genetics of Obesity-Related Liver Disease (GOLD) Consortium; Nguyen, T.; Kamel, I.R.; et al. Association between variants in or near PNPLA3, GCKR, and PPP1R3B with ultrasound-defined steatosis based on data from the third national health and nutrition examination survey. Clin. Gastroenterol. Hepatol. 2013, 11, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Santoro, N.; Kursawe, R.; D’Adamo, E.; Dykas, D.J.; Zhang, C.K.; Bale, A.E.; Cali, A.M.; Narayan, D.; Shaw, M.M.; Pierpont, B.; et al. A common variant in the patatin-like phospholipase 3 gene (PNPLA3) is associated with fatty liver disease in obese children and adolescents. Hepatology 2010, 52, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Mangge, H.; Baumgartner, B.G.; Zelzer, S.; Pruller, F.; Schnedl, W.J.; Reininghaus, E.Z.; Haybaeck, J.; Lackner, C.; Stauber, R.; Aigner, E.; et al. Patatin-like phospholipase 3 (rs738409) gene polymorphism is associated with increased liver enzymes in obese adolescents and metabolic syndrome in all ages. Aliment. Pharmacol. Ther. 2015, 42, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Shang, X.R.; Liu, F.H.; Song, J.Y.; Ma, J.; Wang, H.J. Association of INSIG2 rs9308762 with alt level independent of BMI. J. Pediatr. Gastroenterol. Nutr. 2014, 58, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Bellini, G.; Alisi, A.; Alterio, A.; Maione, S.; Perrone, L.; Locatelli, F.; Miraglia del Giudice, E.; Nobili, V. Cannabinoid receptor type 2 functional variant influences liver damage in children with non-alcoholic fatty liver disease. PLoS ONE 2012, 7, e42259. [Google Scholar] [CrossRef] [PubMed]
- Hyysalo, J.; Mannisto, V.T.; Zhou, Y.; Arola, J.; Karja, V.; Leivonen, M.; Juuti, A.; Jaser, N.; Lallukka, S.; Kakela, P.; et al. A population-based study on the prevalence of NASH using scores validated against liver histology. J. Hepatol. 2014, 60, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Qiao, A.; Liang, J.; Ke, Y.; Li, C.; Cui, Y.; Shen, L.; Zhang, H.; Cui, A.; Liu, X.; Liu, C.; et al. Mouse patatin-like phospholipase domain-containing 3 influences systemic lipid and glucose homeostasis. Hepatology 2011, 54, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Weltman, M.D.; Farrell, G.C.; Hall, P.; Ingelman-Sundberg, M.; Liddle, C. Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology 1998, 27, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Weltman, M.D.; Farrell, G.C.; Liddle, C. Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology 1996, 111, 1645–1653. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Wakai, K.; Nakagawa, H.; Suma, S.; Sasakabe, T.; Sakamoto, T.; Takashima, N.; Suzuki, S.; Ogawa, S.; Ohnaka, K.; et al. Associations between polymorphisms of interleukin-6 and related cytokine genes and serum liver damage markers: A cross-sectional study in the Japan Multi-Institutional Collaborative Cohort (J-MICC) study. Gene 2015, 557, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Chang, P.F.; Hu, F.C.; Chang, M.H.; Ni, Y.H. Variants in the UGT1A1 gene and the risk of pediatric nonalcoholic fatty liver disease. Pediatrics 2009, 124, e1221–e1227. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Bellini, G.; Nobili, B.; Maione, S.; Perrone, L.; del Giudice, E.M. Association of the cannabinoid receptor 2 (Cb2) Gln63Arg polymorphism with indices of liver damage in obese children: An alternative way to highlight the CB2 hepatoprotective properties. Hepatology 2011, 54, 1102. [Google Scholar] [CrossRef] [PubMed]
- Cordero, P.; Milagro, F.I.; Campion, J.; Martinez, J.A. Supplementation with methyl donors during lactation to high-fat-sucrose-fed dams protects offspring against liver fat accumulation when consuming an obesogenic diet. J. Dev. Orig. Health Dis. 2014, 5, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Cordero, P.; Milagro, F.I.; Campion, J.; Martinez, J.A. Maternal methyl donors supplementation during lactation prevents the hyperhomocysteinemia induced by a high-fat-sucrose intake by dams. Int. J. Mol. Sci. 2013, 14, 24422–24437. [Google Scholar] [CrossRef] [PubMed]
- Mouralidarane, A.; Soeda, J.; Sugden, D.; Bocianowska, A.; Carter, R.; Ray, S.; Saraswati, R.; Cordero, P.; Novelli, M.; Fusai, G.; et al. Maternal obesity programs offspring non-alcoholic fatty liver disease through disruption of 24-h rhythms in mice. Int. J. Obes. 2015, 39, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Cordero, P.; Li, J.; Oben, J.A. Epigenetics of obesity: Beyond the genome sequence. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Cordero, P.; Li, J.; Temple, J.L.; Nguyen, V.; Oben, J.A. Epigenetic mechanisms of maternal obesity effects on the descendants. In Parental Obesity: Intergenerational Programming and Consequences; Green, L.R., Hester, R.L., Eds.; Springer-Verlag New York: New York, NY, USA, 2016. [Google Scholar]
- Alisi, A.; Panera, N.; Agostoni, C.; Nobili, V. Intrauterine growth retardation and nonalcoholic fatty liver disease in children. Int. J. Endocrinol. 2011, 2011, 269853. [Google Scholar] [CrossRef] [PubMed]
- Breij, L.M.; Kerkhof, G.F.; Hokken-Koelega, A.C. Accelerated infant weight gain and risk for nonalcoholic fatty liver disease in early adulthood. J. Clin. Endocrinol. Metab. 2014, 99, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Amarapurkar, D.; Kamani, P.; Patel, N.; Gupte, P.; Kumar, P.; Agal, S.; Baijal, R.; Lala, S.; Chaudhary, D.; Deshpande, A. Prevalence of non-alcoholic fatty liver disease: Population based study. Ann. Hepatol. 2007, 6, 161–163. [Google Scholar] [PubMed]
- Hanada, S.; Snider, N.T.; Brunt, E.M.; Hollenberg, P.F.; Omary, M.B. Gender dimorphic formation of mouse mallory-denk bodies and the role of xenobiotic metabolism and oxidative stress. Gastroenterology 2010, 138, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.V.; Duvigneau, J.C.; Hyatt, T.C.; Raju, R.; Behling, T.; Hartl, R.T.; Staniek, K.; Miller, I.; Gregor, W.; Redl, H.; et al. Effect of estrogen on mitochondrial function and intracellular stress markers in rat liver and kidney following trauma-hemorrhagic shock and prolonged hypotension. Mol. Med. 2010, 16, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Shimizu, I.; Shiba, M.; Ito, S. Suppressive effects of estradiol on dimethylnitrosamine-induced fibrosis of the liver in rats. Hepatology 1999, 29, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.B.; Pagadala, M.R.; Dasarathy, J.; Unalp-Arida, A.; Pai, R.K.; Yerian, L.; Khiyami, A.; Sourianarayanane, A.; Sargent, R.; Hawkins, C.; et al. Age impacts ability of aspartate-alanine aminotransferase ratio to predict advanced fibrosis in nonalcoholic fatty liver disease. Dig. Dis. Sci. 2015, 60, 1825–1831. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Abdelmalek, M.F.; Schwimmer, J.B.; Lavine, J.E.; Scheimann, A.O.; Unalp-Arida, A.; Yates, K.P.; Sanyal, A.J.; Guy, C.D.; Diehl, A.M.; et al. Association between puberty and features of nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2012, 10, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, F.; Guy, C.D.; Lu, J.; Suzuki, A.; Burchette, J.L.; Abdelmalek, M.F.; Chen, W.; Diehl, A.M. Increased production of sonic hedgehog by ballooned hepatocytes. J. Pathol. 2011, 224, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Leng, X.S.; Zhu, J.Y.; Wang, G. Suppression of hedgehog signaling regulates hepatic stellate cell activation and collagen secretion. Int. J. Clin. Exp. Pathol. 2015, 8, 14574–14579. [Google Scholar] [PubMed]
- Hernaez, R.; Yeung, E.; Clark, J.M.; Kowdley, K.V.; Brancati, F.L.; Kao, W.H. Hemochromatosis gene and nonalcoholic fatty liver disease: A systematic review and meta-analysis. J. Hepatol. 2011, 55, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Tu, R.; Liu, G.; Huang, L.; Guan, Y.; Zheng, E. Focal fatty sparing usually does not arise in preexisting nonalcoholic diffuse homogeneous fatty liver. J. Ultrasound Med. 2014, 33, 1447–1452. [Google Scholar] [CrossRef] [PubMed]
- Bellentani, S.; Saccoccio, G.; Masutti, F.; Croce, L.S.; Brandi, G.; Sasso, F.; Cristanini, G.; Tiribelli, C. Prevalence of and risk factors for hepatic steatosis in Northern Italy. Ann. Intern. Med. 2000, 132, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Patton, H.M.; Lavine, J.E.; van Natta, M.L.; Schwimmer, J.B.; Kleiner, D.; Molleston, J.; Nonalcoholic Steatohepatitis Clinical Research Network. Clinical correlates of histopathology in pediatric nonalcoholic steatohepatitis. Gastroenterology 2008, 135, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Fracanzani, A.L.; Valenti, L.; Bugianesi, E.; Andreoletti, M.; Colli, A.; Vanni, E.; Bertelli, C.; Fatta, E.; Bignamini, D.; Marchesini, G.; et al. Risk of severe liver disease in nonalcoholic fatty liver disease with normal aminotransferase levels: A role for insulin resistance and diabetes. Hepatology 2008, 48, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Mofrad, P.; Contos, M.J.; Haque, M.; Sargeant, C.; Fisher, R.A.; Luketic, V.A.; Sterling, R.K.; Shiffman, M.L.; Stravitz, R.T.; Sanyal, A.J. Clinical and histologic spectrum of nonalcoholic fatty liver disease associated with normal ALT values. Hepatology 2003, 37, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Wilson, H.K.; Monster, A.C. New technologies in the use of exhaled breath analysis for biological monitoring. Occup. Environ. Med. 1999, 56, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Alkhouri, N.; de Vito, R.; Alisi, A.; Lopez, R.; Nobili, V. Serum cytokeratin-18 fragment levels are useful biomarkers for nonalcoholic steatohepatitis in children. Am. J. Gastroenterol. 2013, 108, 1526–1531. [Google Scholar] [CrossRef] [PubMed]
- Wieckowska, A.; Zein, N.N.; Yerian, L.M.; Lopez, A.R.; McCullough, A.J.; Feldstein, A.E. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology 2006, 44, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. Meta-analysis: Natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann. Med. 2011, 43, 617–649. [Google Scholar] [CrossRef] [PubMed]
- Puri, K.; Nobili, V.; Melville, K.; Corte, C.D.; Sartorelli, M.R.; Lopez, R.; Feldstein, A.E.; Alkhouri, N. Serum bilirubin level is inversely associated with nonalcoholic steatohepatitis in children. J. Pediatr. Gastroenterol. Nutr. 2013, 57, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Eng, K.; Lopez, R.; Liccardo, D.; Nobili, V.; Alkhouri, N. A non-invasive prediction model for non-alcoholic steatohepatitis in paediatric patients with non-alcoholic fatty liver disease. Dig. Liver Dis. 2014, 46, 1008–1013. [Google Scholar] [CrossRef] [PubMed]
- Oresic, M.; Hyotylainen, T.; Kotronen, A.; Gopalacharyulu, P.; Nygren, H.; Arola, J.; Castillo, S.; Mattila, I.; Hakkarainen, A.; Borra, R.J.; et al. Prediction of non-alcoholic fatty-liver disease and liver fat content by serum molecular lipids. Diabetologia 2013, 56, 2266–2274. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Y.; Ergelen, R.; Akin, H.; Imeryuz, N. Noninvasive detection of hepatic steatosis in patients without ultrasonographic evidence of fatty liver using the controlled attenuation parameter evaluated with transient elastography. Eur. J. Gastroenterol. Hepatol. 2013, 25, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Middleton, M.S.; Behling, C.; Newton, K.P.; Awai, H.I.; Paiz, M.N.; Lam, J.; Hooker, J.C.; Hamilton, G.; Fontanesi, J.; et al. Magnetic resonance imaging and liver histology as biomarkers of hepatic steatosis in children with nonalcoholic fatty liver disease. Hepatology 2015, 61, 1887–1895. [Google Scholar] [CrossRef] [PubMed]
- Tovo, C.V.; de Mattos, A.Z.; Coral, G.P.; Branco, F.S.; Suwa, E.; de Mattos, A.A. Noninvasive imaging assessment of non-alcoholic fatty liver disease: Focus on liver scintigraphy. World J. Gastroenterol. 2015, 21, 4432–4439. [Google Scholar] [PubMed]
- Kim, K.M.; Choi, W.B.; Park, S.H.; Yu, E.; Lee, S.G.; Lim, Y.S.; Lee, H.C.; Chung, Y.H.; Lee, Y.S.; Suh, D.J. Diagnosis of hepatic steatosis and fibrosis by transient elastography in asymptomatic healthy individuals: A prospective study of living related potential liver donors. J. Gastroenterol. 2007, 42, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Vizzutti, F.; Arena, U.; Abraldes, J.G.; Marra, F.; Pietrobattista, A.; Fruhwirth, R.; Marcellini, M.; Pinzani, M. Accuracy and reproducibility of transient elastography for the diagnosis of fibrosis in pediatric nonalcoholic steatohepatitis. Hepatology 2008, 48, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Ovchinsky, N.; Moreira, R.K.; Lefkowitch, J.H.; Lavine, J.E. Liver biopsy in modern clinical practice: A pediatric point-of-view. Adv. Anat. Pathol. 2012, 19, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Behling, C.; Newbury, R.; Deutsch, R.; Nievergelt, C.; Schork, N.J.; Lavine, J.E. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology 2005, 42, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Mansoor, S.; Collyer, E.; Alkhouri, N. A comprehensive review of noninvasive liver fibrosis tests in pediatric nonalcoholic fatty liver disease. Curr. Gastroenterol. Rep. 2015, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Parkes, J.; Bottazzo, G.; Marcellini, M.; Cross, R.; Newman, D.; Vizzutti, F.; Pinzani, M.; Rosenberg, W.M. Performance of ELF serum markers in predicting fibrosis stage in pediatric non-alcoholic fatty liver disease. Gastroenterology 2009, 136, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. A meta-analysis of randomized trials for the treatment of nonalcoholic fatty liver disease. Hepatology 2010, 52, 79–104. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Marcellini, M.; Devito, R.; Ciampalini, P.; Piemonte, F.; Comparcola, D.; Sartorelli, M.R.; Angulo, P. Nafld in children: A prospective clinical-pathological study and effect of lifestyle advice. Hepatology 2006, 44, 458–465. [Google Scholar] [CrossRef] [PubMed]
- DeVore, S.; Kohli, R.; Lake, K.; Nicholas, L.; Dietrich, K.; Balistreri, W.F.; Xanthakos, S.A. A multidisciplinary clinical program is effective in stabilizing BMI and reducing transaminase levels in pediatric patients with NAFLD. J. Pediatr. Gastroenterol. Nutr. 2013, 57, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Manco, M.; Devito, R.; Di Ciommo, V.; Comparcola, D.; Sartorelli, M.R.; Piemonte, F.; Marcellini, M.; Angulo, P. Lifestyle intervention and antioxidant therapy in children with nonalcoholic fatty liver disease: A randomized, controlled trial. Hepatology 2008, 48, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Oddy, W.H.; Herbison, C.E.; Jacoby, P.; Ambrosini, G.L.; O’Sullivan, T.A.; Ayonrinde, O.T.; Olynyk, J.K.; Black, L.J.; Beilin, L.J.; Mori, T.A.; et al. The western dietary pattern is prospectively associated with nonalcoholic fatty liver disease in adolescence. Am. J. Gastroenterol. 2013, 108, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Manco, M.; Devito, R.; Ciampalini, P.; Piemonte, F.; Marcellini, M. Effect of Vitamin E on aminotransferase levels and insulin resistance in children with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2006, 24, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Gronbaek, H.; Lange, A.; Birkebaek, N.H.; Holland-Fischer, P.; Solvig, J.; Horlyck, A.; Kristensen, K.; Rittig, S.; Vilstrup, H. Effect of a 10-week weight loss camp on fatty liver disease and insulin sensitivity in obese Danish children. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Zelber-Sagi, S.; Nitzan-Kaluski, D.; Goldsmith, R.; Webb, M.; Zvibel, I.; Goldiner, I.; Blendis, L.; Halpern, Z.; Oren, R. Role of leisure-time physical activity in nonalcoholic fatty liver disease: A population-based study. Hepatology 2008, 48, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Barlow, S.E.; Expert, C. Expert committee recommendations regarding the prevention, assessment, and treatment of child and adolescent overweight and obesity: Summary report. Pediatrics 2007, 120, S164–S192. [Google Scholar] [CrossRef] [PubMed]
- Haukeland, J.W.; Konopski, Z.; Eggesbo, H.B.; von Volkmann, H.L.; Raschpichler, G.; Bjoro, K.; Haaland, T.; Loberg, E.M.; Birkeland, K. Metformin in patients with non-alcoholic fatty liver disease: A randomized, controlled trial. Scand. J. Gastroenterol. 2009, 44, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Krasnoff, J.B.; Painter, P.L.; Wallace, J.P.; Bass, N.M.; Merriman, R.B. Health-related fitness and physical activity in patients with nonalcoholic fatty liver disease. Hepatology 2008, 47, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Kistler, K.D.; Brunt, E.M.; Clark, J.M.; Diehl, A.M.; Sallis, J.F.; Schwimmer, J.B.; Group, N.C.R. Physical activity recommendations, exercise intensity, and histological severity of nonalcoholic fatty liver disease. Am. J. Gastroenterol. 2011, 106, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Niblett, P. Statistics on Obesity, Physical Activity and Diet—England, 2015. Available online: http://www.hscic.gov.uk/catalogue/PUB16988 (accessed on 14 June 2016).
- Jin, R.; Le, N.A.; Liu, S.; Farkas Epperson, M.; Ziegler, T.R.; Welsh, J.A.; Jones, D.P.; McClain, C.J.; Vos, M.B. Children with nafld are more sensitive to the adverse metabolic effects of fructose beverages than children without NAFLD. J. Clin. Endocrinol. Metab. 2012, 97, E1088–E1098. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, T.A.; Oddy, W.H.; Bremner, A.P.; Sherriff, J.L.; Ayonrinde, O.T.; Olynyk, J.K.; Beilin, L.J.; Mori, T.A.; Adams, L.A. Lower fructose intake may help protect against development of nonalcoholic fatty liver in adolescents with obesity. J. Pediatr. Gastroenterol. Nutr. 2014, 58, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Spruss, A.; Bergheim, I. Dietary fructose and intestinal barrier: Potential risk factor in the pathogenesis of nonalcoholic fatty liver disease. J. Nutr. Biochem. 2009, 20, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Igarashi, K.; Koeda, T.; Sugimoto, K.; Nakagawa, K.; Hayashi, S.; Yamaji, R.; Inui, H.; Fukusato, T.; Yamanouchi, T. Rats fed fructose-enriched diets have characteristics of nonalcoholic hepatic steatosis. J. Nutr. 2009, 139, 2067–2071. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Khandelwal, B. Fast foods and physical inactivity are risk factors for obesity and hypertension among adolescent school children in east district of sikkim, india. J. Nat. Sci. Biol. Med. 2015, 6, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Black, L.J.; Jacoby, P.; Ping-Delfos, S.; Chan, W.; Mori, T.A.; Beilin, L.J.; Olynyk, J.K.; Ayonrinde, O.T.; Huang, R.C.; Holt, P.G.; et al. Low serum 25-hydroxyvitamin D concentrations associate with non-alcoholic fatty liver disease in adolescents independent of adiposity. J. Gastroenterol. Hepatol. 2014, 29, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Giorgio, V.; Liccardo, D.; Bedogni, G.; Morino, G.; Alisi, A.; Cianfarani, S. Vitamin D levels and liver histological alterations in children with nonalcoholic fatty liver disease. Eur. J. Endocrinol. 2014, 170, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cordero, P.; Nguyen, V.; Oben, J.A. The role of vitamins in the pathogenesis of non-alcoholic fatty liver disease. Integr. Med. Insights 2016, 11, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.L.; Elfers, C.T.; Figlewicz, D.P.; Melhorn, S.J.; Morton, G.J.; Hoofnagle, A.; Yeh, M.M.; Nelson, J.E.; Kowdley, K.V. Vitamin D deficiency in obese rats exacerbates nonalcoholic fatty liver disease and increases hepatic resistin and toll-like receptor activation. Hepatology 2012, 55, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- George, P.S.; Pearson, E.R.; Witham, M.D. Effect of Vitamin D supplementation on glycaemic control and insulin resistance: A systematic review and meta-analysis. Diabet. Med. 2012, 29, e142–e150. [Google Scholar] [CrossRef] [PubMed]
- Flachs, P.; Rossmeisl, M.; Bryhn, M.; Kopecky, J. Cellular and molecular effects of n-3 polyunsaturated fatty acids on adipose tissue biology and metabolism. Clin. Sci. 2009, 116, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Masterton, G.S.; Plevris, J.N.; Hayes, P.C. Review article: ω-3 fatty acids—A promising novel therapy for non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2010, 31, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Janczyk, W.; Socha, P.; Lebensztejn, D.; Wierzbicka, A.; Mazur, A.; Neuhoff-Murawska, J.; Matusik, P. ω-3 fatty acids for treatment of non-alcoholic fatty liver disease: Design and rationale of randomized controlled trial. BMC Pediatr. 2013, 13, 85. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Carpino, G.; Alisi, A.; de Vito, R.; Franchitto, A.; Alpini, G.; Onori, P.; Gaudio, E. Role of docosahexaenoic acid treatment in improving liver histology in pediatric nonalcoholic fatty liver disease. PLoS ONE 2014, 9, e88005. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Bedogni, G.; Alisi, A.; Pietrobattista, A.; Rise, P.; Galli, C.; Agostoni, C. Docosahexaenoic acid supplementation decreases liver fat content in children with non-alcoholic fatty liver disease: Double-blind randomised controlled clinical trial. Arch. Dis. Child. 2011, 96, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Alkhouri, N.; Alisi, A.; Della Corte, C.; Fitzpatrick, E.; Raponi, M.; Dhawan, A. Nonalcoholic fatty liver disease: A challenge for pediatricians. JAMA Pediatr. 2015, 169, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Loy, J.J.; Youn, H.A.; Schwack, B.; Kurian, M.; Ren Fielding, C.; Fielding, G.A. Improvement in nonalcoholic fatty liver disease and metabolic syndrome in adolescents undergoing bariatric surgery. Surg. Obes. Relat. Dis. 2015, 11, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Peterli, R.; Steinert, R.E.; Woelnerhanssen, B.; Peters, T.; Christoffel-Courtin, C.; Gass, M.; Kern, B.; von Fluee, M.; Beglinger, C. Metabolic and hormonal changes after laparoscopic Roux-en-Y gastric bypass and sleeve gastrectomy: A randomized, prospective trial. Obes. Surg. 2012, 22, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Nitta, H.; Otsuka, K.; Umemura, A.; Baba, S.; Obuchi, T.; Wakabayashi, G. Bariatric surgery and non-alcoholic fatty liver disease: Current and potential future treatments. Front. Endocrinol. 2014, 5, 164. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Tapia, N.C.; Tellez-Avila, F.I.; Barrientos-Gutierrez, T.; Mendez-Sanchez, N.; Lizardi-Cervera, J.; Uribe, M. Bariatric surgery for non-alcoholic steatohepatitis in obese patients. Cochrane Database Syst. Rev. 2010, 1. [Google Scholar] [CrossRef]
- Lassailly, G.; Caiazzo, R.; Buob, D.; Pigeyre, M.; Verkindt, H.; Labreuche, J.; Raverdy, V.; Leteurtre, E.; Dharancy, S.; Louvet, A.; et al. Bariatric surgery reduces features of nonalcoholic steatohepatitis in morbidly obese patients. Gastroenterology 2015, 149, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Nanda, K. Non-alcoholic steatohepatitis in children. Pediatr. Transpl. 2004, 8, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Middleton, M.S.; Deutsch, R.; Lavine, J.E. A phase 2 clinical trial of metformin as a treatment for non-diabetic paediatric non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2005, 21, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Lavine, J.E.; Schwimmer, J.B.; van Natta, M.L.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Abrams, S.H.; Scheimann, A.O.; Sanyal, A.J.; Chalasani, N.; et al. Effect of Vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: The tonic randomized controlled trial. JAMA 2011, 305, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldelli, A.; Tio, F.; Pulcini, J.; et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, Vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hong, S.W.; Rhee, E.J.; Lee, W.Y. GLP-1 receptor agonist and non-alcoholic fatty liver disease. Diabetes Metab. J. 2012, 36, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Duvnjak, M.; Tomasic, V.; Gomercic, M.; Smircic Duvnjak, L.; Barsic, N.; Lerotic, I. Therapy of nonalcoholic fatty liver disease: Current status. J. Physiol. Pharmacol. 2009, 60, 57–66. [Google Scholar] [PubMed]
- Tilg, H.; Moschen, A. Update on nonalcoholic fatty liver disease: Genes involved in nonalcoholic fatty liver disease and associated inflammation. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, E.F. Angiotensin receptor blockers in the treatment of NASH/NAFLD: Could they be a first-class option? Adv. Ther. 2008, 25, 1141–1174. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.; Mouralidarane, A.; Ray, S.; Soeda, J.; Oben, J. Recent advancements in drug treatment of obesity. Clin. Med. 2012, 12, 456–460. [Google Scholar] [CrossRef]
- Harrison, S.A.; Fecht, W.; Brunt, E.M.; Neuschwander-Tetri, B.A. Orlistat for overweight subjects with nonalcoholic steatohepatitis: A randomized, prospective trial. Hepatology 2009, 49, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicki, A.S.; Oben, J. Nonalcoholic fatty liver disease and lipids. Curr. Opin. Lipidol. 2012, 23, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Tziomalos, K.; Athyros, V.G.; Paschos, P.; Karagiannis, A. Nonalcoholic fatty liver disease and statins. Metabolism 2015, 64, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.; Baker, S.S.; Liu, W.; Moya, D.A.; Browne, R.W.; Mastrandrea, L.; Baker, R.D.; Zhu, L. Paraoxonase 1 and oxidative stress in paediatric non-alcoholic steatohepatitis. Liver Int. 2014, 34, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Berk, M.; McIntyre, T.M.; Gores, G.J.; Feldstein, A.E. The lysosomal-mitochondrial axis in free fatty acid-induced hepatic lipotoxicity. Hepatology 2008, 47, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhong, R.; Liu, Y.; Jiang, X.; Tang, X.; Li, Z.; Xia, M.; Ling, W. Effects of bayberry juice on inflammatory and apoptotic markers in young adults with features of non-alcoholic fatty liver disease. Nutrition 2014, 30, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.F.; Sun, Y.; Shen, L. Effect of Vitamin E supplementation on aminotransferase levels in patients with nafld, nash, and chc: Results from a meta-analysis. Nutrition 2014, 30, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, C. Antioxidant supplements and mortality. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V. Treatment of NASH with ursodeoxycholic acid: Pro. Clin. Res. Hepatol. Gastroenterol. 2012, 36, S41–S45. [Google Scholar] [CrossRef]
- Xiang, Z.; Chen, Y.P.; Ma, K.F.; Ye, Y.F.; Zheng, L.; Yang, Y.D.; Li, Y.M.; Jin, X. The role of ursodeoxycholic acid in non-alcoholic steatohepatitis: A systematic review. BMC Gastroenterol. 2013, 13, 140. [Google Scholar] [CrossRef] [PubMed]
- Pietu, F.; Guillaud, O.; Walter, T.; Vallin, M.; Hervieu, V.; Scoazec, J.Y.; Dumortier, J. Ursodeoxycholic acid with Vitamin E in patients with nonalcoholic steatohepatitis: Long-term results. Clin. Res. Hepatol. Gastroenterol. 2012, 36, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Vajro, P.; Franzese, A.; Valerio, G.; Iannucci, M.P.; Aragione, N. Lack of efficacy of ursodeoxycholic acid for the treatment of liver abnormalities in obese children. J. Pediatr. 2000, 136, 739–743. [Google Scholar] [CrossRef]
- Van de Meeberg, P.C.; Houwen, R.H.; Sinaasappel, M.; Heijerman, H.G.; Bijleveld, C.M.; Vanberge-Henegouwen, G.P. Low-dose versus high-dose ursodeoxycholic acid in cystic fibrosis-related cholestatic liver disease. Results of a randomized study with 1-year follow-up. Scand. J. Gastroenterol. 1997, 32, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.F.; Oneta, C.M.; Gonvers, J.J.; Bihl, F.; Cerny, A.; Cereda, J.M.; Zala, J.F.; Helbling, B.; Steuerwald, M.; Zimmermann, A.; et al. Randomized placebo-controlled trial of ursodeoxycholic acid with Vitamin E in nonalcoholic steatohepatitis. Clin. Gastroenterol. Hepatol. 2006, 4, 1537–1543. [Google Scholar] [CrossRef] [PubMed]
- Loguercio, C.; Federico, A.; Tuccillo, C.; Terracciano, F.; D’Auria, M.V.; de Simone, C.; del Vecchio Blanco, C. Beneficial effects of a probiotic VSL#3 on parameters of liver dysfunction in chronic liver diseases. J. Clin. Gastroenterol. 2005, 39, 540–543. [Google Scholar] [PubMed]
- Vajro, P.; Mandato, C.; Licenziati, M.R.; Franzese, A.; Vitale, D.F.; Lenta, S.; Caropreso, M.; Vallone, G.; Meli, R. Effects of lactobacillus rhamnosus strain GG in pediatric obesity-related liver disease. J. Pediatr. Gastroenterol. Nutr. 2011, 52, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Cortez-Pinto, H. Diet, microbiota, obesity, and NAFLD: A dangerous quartet. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, M. Non-alcoholic fatty liver disease: The bile acid-activated farnesoid x receptor as an emerging treatment target. J. Lipids 2012, 2012, 934396. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Temple, J.L.; Cordero, P.; Li, J.; Nguyen, V.; Oben, J.A. A Guide to Non-Alcoholic Fatty Liver Disease in Childhood and Adolescence. Int. J. Mol. Sci. 2016, 17, 947. https://doi.org/10.3390/ijms17060947
Temple JL, Cordero P, Li J, Nguyen V, Oben JA. A Guide to Non-Alcoholic Fatty Liver Disease in Childhood and Adolescence. International Journal of Molecular Sciences. 2016; 17(6):947. https://doi.org/10.3390/ijms17060947
Chicago/Turabian StyleTemple, Jonathan L., Paul Cordero, Jiawei Li, Vi Nguyen, and Jude A. Oben. 2016. "A Guide to Non-Alcoholic Fatty Liver Disease in Childhood and Adolescence" International Journal of Molecular Sciences 17, no. 6: 947. https://doi.org/10.3390/ijms17060947
APA StyleTemple, J. L., Cordero, P., Li, J., Nguyen, V., & Oben, J. A. (2016). A Guide to Non-Alcoholic Fatty Liver Disease in Childhood and Adolescence. International Journal of Molecular Sciences, 17(6), 947. https://doi.org/10.3390/ijms17060947