
Neuroprotective and Therapeutic Strategies against Parkinson’s Disease: Recent Perspectives


Abstract
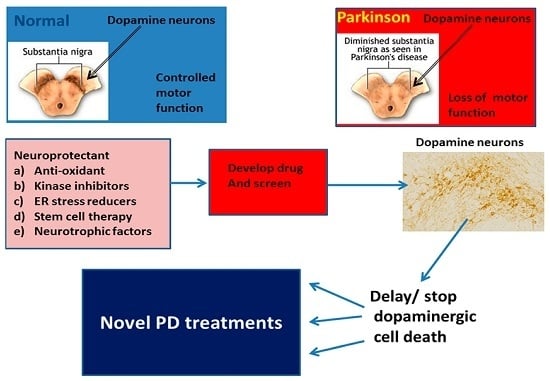
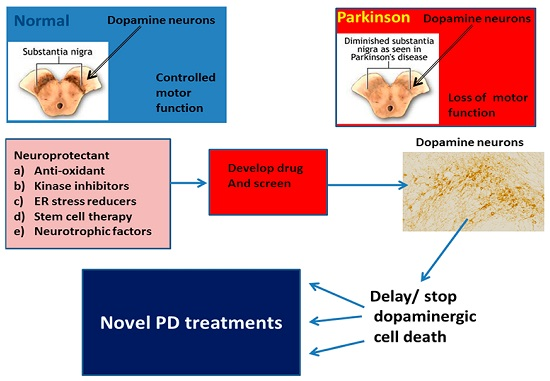
:
1. Introduction
2. Neuropathology of PD
3. Striatal Pathophysiology of PD
4. Mechanisms of PD Pathogenesis and Possible Targets for Neuroprotection
4.1. Oxidative Stress and Mitochondrial Dysfunction
4.2. Protein Aggregation and Misfolding
4.3. Neuroinflammation
4.4. Excitotoxicity
4.5. Apoptosis
4.6. Loss of Trophic Factors
5. Neuroprotection in Clinical Trials: Recent Updates
5.1. Levo-Dopa as a Neuroprotective Agent
5.2. Neuroprotection by Using Dopamine Receptor Agonists
5.3. Therapeutic Potential of Antioxidants
5.4. Apoptotic Inhibitory Factors
5.5. Trophic Factors
6. Drawbacks of Neuroprotective Therapy
6.1. Limitations of Testing Methods
6.2. Shortcomings of Animal Models
6.3. Diversity of the Disease
7. The Future of Neuroprotection
7.1. Adenosine Receptor Antagonists
7.2. Anti-Inflammatory Agents
7.3. Other Antioxidants
7.4. Strategies to Use α-Synuclein as Neuroprotectant
7.5. Kinase Inhibitors
7.6. Trophic Factors
8. Possible Therapeutics for Nonmotor Symptoms of PD
8.1. Surgical Therapies for PD
8.2. Are There Any Non-Motor Side Effects of STN and GPi-DBS?
8.3. Is STN a Better Target than GPi for DBS to Treat PD?
8.4. Possible Neural Transplantation for PD
8.5. Gene Therapy for PD
9. Future Directions: Search for Biomarkers and Neuroprotective Therapies
9.1. Can Olfactory Dysfunction Used as an Early Biomarker?
9.2. Can Neuroimaging Possibly Be Used an Early Biomarker?
9.3. Can Proteomics and “Omics” Approach Used as Biomarker?
9.4. Stem Cell Approach to Treat Parkinson’s Disease
10. Conclusions
Acknowledgments
Conflicts of Interest
References
- Prkinson, J. An Essay On The Shaking Palsy; Whiitngham and Rowland: London, UK, 1817. [Google Scholar]
- Carlsson, A.; Lindqvist, M.; Magnusson, T. 3,4-Dihydroxyphenylalanine and 5-Hydroxytryptophan as Reserpine Antagonists. Nature 1957, 180, 1200. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, A.; Waldeck, B. A Fluorimetric Method for the Determination of Dopamine (3-Hydroxytyramine). Acta Physiol. Scand. 1958, 44, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Barbeau, A. Treatment of Parkinson’s Disease by l-Dopa. Union Med. Can. 1969, 98, 183–186. [Google Scholar] [PubMed]
- Birkmayer, W.; Hornykiewicz, O. The l-3,4-Dioxyphenylalanine (Dopa)-Effect in Parkinson-Akinesia. Wien. Klin. Wochenschr. 1961, 73, 787–788. [Google Scholar] [PubMed]
- Godwin-Austen, R.B.; Smith, N.J. Comparison of the Effects of Bromocriptine and Levodopa in Parkinson’s Disease. J. Neurol. Neurosurg. Psychiatry 1977, 40, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Cotzias, G.C.; Papavasiliou, P.S.; Gellene, R. Modification of Parkinsonism—Chronic Treatment with l-Dopa. N. Engl. J. Med. 1969, 280, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Simuni, T.; Jaggi, J.L.; Mulholland, H.; Hurtig, H.I.; Colcher, A.; Siderowf, A.D.; Ravina, B.; Skolnick, B.E.; Goldstein, R.; Stern, M.B.; et al. Bilateral Stimulation of the Subthalamic Nucleus in Patients with Parkinson Disease: A Study of Efficacy and Safety. J. Neurosurg. 2002, 96, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Calne, D.B.; Teychenne, P.F.; Claveria, L.E.; Eastman, R.; Greenacre, J.K.; Petrie, A. Bromocriptine in Parkinsonism. Br. Med. J. 1974, 4, 442–444. [Google Scholar] [CrossRef] [PubMed]
- Calne, D.B.; Teychenne, P.F.; Leigh, P.N.; Bamji, A.N.; Greenacre, J.K. Treatment of Parkinsonism with Bromocriptine. Lancet 1974, 2, 1355–1356. [Google Scholar] [CrossRef]
- Corrodi, H.; Fuxe, K.; Hokfelt, T.; Lidbrink, P.; Ungerstedt, U. Effect of Ergot Drugs on Central Catecholamine Neurons: Evidence for a Stimulation of Central Dopamine Neurons. J. Pharm. Pharmacol. 1973, 25, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S.; Parkinson Study Group. Does Levodopa Slow or Hasten the Rate of Progression of Parkinson’s Disease? J. Neurol. 2005, 252 (Suppl. S4), Iv37–Iv42. [Google Scholar] [CrossRef] [PubMed]
- Rascol, O.; Brooks, D.J.; Korczyn, A.D.; de Deyn, P.P.; Clarke, C.E.; Lang, A.E. A Five-Year Study of the Incidence of Dyskinesia in Patients with Early Parkinson’s Disease Who Were Treated with Ropinirole or Levodopa. N. Engl. J. Med. 2000, 342, 1484–1491. [Google Scholar] [CrossRef] [PubMed]
- Korczyn, A.D. Dopaminergic Drugs in Development for Parkinson’s Disease. Adv. Neurol. 2003, 91, 267–271. [Google Scholar] [PubMed]
- Albin, R.L.; Young, A.B.; Penney, J.B. The Functional Anatomy of Basal Ganglia Disorders. Trends Neurosci. 1989, 12, 366–375. [Google Scholar] [CrossRef]
- Crossman, A.R. Neural Mechanisms in Disorders of Movement. Comp. Biochem. Physiol. Comp. Physiol. 1989, 93, 141–149. [Google Scholar] [CrossRef]
- Delong, M.R. Primate Models of Movement Disorders of Basal Ganglia Origin. Trends Neurosci. 1990, 13, 281–285. [Google Scholar] [CrossRef]
- Benabid, A.L. Targeting the Caudal Intralaminar Nuclei for Functional Neurosurgery of Movement Disorders. Brain Res. Bull. 2009, 78, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Benabid, A.L.; Chabardes, S.; Mitrofanis, J.; Pollak, P. Deep Brain Stimulation of the Subthalamic Nucleus for the Treatment of Parkinson’s Disease. Lancet Neurol. 2009, 8, 67–81. [Google Scholar] [CrossRef]
- Brundin, P.; Barker, R.A.; Parmar, M. Neural Grafting in Parkinson’s Disease Problems and Possibilities. Prog. Brain Res. 2010, 184, 265–294. [Google Scholar] [PubMed]
- Freed, C.R.; Greene, P.E.; Breeze, R.E.; Tsai, W.Y.; Dumouchel, W.; Kao, R.; Dillon, S.; Winfield, H.; Culver, S.; Trojanowski, J.Q.; et al. Transplantation of Embryonic Dopamine Neurons for Severe Parkinson’s Disease. N. Engl. J. Med. 2001, 344, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Goetz, C.G.; Kordower, J.H.; Stoessl, A.J.; Sossi, V.; Brin, M.F.; Shannon, K.M.; Nauert, G.M.; Perl, D.P.; Godbold, J.; et al. A Double-Blind Controlled Trial of Bilateral Fetal Nigral Transplantation in Parkinson’s Disease. Ann. Neurol. 2003, 54, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Christophersen, N.S.; Hall, V.; Soulet, D.; Brundin, P. Critical Issues of Clinical Human Embryonic Stem Cell Therapy for Brain Repair. Trends Neurosci. 2008, 31, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, K.R.; Healy, D.G.; Schapira, A.H.V. Non-Motor Symptoms of Parkinson’s Disease: Diagnosis and Management. Lancet Neurol. 2006, 5, 235–245. [Google Scholar] [CrossRef]
- Chaudhuri, K.R.; Odin, P.; Antonini, A.; Martinez-Martin, P. Parkinson’s Disease: The Non-Motor Issues. Parkinsonism Relat. Disord. 2011, 17, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, K.R.; Schapira, A.H. Non-Motor Symptoms of Parkinson’s Disease: Dopaminergic Pathophysiology and Treatment. Lancet Neurol. 2009, 8, 464–474. [Google Scholar] [CrossRef]
- Kasten, M.; Kertelge, L.; Bruggemann, N.; van der Vegt, J.; Schmidt, A.; Tadic, V.; Buhmann, C.; Steinlechner, S.; Behrens, M.I.; Ramirez, A.; et al. Nonmotor Symptoms in Genetic Parkinson Disease. Arch. Neurol. 2010, 67, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Lang, A.E. The Nonmotor Symptoms of Parkinson’s Disease—An Overview. Mov. Disord. 2010, 25 (Suppl. S1), S123–S130. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W. Parkinson Disease: Treatment of the Nonmotor Symptoms of Parkinson Disease. Nat. Rev. Neurol. 2010, 6, 417–418. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D. Clinical Review and Treatment of Select Adverse Effects of Dopamine Receptor Agonists in Parkinson’s Disease. Drugs Aging 2010, 27, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Etiology and Pathogenesis of Parkinson Disease. Neurol. Clin. 2009, 27, 583–603. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Molecular and Clinical Pathways to Neuroprotection of Dopaminergic Drugs in Parkinson Disease. Neurology 2009, 72, S44–S50. [Google Scholar] [CrossRef] [PubMed]
- Siderowf, A.; Stern, M.B. Preclinical Diagnosis of Parkinson’s Disease: Are We There Yet? Curr. Neurol. Neurosci. Rep. 2006, 6, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.; Olanow, C.W. Neuroprotection in Parkinson Disease: Mysteries, Myths, and Misconceptions. JAMA 2004, 291, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, R.; Siderowf, A.; Stern, M.B. Premotor Parkinson’s Disease: Clinical Features and Detection Strategies. Mov. Disord. 2009, 24, S665–S670. [Google Scholar] [CrossRef] [PubMed]
- Wirdefeldt, K.; Adami, H.O.; Cole, P.; Trichopoulos, D.; Mandel, J. Epidemiology and Etiology of Parkinson’s Disease: A Review of the Evidence. Eur. J. Epidemiol. 2011, 26, S1–S58. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, M.C.; Growdon, W.; Gomez-Isla, T.; Newell, K.; George, J.M.; Clayton, D.F.; Hyman, B.T. Nigral and Cortical Lewy Bodies and Dystrophic Nigral Neurites in Parkinson’s Disease and Cortical Lewy Body Disease Contain Alpha-Synuclein Immunoreactivity. J. Neuropathol. Exp. Neurol. 1998, 57, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. Alpha-Synuclein in Filamentous Inclusions of Lewy Bodies from Parkinson’s Disease and Dementia with Lewy Bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Engelender, S.; Yoshimoto, M.; Tsuji, S.; Ross, C.A.; Takahashi, H. Synphilin-1 Is Present in Lewy Bodies in Parkinson’s Disease. Ann. Neurol. 2000, 47, 521–523. [Google Scholar] [CrossRef]
- Kuzuhara, S.; Mori, H.; Izumiyama, N.; Yoshimura, M.; Ihara, Y. Lewy Bodies Are Ubiquitinated. A Light and Electron Microscopic Immunocytochemical Study. Acta Neuropathol. 1988, 75, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Chesselet, M.F.; Richter, F. Modelling of Parkinson’s Disease in Mice. Lancet Neurol. 2011, 10, 1108–1118. [Google Scholar] [CrossRef]
- Calabresi, P.; Picconi, B.; Tozzi, A.; Ghiglieri, V.; di Filippo, M. Direct and Indirect Pathways of Basal Ganglia: A Critical Reappraisal. Nat. Neurosci. 2014, 17, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Kravitz, A.V.; Kreitzer, A.C. Striatal Mechanisms Underlying Movement, Reinforcement, and Punishment. Physiology 2012, 27, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, D.; Comella, C.L.; Horn, S. Parkinson’s Disease—Part 2: Treatment of Motor Symptoms. Am. J. Manag. Care 2008, 14, S49–S58. [Google Scholar] [PubMed]
- Weintraub, D.; Comella, C.L.; Horn, S. Parkinson’s Disease—Part 3: Neuropsychiatric Symptoms. Am. J. Manag. Care 2008, 14, S59–S69. [Google Scholar] [PubMed]
- Bolam, J.P.; Pissadaki, E.K. Living on the Edge with Too Many Mouths to Feed: Why Dopamine Neurons Die. Mov. Disord. 2012, 27, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Milnerwood, A.J.; Raymond, L.A. Early Synaptic Pathophysiology in Neurodegeneration: Insights from Huntington’s Disease. Trends Neurosci. 2010, 33, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.E.; O’malley, K. Axon Degeneration in Parkinson’s Disease. Exp. Neurol. 2013, 246, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Yacoubian, T.A.; Standaert, D.G. Targets for Neuroprotection in Parkinson’s Disease. Biochim. Biophys. Acta 2009, 1792, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Perez, E.; Liu, R.; Yan, L.J.; Mallet, R.T.; Yang, S.H. Pyruvate Protects Mitochondria from Oxidative Stress in Human Neuroblastoma Sk-N-Sh Cells. Brain Res. 2007, 1132, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free Radicals and Antioxidants in Normal Physiological Functions and Human Disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Alam, Z.I.; Jenner, A.; Daniel, S.E.; Lees, A.J.; Cairns, N.; Marsden, C.D.; Jenner, P.; Halliwell, B. Oxidative Dna Damage in the Parkinsonian Brain: An Apparent Selective Increase in 8-Hydroxyguanine Levels in Substantia Nigra. J. Neurochem. 1997, 69, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Sian, J.; Rose, S.; Hindmarsh, J.G.; Mann, V.M.; Cooper, J.M.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Schapira, A.H.; et al. Indices of Oxidative Stress and Mitochondrial Function in Individuals with Incidental Lewy Body Disease. Ann. Neurol. 1994, 35, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial Complex I Deficiency in Parkinson’s Disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Betarbet, R.; Sherer, T.B.; Mackenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic Systemic Pesticide Exposure Reproduces Features of Parkinson’s Disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [PubMed]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in Humans Due to a Product of Meperidine-Analog Synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Forno, L.S.; Rebert, C.S.; Irwin, I. Selective Nigral Toxicity after Systemic Administration of 1-Methyl-4-Phenyl-1,2,5,6-Tetrahydropyrine (Mptp) in the Squirrel Monkey. Brain Res. 1984, 292, 390–394. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, I.; Lin, M.T.; Zheng, K.; Keller-Mcgandy, C.E.; Betensky, R.A.; Johns, D.R.; Beal, M.F.; Standaert, D.G.; Simon, D.K. Somatic Mitochondrial Dna Mutations in Single Neurons and Glia. Neurobiol. Aging 2005, 26, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Mitochondria in the Aetiology and Pathogenesis of Parkinson’s Disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Wooten, G.F.; Currie, L.J.; Bennett, J.P.; Harrison, M.B.; Trugman, J.M.; Parker, W.D., Jr. Maternal Inheritance in Parkinson’s Disease. Ann. Neurol. 1997, 41, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Sofic, E.; Rausch, W.D.; Schmidt, B.; Reynolds, G.P.; Jellinger, K.; Youdim, M.B. Transition Metals, Ferritin, Glutathione, and Ascorbic Acid in Parkinsonian Brains. J. Neurochem. 1989, 52, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.L.; Yong, V.W. Idiopathic Parkinson’s Disease, Progressive Supranuclear Palsy and Glutathione Metabolism in the Substantia Nigra of Patients. Neurosci. Lett. 1986, 67, 269–274. [Google Scholar] [CrossRef]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in Glutathione Levels in Parkinson’s Disease and Other Neurodegenerative Disorders Affecting Basal Ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Sofic, E.; Lange, K.W.; Jellinger, K.; Riederer, P. Reduced and Oxidized Glutathione in the Substantia Nigra of Patients with Parkinson’s Disease. Neurosci. Lett. 1992, 142, 128–130. [Google Scholar] [CrossRef]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila Pink1 Is Required for Mitochondrial Function and Interacts Genetically with Parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Smith, P.D.; Aleyasin, H.; Hayley, S.; Mount, M.P.; Pownall, S.; Wakeham, A.; You-Ten, A.J.; Kalia, S.K.; Horne, P.; et al. Hypersensitivity of Dj-1-Deficient Mice to 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyrindine (Mptp) and Oxidative Stress. Proc. Natl. Acad. Sci. USA 2005, 102, 5215–5220. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.M.; et al. Mitochondrial Dysfunction in Drosophila Pink1 Mutants Is Complemented by Parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Neuroprotection in Parkinson’s Disease. Parkinsonism Relat. Disord. 2009, 15, S41–S43. [Google Scholar] [CrossRef]
- Hoozemans, J.J.; Van Haastert, E.S.; Eikelenboom, P.; de Vos, R.A.; Rozemuller, J.M.; Scheper, W. Activation of the Unfolded Protein Response in Parkinson’s Disease. Biochem. Biophys. Res. Commun. 2007, 354, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the Parkin Gene Cause Autosomal Recessive Juvenile Parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [PubMed]
- Imai, Y.; Soda, M.; Inoue, H.; Hattori, N.; Mizuno, Y.; Takahashi, R. An Unfolded Putative Transmembrane Polypeptide, Which Can Lead to Endoplasmic Reticulum Stress, Is a Substrate of Parkin. Cell 2001, 105, 891–902. [Google Scholar] [CrossRef]
- Sarkar, S.; Chigurupati, S.; Raymick, J.; Mann, D.; Bowyer, J.F.; Schmitt, T.; Beger, R.D.; Hanig, J.P.; Schmued, L.C.; Paule, M.G. Neuroprotective Effect of the Chemical Chaperone, Trehalose in a Chronic Mptp-Induced Parkinson’s Disease Mouse Model. Neurotoxicology 2014, 44, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Leroy, E.; Boyer, R.; Auburger, G.; Leube, B.; Ulm, G.; Mezey, E.; Harta, G.; Brownstein, M.J.; Jonnalagada, S.; Chernova, T.; et al. The Ubiquitin Pathway in Parkinson’s Disease. Nature 1998, 395, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Klucken, J.; Shin, Y.; Masliah, E.; Hyman, B.T.; Mclean, P.J. Hsp70 Reduces Alpha-Synuclein Aggregation and Toxicity. J. Biol. Chem. 2004, 279, 25497–25502. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.J.; West, A.B.; Dikeman, D.A.; Dawson, V.L.; Dawson, T.M. Parkin Mediates the Degradation-Independent Ubiquitination of Hsp70. J. Neurochem. 2008, 105, 1806–1819. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.K.; Dawson, V.L.; Dawson, T.M. The Role of the Ubiquitin-Proteasomal Pathway in Parkinson’s Disease and Other Neurodegenerative Disorders. Trends Neurosci. 2001, 24, S7–S14. [Google Scholar] [CrossRef]
- Mcnaught, K.S.; Mytilineou, C.; Jnobaptiste, R.; Yabut, J.; Shashidharan, P.; Jennert, P.; Olanow, C.W. Impairment of the Ubiquitin-Proteasome System Causes Dopaminergic Cell Death and Inclusion Body Formation in Ventral Mesencephalic Cultures. J. Neurochem. 2002, 81, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, K.; Li, H.; Kawamura, R.; Osaka, H.; Wang, Y.L.; Hara, Y.; Hirokawa, T.; Manago, Y.; Amano, T.; Noda, M.; et al. Alterations of Structure and Hydrolase Activity of Parkinsonism-Associated Human Ubiquitin Carboxyl-Terminal Hydrolase L1 Variants. Biochem. Biophys. Res. Commun. 2003, 304, 176–183. [Google Scholar] [CrossRef]
- Esposito, E.; Di Matteo, V.; Benigno, A.; Pierucci, M.; Crescimanno, G.; Di Giovanni, G. Non-Steroidal Anti-Inflammatory Drugs in Parkinson’s Disease. Exp. Neurol. 2007, 205, 295–312. [Google Scholar] [CrossRef] [PubMed]
- Mcgeer, E.G.; Mcgeer, P.L. The Role of Anti-Inflammatory Agents in Parkinson’s Disease. CNS Drugs 2007, 21, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Mccoy, M.K.; Frank-Cannon, T.C. Neuroinflammatory Mechanisms in Parkinson’s Disease: Potential Environmental Triggers, Pathways, and Targets for Early Therapeutic Intervention. Exp. Neurol. 2007, 208, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Portwood, N.; Brodin, D.; Grill, V.; Bjorklund, A. Effects of Diazoxide on Gene Expression in Rat Pancreatic Islets Are Largely Linked to Elevated Glucose and Potentially Serve to Enhance Beta-Cell Sensitivity. Diabetes 2007, 56, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1 Beta, Interleukin-6, Epidermal Growth Factor and Transforming Growth Factor-Alpha Are Elevated in the Brain from Parkinsonian Patients. Neurosci. Lett. 1994, 180, 147–150. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor Necrosis Factor-Alpha (Tnf-Alpha) Increases Both in the Brain and in the Cerebrospinal Fluid from Parkinsonian Patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Goldknopf, I.L.; Bryson, J.K.; Strelets, I.; Quintero, S.; Sheta, E.A.; Mosqueda, M.; Park, H.R.; Appel, S.H.; Shill, H.; Sabbagh, M.; et al. Abnormal Serum Concentrations of Proteins in Parkinson’s Disease. Biochem. Biophys. Res. Commun. 2009, 389, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Mcgeer, P.L.; Mcgeer, E.G. Lewy Bodies in Parkinson’s Disease Are Recognized by Antibodies to Complement Proteins. Acta Neuropathol. 1992, 84, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.D.; Banerjee, R.; Liu, J.; Gendelman, H.E.; Mosley, R.L. Neuroprotective Activities of Cd4+Cd25+ Regulatory T Cells in an Animal Model of Parkinson’s Disease. J. Leukoc. Biol. 2007, 82, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.D.; Glanzer, J.G.; Kadiu, I.; Ricardo-Dukelow, M.; Chaudhuri, A.; Ciborowski, P.; Cerny, R.; Gelman, B.; Thomas, M.P.; Mosley, R.L.; et al. Nitrated Alpha-Synuclein-Activated Microglial Profiling for Parkinson’s Disease. J. Neurochem. 2008, 104, 1504–1525. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.D.; Kadiu, I.; Garg, S.K.; Glanzer, J.G.; Nordgren, T.; Ciborowski, P.; Banerjee, R.; Gendelman, H.E. Nitrated Alpha-Synuclein and Microglial Neuroregulatory Activities. J. Neuroimmune Pharmacol. 2008, 3, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated Alpha-Synuclein Activates Microglia: A Process Leading to Disease Progression in Parkinson’s Disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Benner, E.J.; Banerjee, R.; Reynolds, A.D.; Sherman, S.; Pisarev, V.M.; Tsiperson, V.; Nemachek, C.; Ciborowski, P.; Przedborski, S.; Mosley, R.L.; et al. Nitrated Alpha-Synuclein Immunity Accelerates Degeneration of Nigral Dopaminergic Neurons. PLoS ONE 2008, 3, e1376. [Google Scholar] [CrossRef] [PubMed]
- Theodore, S.; Cao, S.; Mclean, P.J.; Standaert, D.G. Targeted Overexpression of Human Alpha-Synuclein Triggers Microglial Activation and an Adaptive Immune Response in a Mouse Model of Parkinson Disease. J. Neuropathol. Exp. Neurol. 2008, 67, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Roy, A.; Liu, X.; Kordower, J.H.; Mufson, E.J.; Hartley, D.M.; Ghosh, S.; Mosley, R.L.; Gendelman, H.E.; Pahan, K. Selective Inhibition of Nf-Kappab Activation Prevents Dopaminergic Neuronal Loss in a Mouse Model of Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2007, 104, 18754–18759. [Google Scholar] [CrossRef] [PubMed]
- Bower, J.H.; Maraganore, D.M.; Peterson, B.J.; Ahlskog, J.E.; Rocca, W.A. Immunologic Diseases, Anti-Inflammatory Drugs, and Parkinson Disease: A Case-Control Study. Neurology 2006, 67, 494–496. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, S.M.; Hernan, M.A.; Schwarzschild, M.A.; Willett, W.C.; Colditz, G.A.; Speizer, F.E.; Ascherio, A. Nonsteroidal Anti-Inflammatory Drugs and the Risk of Parkinson Disease. Arch. Neurol. 2003, 60, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Hernan, M.A.; Logroscino, G.; Garcia Rodriguez, L.A. Nonsteroidal Anti-Inflammatory Drugs and the Incidence of Parkinson Disease. Neurology 2006, 66, 1097–1099. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B.; Wang, S.W.; Li, N.C.; Lee, A.; Lee, T.A.; Kazis, L.E. Simvastatin Is Associated with a Reduced Incidence of Dementia and Parkinson’s Disease. BMC Med. 2007, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- The NINDS NET-PD Investigators. A Randomized, Double-Blind, Futility Clinical Trial of Creatine and Minocycline in Early Parkinson Disease. Neurology 2006, 66, 664–671. [Google Scholar]
- Mody, I.; Macdonald, J.F. Nmda Receptor-Dependent Excitotoxicity: The Role of Intracellular Ca2+ Release. Trends Pharmacol. Sci. 1995, 16, 356–359. [Google Scholar] [CrossRef]
- Dawson, V.L.; Dawson, T.M. Nitric Oxide Neurotoxicity. J. Chem. Neuroanat. 1996, 10, 179–190. [Google Scholar] [CrossRef]
- Good, P.F.; Hsu, A.; Werner, P.; Perl, D.P.; Olanow, C.W. Protein Nitration in Parkinson’s Disease. J. Neuropathol. Exp. Neurol. 1998, 57, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Brouillet, E.; Beal, M.F. Nmda Antagonists Partially Protect Against Mptp Induced Neurotoxicity in Mice. Neuroreport 1993, 4, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Turski, L.; Bressler, K.; Rettig, K.J.; Loschmann, P.A.; Wachtel, H. Protection of Substantia Nigra from Mpp+ Neurotoxicity by N-Methyl-d-Aspartate Antagonists. Nature 1991, 349, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Hunter, C. A Double-Blind, Placebo-Controlled and Longitudinal Study of Riluzole in Early Parkinson’s Disease. Parkinsonism Relat. Disord. 2002, 8, 271–276. [Google Scholar] [CrossRef]
- Kornhuber, J.; Weller, M.; Schoppmeyer, K.; Riederer, P. Amantadine and Memantine Are Nmda Receptor Antagonists with Neuroprotective Properties. J. Neural. Transm. Suppl. 1994, 43, 91–104. [Google Scholar] [PubMed]
- Hallett, P.J.; Standaert, D.G. Rationale for and Use of Nmda Receptor Antagonists in Parkinson’s Disease. Pharmacol. Ther. 2004, 102, 155–174. [Google Scholar] [CrossRef] [PubMed]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and Autophagy in Nigral Neurons of Patients with Parkinson’s Disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar] [PubMed]
- Hirsch, E.C.; Hunot, S.; Damier, P.; Brugg, B.; Faucheux, B.A.; Michel, P.P.; Ruberg, M.; Muriel, M.P.; Mouatt-Prigent, A.; Agid, Y. Glial Cell Participation in the Degeneration of Dopaminergic Neurons in Parkinson’s Disease. Adv. Neurol. 1999, 80, 9–18. [Google Scholar] [PubMed]
- Tompkins, M.M.; Basgall, E.J.; Zamrini, E.; Hill, W.D. Apoptotic-Like Changes in Lewy-Body-Associated Disorders and Normal Aging in Substantia Nigral Neurons. Am. J. Pathol. 1997, 150, 119–131. [Google Scholar] [PubMed]
- Blum, D.; Torch, S.; Lambeng, N.; Nissou, M.; Benabid, A.L.; Sadoul, R.; Verna, J.M. Molecular Pathways Involved in the Neurotoxicity of 6-Ohda, Dopamine and Mptp: Contribution to the Apoptotic Theory in Parkinson’s Disease. Prog. Neurobiol. 2001, 65, 135–172. [Google Scholar] [CrossRef]
- Mattson, M.P. Neuronal Life-and-Death Signaling, Apoptosis, and Neurodegenerative Disorders. Antioxid. Redox Signal. 2006, 8, 1997–2006. [Google Scholar] [CrossRef] [PubMed]
- Parkinson Study Group PRECEPT Investigators. Mixed Lineage Kinase Inhibitor Cep-1347 Fails to Delay Disability in Early Parkinson Disease. Neurology 2007, 69, 1480–1490. [Google Scholar]
- Mogi, M.; Togari, A.; Kondo, T.; Mizuno, Y.; Komure, O.; Kuno, S.; Ichinose, H.; Nagatsu, T. Brain-Derived Growth Factor and Nerve Growth Factor Concentrations Are Decreased in the Substantia Nigra in Parkinson’s Disease. Neurosci. Lett. 1999, 270, 45–48. [Google Scholar] [CrossRef]
- Chauhan, N.B.; Siegel, G.J.; Lee, J.M. Depletion of Glial Cell Line-Derived Neurotrophic Factor in Substantia Nigra Neurons of Parkinson’s Disease Brain. J. Chem. Neuroanat. 2001, 21, 277–288. [Google Scholar] [CrossRef]
- Eslamboli, A.; Georgievska, B.; Ridley, R.M.; Baker, H.F.; Muzyczka, N.; Burger, C.; Mandel, R.J.; Annett, L.; Kirik, D. Continuous Low-Level Glial Cell Line-Derived Neurotrophic Factor Delivery Using Recombinant Adeno-Associated Viral Vectors Provides Neuroprotection and Induces Behavioral Recovery in a Primate Model of Parkinson’s Disease. J. Neurosci. 2005, 25, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Gasmi, M.; Herzog, C.D.; Brandon, E.P.; Cunningham, J.J.; Ramirez, G.A.; Ketchum, E.T.; Bartus, R.T. Striatal Delivery of Neurturin by Cere-120, an AAV2 Vector for the Treatment of Dopaminergic Neuron Degeneration in Parkinson’s Disease. Mol. Ther. 2007, 15, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Olanow, C.W.; Freeman, T.B. Transplanted Dopaminergic Neurons Develop PD Pathologic Changes: A Second Case Report. Mov. Disord. 2008, 23, 2303–2306. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.S.; Patel, N.K.; Hotton, G.R.; O’sullivan, K.; Mccarter, R.; Bunnage, M.; Brooks, D.J.; Svendsen, C.N.; Heywood, P. Direct Brain Infusion of Glial Cell Line-Derived Neurotrophic Factor in Parkinson Disease. Nat. Med. 2003, 9, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.E.; Gill, S.; Patel, N.K.; Lozano, A.; Nutt, J.G.; Penn, R.; Brooks, D.J.; Hotton, G.; Moro, E.; Heywood, P.; et al. Randomized Controlled Trial of Intraputamenal Glial Cell Line-Derived Neurotrophic Factor Infusion in Parkinson Disease. Ann. Neurol. 2006, 59, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Tatton, W.G. Etiology and Pathogenesis of Parkinson’s Disease. Annu. Rev. Neurosci. 1999, 22, 123–144. [Google Scholar] [CrossRef] [PubMed]
- Yahr, M.D.; Duvoisin, R.C.; Schear, M.J.; Barrett, R.E.; Hoehn, M.M. Treatment of Parkinsonism with Levodopa. Arch. Neurol. 1969, 21, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, A.N.; Pedersen, B. Levodopa and Adventitious Movements. Lancet 1970, 2, 985. [Google Scholar] [CrossRef]
- Celesia, G.G.; Barr, A.N. Psychosis and Other Psychiatric Manifestations of Levodopa Therapy. Arch. Neurol. 1970, 23, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Mcdowell, F.H.; Lee, J.E. Levodopa, Parkinson’s Disease, and Hypotension. Ann. Intern. Med. 1970, 72, 751–752. [Google Scholar] [CrossRef] [PubMed]
- Agid, Y.; Olanow, C.W.; Mizuno, Y. Levodopa: Why the Controversy? Lancet 2002, 360, 575. [Google Scholar] [CrossRef]
- Fahn, S.; Oakes, D.; Shoulson, I.; Kieburtz, K.; Rudolph, A.; Lang, A.; Olanow, C.W.; Tanner, C.; Marek, K. Levodopa and the Progression of Parkinson’s Disease. N. Engl. J. Med. 2004, 351, 2498–2508. [Google Scholar] [PubMed]
- Alonso Canovas, A.; Luquin Piudo, R.; Garcia Ruiz-Espiga, P.; Burguera, J.A.; Campos Arillo, V.; Castro, A.; Linazasoro, G.; Lopez Del Val, J.; Vela, L.; Martinez Castrillo, J.C. Dopaminergic Agonists in Parkinson’s Disease. Neurologia 2014, 29, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Guay, D.R. Rasagiline (Tvp-1012): A New Selective Monoamine Oxidase Inhibitor for Parkinson’s Disease. Am. J. Geriatr. Pharmacother. 2006, 4, 330–346. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, R. Update on the Use of Pramipexole in the Treatment of Parkinson’s Disease. Neuropsychiatr. Dis. Treat. 2008, 4, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Odin, P.; Wolters, E.; Antonini, A. Continuous Dopaminergic Stimulation Achieved by Duodenal Levodopa Infusion. Neurol. Sci. 2008, 29 (Suppl. S5), S387–S388. [Google Scholar] [CrossRef] [PubMed]
- Brocks, D.R. Anticholinergic Drugs Used in Parkinson’s Disease: An Overlooked Class of Drugs from a Pharmacokinetic Perspective. J. Pharm. Pharm. Sci. 1999, 2, 39–46. [Google Scholar] [PubMed]
- Schapira, A.H. Present and Future Drug Treatment for Parkinson’s Disease. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.A.; Lobb, B.M.; Nutt, J.G.; Horak, F.B. Effects of a Central Cholinesterase Inhibitor on Reducing Falls in Parkinson Disease. Neurology 2010, 75, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Henderson, E.J.; Lord, S.R.; Close, J.C.; Lawrence, A.D.; Whone, A.; Ben-Shlomo, Y. The Respond Trial—Rivastigmine to Stabilise Gait in Parkinson’s Disease a Phase II, Randomised, Double Blind, Placebo Controlled Trial to Evaluate the Effect of Rivastigmine on Gait in Patients with Parkinson’s Disease Who Have Fallen. BMC Neurol. 2013, 13, 188. [Google Scholar] [CrossRef] [PubMed]
- Carvey, P.M.; Pieri, S.; Ling, Z.D. Attenuation of Levodopa-Induced Toxicity in Mesencephalic Cultures by Pramipexole. J. Neural. Transm. 1997, 104, 209–228. [Google Scholar] [CrossRef] [PubMed]
- Cassarino, D.S.; Fall, C.P.; Smith, T.S.; Bennett, J.P., Jr. Pramipexole Reduces Reactive Oxygen Species Production in Vivo and in Vitro and Inhibits the Mitochondrial Permeability Transition Produced by the Parkinsonian Neurotoxin Methylpyridinium Ion. J. Neurochem. 1998, 71, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Iravani, M.M.; Haddon, C.O.; Cooper, J.M.; Jenner, P.; Schapira, A.H. Pramipexole Protects Against Mptp Toxicity in Non-Human Primates. J. Neurochem. 2006, 96, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Kosaka, T.; Kakimura, J.I.; Matsuoka, Y.; Kohno, Y.; Nomura, Y.; Taniguchi, T. Protective Effects of the Antiparkinsonian Drugs Talipexole and Pramipexole Against 1-Methyl-4-Phenylpyridinium-Induced Apoptotic Death in Human Neuroblastoma Sh-Sy5y Cells. Mol. Pharmacol. 1998, 54, 1046–1054. [Google Scholar] [PubMed]
- Ogawa, N.; Tanaka, K.; Asanuma, M.; Kawai, M.; Masumizu, T.; Kohno, M.; Mori, A. Bromocriptine Protects Mice Against 6-Hydroxydopamine and Scavenges Hydroxyl Free Radicals in Vitro. Brain Res. 1994, 657, 207–213. [Google Scholar] [CrossRef]
- Parkinson Study Group. Dopamine Transporter Brain Imaging to Assess the Effects of Pramipexole vs. Levodopa on Parkinson Disease Progression. JAMA 2002, 287, 1653–1661. [Google Scholar]
- Allain, H.; Bentue-Ferrer, D.; Akwa, Y. Disease-Modifying Drugs and Parkinson’s Disease. Prog. Neurobiol. 2008, 84, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Ravina, B.; Eidelberg, D.; Ahlskog, J.E.; Albin, R.L.; Brooks, D.J.; Carbon, M.; Dhawan, V.; Feigin, A.; Fahn, S.; Guttman, M.; et al. The Role of Radiotracer Imaging in Parkinson Disease. Neurology 2005, 64, 208–215. [Google Scholar] [CrossRef] [PubMed]
- The Parkinson Study Group. Effect of Deprenyl on the Progression of Disability in Early Parkinson’s Disease. N. Engl. J. Med. 1989, 321, 1364–1371. [Google Scholar]
- Parkinson Study Group. Effects of Tocopherol and Deprenyl on the Progression of Disability in Early Parkinson’s Disease. N. Engl. J. Med. 1993, 328, 176–183. [Google Scholar]
- Parkinson Study Group. A Controlled, Randomized, Delayed-Start Study of Rasagiline in Early Parkinson Disease. Arch. Neurol. 2004, 61, 561–566. [Google Scholar]
- Beal, M.F.; Matthews, R.T.; Tieleman, A.; Shults, C.W. Coenzyme Q10 Attenuates the 1-Methyl-4-Phenyl-1,2,3,Tetrahydropyridine (Mptp) Induced Loss of Striatal Dopamine and Dopaminergic Axons in Aged Mice. Brain Res. 1998, 783, 109–114. [Google Scholar] [CrossRef]
- Shults, C.W.; Oakes, D.; Kieburtz, K.; Beal, M.F.; Haas, R.; Plumb, S.; Juncos, J.L.; Nutt, J.; Shoulson, I.; Carter, J.; et al. Effects of Coenzyme Q10 in Early Parkinson Disease: Evidence of Slowing of the Functional Decline. Arch. Neurol. 2002, 59, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.T.; Ferrante, R.J.; Klivenyi, P.; Yang, L.; Klein, A.M.; Mueller, G.; Kaddurah-Daouk, R.; Beal, M.F. Creatine and Cyclocreatine Attenuate Mptp Neurotoxicity. Exp. Neurol. 1999, 157, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Schapira, A.H.; Lewitt, P.A.; Kieburtz, K.; Sauer, D.; Olivieri, G.; Pohlmann, H.; Hubble, J. Tch346 As a Neuroprotective Drug in Parkinson’s Disease: A Double-Blind, Randomised, Controlled Trial. Lancet Neurol. 2006, 5, 1013–1020. [Google Scholar] [CrossRef]
- Lotharius, J.; Falsig, J.; van Beek, J.; Payne, S.; Dringen, R.; Brundin, P.; Leist, M. Progressive Degeneration of Human Mesencephalic Neuron-Derived Cells Triggered by Dopamine-Dependent Oxidative Stress Is Dependent on the Mixed-Lineage Kinase Pathway. J. Neurosci. 2005, 25, 6329–6342. [Google Scholar] [CrossRef] [PubMed]
- Mathiasen, J.R.; Mckenna, B.A.; Saporito, M.S.; Ghadge, G.D.; Roos, R.P.; Holskin, B.P.; Wu, Z.L.; Trusko, S.P.; Connors, T.C.; Maroney, A.C.; et al. Inhibition of Mixed Lineage Kinase 3 Attenuates Mpp+-Induced Neurotoxicity in Sh-Sy5y Cells. Brain Res. 2004, 1003, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Saporito, M.S.; Brown, E.M.; Miller, M.S.; Carswell, S. Cep-1347/Kt-7515, an Inhibitor of C-Jun N-Terminal Kinase Activation, Attenuates the 1-Methyl-4-Phenyl Tetrahydropyridine-Mediated Loss of Nigrostriatal Dopaminergic Neurons in Vivo. J. Pharmacol. Exp. Ther. 1999, 288, 421–427. [Google Scholar] [PubMed]
- Kordower, J.H.; Emborg, M.E.; Bloch, J.; Ma, S.Y.; Chu, Y.; Leventhal, L.; Mcbride, J.; Chen, E.Y.; Palfi, S.; Roitberg, B.Z.; et al. Neurodegeneration Prevented by Lentiviral Vector Delivery of Gdnf in Primate Models of Parkinson’s Disease. Science 2000, 290, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Akerud, P.; Alberch, J.; Eketjall, S.; Wagner, J.; Arenas, E. Differential Effects of Glial Cell Line-Derived Neurotrophic Factor and Neurturin on Developing and Adult Substantia Nigra Dopaminergic Neurons. J. Neurochem. 1999, 73, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Horger, B.A.; Nishimura, M.C.; Armanini, M.P.; Wang, L.C.; Poulsen, K.T.; Rosenblad, C.; Kirik, D.; Moffat, B.; Simmons, L.; Johnson, E., Jr.; et al. Neurturin Exerts Potent Actions on Survival and Function of Midbrain Dopaminergic Neurons. J. Neurosci. 1998, 18, 4929–4937. [Google Scholar] [PubMed]
- Kordower, J.H.; Herzog, C.D.; Dass, B.; Bakay, R.A.; Stansell, J., 3rd; Gasmi, M.; Bartus, R.T. Delivery of Neurturin by AAV2 (Cere-120)-Mediated Gene Transfer Provides Structural and Functional Neuroprotection and Neurorestoration in Mptp-Treated Monkeys. Ann. Neurol. 2006, 60, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Marks, W.J., Jr.; Ostrem, J.L.; Verhagen, L.; Starr, P.A.; Larson, P.S.; Bakay, R.A.; Taylor, R.; Cahn-Weiner, D.A.; Stoessl, A.J.; Olanow, C.W.; et al. Safety and Tolerability of Intraputaminal Delivery of Cere-120 (Adeno-Associated Virus Serotype 2-Neurturin) to Patients with Idiopathic Parkinson’s Disease: An Open-Label, Phase I Trial. Lancet Neurol. 2008, 7, 400–408. [Google Scholar] [CrossRef]
- Guo, X.; Dawson, V.L.; Dawson, T.M. Neuroimmunophilin Ligands Exert Neuroregeneration and Neuroprotection in Midbrain Dopaminergic Neurons. Eur. J. Neurosci. 2001, 13, 1683–1693. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Ferrari, G.; Kasper, M.; Tonge, D.A.; Steiner, J.P.; Hamilton, G.S.; Gordon-Weeks, P.R. The Non-Immunosuppressive Immunophilin Ligand GPi-1046 Potently Stimulates Regenerating Axon Growth from Adult Mouse Dorsal Root Ganglia Cultured in Matrigel. Neuroscience 2002, 114, 601–609. [Google Scholar] [CrossRef]
- Steiner, J.P.; Connolly, M.A.; Valentine, H.L.; Hamilton, G.S.; Dawson, T.M.; Hester, L.; Snyder, S.H. Neurotrophic Actions of Nonimmunosuppressive Analogues of Immunosuppressive Drugs Fk506, Rapamycin and Cyclosporin A. Nat. Med. 1997, 3, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Fujita, N.; Ogawa, N. Immunosuppressive (Fk506) and Non-Immunosuppressive (GPi1046) Immunophilin Ligands Activate Neurotrophic Factors in the Mouse Brain. Brain Res. 2003, 970, 250–253. [Google Scholar] [CrossRef]
- Poulter, M.O.; Payne, K.B.; Steiner, J.P. Neuroimmunophilins: A Novel Drug Therapy for the Reversal of Neurodegenerative Disease? Neuroscience 2004, 128, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Movement Disorder Society Task Force on Rating Scales for Parkinson’s Disease. The Unified Parkinson’s Disease Rating Scale (Updrs): Status and Recommendations. Mov. Disord. 2003, 18, 738–750. [Google Scholar]
- Ramaker, C.; Marinus, J.; Stiggelbout, A.M.; Van Hilten, B.J. Systematic Evaluation of Rating Scales for Impairment and Disability in Parkinson’s Disease. Mov. Disord. 2002, 17, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Goetz, C.G.; Fahn, S.; Martinez-Martin, P.; Poewe, W.; Sampaio, C.; Stebbins, G.T.; Stern, M.B.; Tilley, B.C.; Dodel, R.; Dubois, B.; et al. Movement Disorder Society-Sponsored Revision of the Unified Parkinson’s Disease Rating Scale (Mds-Updrs): Process, Format, and Clinimetric Testing Plan. Mov. Disord. 2007, 22, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Kieburtz, K. Issues in Neuroprotection Clinical Trials in Parkinson’s Disease. Neurology 2006, 66, S50–S57. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Rockenstein, E.; Masliah, E. Transgenic Models of Alpha-Synuclein Pathology: Past, Present, and Future. Ann. N. Y. Acad. Sci. 2003, 991, 171–188. [Google Scholar] [CrossRef] [PubMed]
- Betarbet, R.; Sherer, T.B.; Greenamyre, J.T. Animal Models of Parkinson’s Disease. Bioessays 2002, 24, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.Y.; Schwarzschild, M.A. Clinical Trials for Neuroprotection in Parkinson’s Disease: Overcoming Angst and Futility? Curr. Opin. Neurol. 2007, 20, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Maries, E.; Dass, B.; Collier, T.J.; Kordower, J.H.; Steece-Collier, K. The Role of Alpha-Synuclein in Parkinson’s Disease: Insights from Animal Models. Nat. Rev. Neurosci. 2003, 4, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, E.; Periquet, M.; Bonifati, V.; Wood, N.W.; de Michele, G.; Bonnet, A.M.; Fraix, V.; Broussolle, E.; Horstink, M.W.; Vidailhet, M.; et al. How Much Phenotypic Variation Can Be Attributed to Parkin Genotype? Ann. Neurol. 2003, 54, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Lucking, C.B.; Durr, A.; Bonifati, V.; Vaughan, J.; de Michele, G.; Gasser, T.; Harhangi, B.S.; Meco, G.; Denefle, P.; Wood, N.W.; et al. Association between Early-Onset Parkinson’s Disease and Mutations in the Parkin Gene. N. Engl. J. Med. 2000, 342, 1560–1567. [Google Scholar] [CrossRef] [PubMed]
- Gilks, W.P.; Abou-Sleiman, P.M.; Gandhi, S.; Jain, S.; Singleton, A.; Lees, A.J.; Shaw, K.; Bhatia, K.P.; Bonifati, V.; Quinn, N.P.; et al. A Common Lrrk2 Mutation in Idiopathic Parkinson’s Disease. Lancet 2005, 365, 415–416. [Google Scholar] [CrossRef]
- Kachergus, J.; Mata, I.F.; Hulihan, M.; Taylor, J.P.; Lincoln, S.; Aasly, J.; Gibson, J.M.; Ross, O.A.; Lynch, T.; Wiley, J.; et al. Identification of a Novel Lrrk2 Mutation Linked to Autosomal Dominant Parkinsonism: Evidence of a Common Founder across European Populations. Am. J. Hum. Genet. 2005, 76, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Brotchie, J.M.; Fox, S.H. Novel Nondopaminergic Targets for Motor Features of Parkinson’s Disease: Review of Recent Trials. Mov. Disord. 2013, 28, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Jacobs, E.; Schwarzschild, M.A.; Mccullough, M.L.; Calle, E.E.; Thun, M.J.; Ascherio, A. Nonsteroidal Antiinflammatory Drug Use and the Risk for Parkinson’s Disease. Ann. Neurol. 2005, 58, 963–967. [Google Scholar] [CrossRef] [PubMed]
- Selley, M.L. Simvastatin Prevents 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Striatal Dopamine Depletion and Protein Tyrosine Nitration in Mice. Brain Res. 2005, 1037, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Di Napoli, P.; Taccardi, A.A.; Oliver, M.; de Caterina, R. Statins and Stroke: Evidence for Cholesterol-Independent Effects. Eur. Heart J. 2002, 23, 1908–1921. [Google Scholar] [CrossRef] [PubMed]
- Wahner, A.D.; Bronstein, J.M.; Bordelon, Y.M.; Ritz, B. Statin Use and the Risk of Parkinson Disease. Neurology 2008, 70, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- De Lau, L.M.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M. Serum Cholesterol Levels and the Risk of Parkinson’s Disease. Am. J. Epidemiol. 2006, 164, 998–1002. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Chen, H.; Miller, W.C.; Mailman, R.B.; Woodard, J.L.; Chen, P.C.; Xiang, D.; Murrow, R.W.; Wang, Y.Z.; Poole, C. Lower Low-Density Lipoprotein Cholesterol Levels Are Associated with Parkinson’s Disease. Mov. Disord. 2007, 22, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Ladenheim, B.; Cutler, R.G.; Kruman, I.I.; Cadet, J.L.; Mattson, M.P. Dietary Folate Deficiency and Elevated Homocysteine Levels Endanger Dopaminergic Neurons in Models of Parkinson’s Disease. J. Neurochem. 2002, 80, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Ma, Z.; Lin, S.; Dodel, R.C.; Gao, F.; Bales, K.R.; Triarhou, L.C.; Chernet, E.; Perry, K.W.; Nelson, D.L.; et al. Minocycline Prevents Nigrostriatal Dopaminergic Neurodegeneration in the Mptp Model of Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2001, 98, 14669–14674. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Appel, S.; Le, W. Minocycline Inhibits Microglial Activation and Protects Nigral Cells after 6-Hydroxydopamine Injection into Mouse Striatum. Brain Res. 2001, 909, 187–193. [Google Scholar] [CrossRef]
- Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Tieu, K.; Teismann, P.; Vadseth, C.; Choi, D.K.; Ischiropoulos, H.; Przedborski, S. Blockade of Microglial Activation Is Neuroprotective in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model of Parkinson Disease. J. Neurosci. 2002, 22, 1763–1771. [Google Scholar] [PubMed]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric Acid Provides an Antioxidant Defense in Humans Against Oxidant- and Radical-Caused Aging and Cancer: A Hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar] [CrossRef] [PubMed]
- Schwarzschild, M.A.; Schwid, S.R.; Marek, K.; Watts, A.; Lang, A.E.; Oakes, D.; Shoulson, I.; Ascherio, A.; Parkinson Study Group PRECEPT Investigators; Hyson, C.; et al. Serum Urate As a Predictor of Clinical and Radiographic Progression in Parkinson Disease. Arch. Neurol. 2008, 65, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Chen, H.; Choi, H.K.; Curhan, G.; Schwarzschild, M.A.; Ascherio, A. Diet, Urate, and Parkinson’s Disease Risk in Men. Am. J. Epidemiol. 2008, 167, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Haywood, A.F.; Staveley, B.E. Parkin Counteracts Symptoms in a Drosophila Model of Parkinson’s Disease. BMC Neurosci. 2004, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Lo Bianco, C.; Schneider, B.L.; Bauer, M.; Sajadi, A.; Brice, A.; Iwatsubo, T.; Aebischer, P. Lentiviral Vector Delivery of Parkin Prevents Dopaminergic Degeneration in an Alpha-Synuclein Rat Model of Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2004, 101, 17510–17515. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Mizuno, Y.; Mochizuki, H. Parkin Gene Therapy for Alpha-Synucleinopathy: A Rat Model of Parkinson’s Disease. Hum. Gene Ther. 2005, 16, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Yacoubian, T.A.; Wilson, S.; Xie, Z.L.; Speake, L.D.; Parks, R.; Crabtree, D.; et al. Lysosomal Enzyme Cathepsin D Protects Against Alpha-Synuclein Aggregation and Toxicity. Mol. Brain 2008, 1, 17. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Rockenstein, E.; Adame, A.; Alford, M.; Crews, L.; Hashimoto, M.; Seubert, P.; Lee, M.; Goldstein, J.; Chilcote, T.; et al. Effects of Alpha-Synuclein Immunization in a Mouse Model of Parkinson’s Disease. Neuron 2005, 46, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Feany, M.B. Alpha-Synuclein Phosphorylation Controls Neurotoxicity and Inclusion Formation in a Drosophila Model of Parkinson Disease. Nat. Neurosci. 2005, 8, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.W.; Margolis, R.L.; Li, X.; Troncoso, J.C.; Lee, M.K.; Dawson, V.L.; Dawson, T.M.; Iwatsubo, T.; Ross, C.A. Alpha-Synuclein Phosphorylation Enhances Eosinophilic Cytoplasmic Inclusion Formation in Sh-Sy5y Cells. J. Neurosci. 2005, 25, 5544–5552. [Google Scholar] [CrossRef] [PubMed]
- Souza, J.M.; Giasson, B.I.; Chen, Q.; Lee, V.M.; Ischiropoulos, H. Dityrosine Cross-Linking Promotes Formation of Stable Alpha-Synuclein Polymers. Implication of Nitrative and Oxidative Stress in the Pathogenesis of Neurodegenerative Synucleinopathies. J. Biol. Chem. 2000, 275, 18344–18349. [Google Scholar] [CrossRef] [PubMed]
- Imam, S.Z.; Zhou, Q.; Yamamoto, A.; Valente, A.J.; Ali, S.F.; Bains, M.; Roberts, J.L.; Kahle, P.J.; Clark, R.A.; Li, S. Novel Regulation of Parkin Function through c-Abl-Mediated Tyrosine Phosphorylation: Implications for Parkinson’s Disease. J. Neurosci. 2011, 31, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Hebron, M.L.; Lonskaya, I.; Moussa, C.E. Nilotinib Reverses Loss of Dopamine Neurons and Improves Motor Behavior Via Autophagic Degradation of Alpha-Synuclein in Parkinson’s Disease Models. Hum. Mol. Genet. 2013, 22, 3315–3328. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.S.; Lee, Y.; Shin, J.H.; Karuppagounder, S.S.; GADad, B.S.; Koleske, A.J.; Pletnikova, O.; Troncoso, J.C.; Dawson, V.L.; Dawson, T.M. Phosphorylation by the c-Abl Protein Tyrosine Kinase Inhibits Parkin’s Ubiquitination and Protective Function. Proc. Natl. Acad. Sci. USA 2010, 107, 16691–16696. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Meng, Y.; Jiang, W.; Roux, B. Explaining Why Gleevec Is a Specific and Potent Inhibitor of Abl Kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 1664–1669. [Google Scholar] [CrossRef] [PubMed]
- Deremer, D.L.; Ustun, C.; Natarajan, K. Nilotinib: A Second-Generation Tyrosine Kinase Inhibitor for the Treatment of Chronic Myelogenous Leukemia. Clin. Ther. 2008, 30, 1956–1975. [Google Scholar] [CrossRef] [PubMed]
- Imam, S.Z.; Trickler, W.; Kimura, S.; Binienda, Z.K.; Paule, M.G.; Slikker, W., Jr.; Li, S.; Clark, R.A.; Ali, S.F. Neuroprotective Efficacy of a New Brain-Penetrating c-Abl Inhibitor in a Murine Parkinson’s Disease Model. PLoS ONE 2013, 8, e65129. [Google Scholar] [CrossRef] [PubMed]
- Hely, M.A.; Morris, J.G.; Reid, W.G.; Trafficante, R. Sydney Multicenter Study of Parkinson’s Disease: Non-l-Dopa-Responsive Problems Dominate at 15 Years. Mov. Disord. 2005, 20, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Neumiller, J.J.; Setter, S.M.; Dobbins, E.K. Clinical Review of Treatment Options for Select Nonmotor Symptoms of Parkinson’s Disease. Am. J. Geriatr. Pharmacother. 2010, 8, 294–315. [Google Scholar] [CrossRef] [PubMed]
- Menza, M.; Dobkin, R.D.; Marin, H.; Mark, M.H.; Gara, M.; Buyske, S.; Bienfait, K.; Dicke, A. A Controlled Trial of Antidepressants in Patients with Parkinson Disease and Depression. Neurology 2009, 72, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Bergman, H.; Wichmann, T.; Delong, M.R. Reversal of Experimental Parkinsonism by Lesions of the Subthalamic Nucleus. Science 1990, 249, 1436–1438. [Google Scholar] [CrossRef] [PubMed]
- Galvan, A.; Kuwajima, M.; Smith, Y. Glutamate and GABA Receptors and Transporters in the Basal Ganglia: What Does Their Subsynaptic Localization Reveal about Their Function? Neuroscience 2006, 143, 351–375. [Google Scholar] [CrossRef] [PubMed]
- Bronstein, J.M.; Tagliati, M.; Alterman, R.L.; Lozano, A.M.; Volkmann, J.; Stefani, A.; Horak, F.B.; Okun, M.S.; Foote, K.D.; Krack, P.; et al. Deep Brain Stimulation for Parkinson Disease: An Expert Consensus and Review of Key Issues. Arch. Neurol. 2011, 68, 165. [Google Scholar] [CrossRef] [PubMed]
- Chang, V.C.; Chou, K.L. Deep Brain Stimulation for Parkinson’s Disease: Patient Selection and Motor Outcomes. Med. Health Rhode Isl. 2006, 89, 142–144. [Google Scholar]
- Anderson, V.C.; Burchiel, K.J.; Hogarth, P.; Favre, J.; Hammerstad, J.P. Pallidal vs. Subthalamic Nucleus Deep Brain Stimulation in Parkinson Disease. Arch. Neurol. 2005, 62, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Portman, A.T.; Van Laar, T.; Staal, M.J.; Rutgers, A.W.; Journee, H.L.; Leenders, K.L. Chronic Stimulation of the Subthalamic Nucleus Increases Daily on-Time without Dyskinesia in Advanced Parkinson’s Disease. Parkinsonism Relat. Disord. 2006, 12, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Oroz, M.C.; Obeso, J.A.; Lang, A.E.; Houeto, J.L.; Pollak, P.; Rehncrona, S.; Kulisevsky, J.; Albanese, A.; Volkmann, J.; Hariz, M.I.; et al. Bilateral Deep Brain Stimulation in Parkinson’s Disease: A Multicentre Study with 4 Years Follow-up. Brain 2005, 128, 2240–2249. [Google Scholar] [CrossRef] [PubMed]
- Weaver, F.; Follett, K.; Hur, K.; Ippolito, D.; Stern, M. Deep Brain Stimulation in Parkinson Disease: A Metaanalysis of Patient Outcomes. J. Neurosurg. 2005, 103, 956–967. [Google Scholar] [CrossRef] [PubMed]
- Castelli, L.; Perozzo, P.; Zibetti, M.; Crivelli, B.; Morabito, U.; Lanotte, M.; Cossa, F.; Bergamasco, B.; Lopiano, L. Chronic Deep Brain Stimulation of the Subthalamic Nucleus for Parkinson’s Disease: Effects on Cognition, Mood, Anxiety and Personality Traits. Eur. Neurol. 2006, 55, 136–144. [Google Scholar] [CrossRef] [PubMed]
- De Gaspari, D.; Siri, C.; Landi, A.; Cilia, R.; Bonetti, A.; Natuzzi, F.; Morgante, L.; Mariani, C.B.; Sganzerla, E.; Pezzoli, G.; et al. Clinical and Neuropsychological Follow up at 12 Months in Patients with Complicated Parkinson’s Disease Treated with Subcutaneous Apomorphine Infusion or Deep Brain Stimulation of the Subthalamic Nucleus. J. Neurol. Neurosurg. Psychiatry 2006, 77, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, K.; Blairy, S.; Defebvre, L.; Krystkowiak, P.; Hess, U.; Blond, S.; Destee, A. Subthalamic Nucleus Stimulation Induces Deficits in Decoding Emotional Facial Expressions in Parkinson’s Disease. J. Neurol. Neurosurg. Psychiatry 2004, 75, 202–208. [Google Scholar] [PubMed]
- Dujardin, K.; Defebvre, L.; Krystkowiak, P.; Blond, S.; Destee, A. Influence of Chronic Bilateral Stimulation of the Subthalamic Nucleus on Cognitive Function in Parkinson’s Disease. J. Neurol. 2001, 248, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Merello, M.; Tenca, E.; Perez Lloret, S.; Martin, M.E.; Bruno, V.; Cavanagh, S.; Antico, J.; Cerquetti, D.; Leiguarda, R. Prospective Randomized 1-Year Follow-up Comparison of Bilateral Subthalamotomy versus Bilateral Subthalamic Stimulation and the Combination of Both in Parkinson’s Disease Patients: A Pilot Study. Br. J. Neurosurg. 2008, 22, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Schupbach, W.M.; Chastan, N.; Welter, M.L.; Houeto, J.L.; Mesnage, V.; Bonnet, A.M.; Czernecki, V.; Maltete, D.; Hartmann, A.; Mallet, L.; et al. Stimulation of the Subthalamic Nucleus in Parkinson’s Disease: A 5 Year Follow up. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1640–1644. [Google Scholar] [CrossRef] [PubMed]
- Smeding, H.M.; Esselink, R.A.; Schmand, B.; Koning-Haanstra, M.; Nijhuis, I.; Wijnalda, E.M.; Speelman, J.D. Unilateral Pallidotomy versus Bilateral Subthalamic Nucleus Stimulation in PD—A Comparison of Neuropsychological Effects. J. Neurol. 2005, 252, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Smeding, H.M.; Speelman, J.D.; Huizenga, H.M.; Schuurman, P.R.; Schmand, B. Predictors of Cognitive and Psychosocial Outcome after STN DBS in Parkinson’s Disease. J. Neurol. Neurosurg. Psychiatry 2011, 82, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Smeding, H.M.; Speelman, J.D.; Koning-Haanstra, M.; Schuurman, P.R.; Nijssen, P.; Van Laar, T.; Schmand, B. Neuropsychological Effects of Bilateral STN Stimulation in Parkinson Disease: A Controlled Study. Neurology 2006, 66, 1830–1836. [Google Scholar] [CrossRef] [PubMed]
- Harik, S.I. Changes in the Glucose Transporter of Brain Capillaries. Can. J. Physiol. Pharmacol. 1992, 70, S113–S117. [Google Scholar] [CrossRef] [PubMed]
- Berney, A.; Vingerhoets, F.; Perrin, A.; Guex, P.; Villemure, J.G.; Burkhard, P.R.; Benkelfat, C.; Ghika, J. Effect on Mood of Subthalamic DBS for Parkinson’s Disease: A Consecutive Series of 24 Patients. Neurology 2002, 59, 1427–1429. [Google Scholar] [CrossRef] [PubMed]
- Doshi, P.K.; Chhaya, N.; Bhatt, M.H. Depression Leading to Attempted Suicide after Bilateral Subthalamic Nucleus Stimulation for Parkinson’s Disease. Mov. Disord. 2002, 17, 1084–1085. [Google Scholar] [CrossRef] [PubMed]
- Funkiewiez, A.; Ardouin, C.; Caputo, E.; Krack, P.; Fraix, V.; Klinger, H.; Chabardes, S.; Foote, K.; Benabid, A.L.; Pollak, P. Long Term Effects of Bilateral Subthalamic Nucleus Stimulation on Cognitive Function, Mood, and Behaviour in Parkinson’s Disease. J. Neurol. Neurosurg. Psychiatry 2004, 75, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Okun, M.S.; Fernandez, H.H.; Wu, S.S.; Kirsch-Darrow, L.; Bowers, D.; Bova, F.; Suelter, M.; Jacobson, C.E.T.; Wang, X.; Gordon, C.W., Jr.; et al. Cognition and Mood in Parkinson’s Disease in Subthalamic Nucleus versus Globus Pallidus Interna Deep Brain Stimulation: The Compare Trial. Ann. Neurol. 2009, 65, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Okun, M.S.; Green, J.; Saben, R.; Gross, R.; Foote, K.D.; Vitek, J.L. Mood Changes with Deep Brain Stimulation of STN and GPi: Results of a Pilot Study. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1584–1586. [Google Scholar] [CrossRef] [PubMed]
- Voon, V.; Krack, P.; Lang, A.E.; Lozano, A.M.; Dujardin, K.; Schupbach, M.; D’ambrosia, J.; Thobois, S.; Tamma, F.; Herzog, J.; et al. A Multicentre Study on Suicide Outcomes Following Subthalamic Stimulation for Parkinson’s Disease. Brain 2008, 131, 2720–2728. [Google Scholar] [CrossRef] [PubMed]
- Moro, E.; Lozano, A.M.; Pollak, P.; Agid, Y.; Rehncrona, S.; Volkmann, J.; Kulisevsky, J.; Obeso, J.A.; Albanese, A.; Hariz, M.I.; et al. Long-Term Results of a Multicenter Study on Subthalamic and Pallidal Stimulation in Parkinson’s Disease. Mov. Disord. 2010, 25, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, J.; Diamond, A.; Hunter, C.; Davidson, A.; Almaguer, M.; Jankovic, J. Impact of STN-DBS on Life and Health Satisfaction in Patients with Parkinson’s Disease. J. Neurol. Neurosurg. Psychiatry 2010, 81, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Englund, E.; Widner, H.; Rehncrona, S.; Bjorklund, A.; Lindvall, O.; Brundin, P. Characterization of Lewy Body Pathology in 12- And 16-Year-Old Intrastriatal Mesencephalic Grafts Surviving in a Patient with Parkinson’s Disease. Mov. Disord. 2010, 25, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Lane, E.L.; Bjorklund, A.; Dunnett, S.B.; Winkler, C. Neural Grafting in Parkinson’s Disease Unraveling the Mechanisms Underlying Graft-Induced Dyskinesia. Prog. Brain Res. 2010, 184, 295–309. [Google Scholar] [PubMed]
- Lane, E.L.; Winkler, C.; Brundin, P.; Cenci, M.A. The Impact of Graft Size on the Development of Dyskinesia Following Intrastriatal Grafting of Embryonic Dopamine Neurons in the Rat. Neurobiol. Dis. 2006, 22, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Politis, M.; Wu, K.; Loane, C.; Quinn, N.P.; Brooks, D.J.; Rehncrona, S.; Bjorklund, A.; Lindvall, O.; Piccini, P. Serotonergic Neurons Mediate Dyskinesia Side Effects in Parkinson’s Patients with Neural Transplants. Sci. Transl. Med. 2010, 2, 38ra46. [Google Scholar] [CrossRef] [PubMed]
- Baekelandt, V.; de Strooper, B.; Nuttin, B.; Debyser, Z. Gene Therapeutic Strategies for Neurodegenerative Diseases. Curr. Opin. Mol. Ther. 2000, 2, 540–554. [Google Scholar] [PubMed]
- Bjorklund, A.; Kirik, D.; Rosenblad, C.; Georgievska, B.; Lundberg, C.; Mandel, R.J. Towards a Neuroprotective Gene Therapy for Parkinson’s Disease: Use of Adenovirus, AAV and Lentivirus Vectors for Gene Transfer of Gdnf to the Nigrostriatal System in the Rat Parkinson Model. Brain Res. 2000, 886, 82–98. [Google Scholar] [CrossRef]
- Feng, L.R.; Maguire-Zeiss, K.A. Gene Therapy in Parkinson’s Disease: Rationale and Current Status. CNS Drugs 2010, 24, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Kaplitt, M.G.; Feigin, A.; Tang, C.; Fitzsimons, H.L.; Mattis, P.; Lawlor, P.A.; Bland, R.J.; Young, D.; Strybing, K.; Eidelberg, D.; et al. Safety and Tolerability of Gene Therapy with an Adeno-Associated Virus (AAV) Borne GAD Gene for Parkinson’s Disease: An Open Label, Phase I Trial. Lancet 2007, 369, 2097–2105. [Google Scholar] [CrossRef]
- Lewitt, P.A.; Rezai, A.R.; Leehey, M.A.; Ojemann, S.G.; Flaherty, A.W.; Eskandar, E.N.; Kostyk, S.K.; Thomas, K.; Sarkar, A.; Siddiqui, M.S.; et al. AAV2-GAD Gene Therapy for Advanced Parkinson’s Disease: A Double-Blind, Sham-Surgery Controlled, Randomised Trial. Lancet Neurol. 2011, 10, 309–319. [Google Scholar] [CrossRef]
- Christine, C.W.; Starr, P.A.; Larson, P.S.; Eberling, J.L.; Jagust, W.J.; Hawkins, R.A.; Vanbrocklin, H.F.; Wright, J.F.; Bankiewicz, K.S.; Aminoff, M.J. Safety and Tolerability of Putaminal Aadc Gene Therapy for Parkinson Disease. Neurology 2009, 73, 1662–1669. [Google Scholar] [CrossRef] [PubMed]
- Eberling, J.L.; Jagust, W.J.; Christine, C.W.; Starr, P.; Larson, P.; Bankiewicz, K.S.; Aminoff, M.J. Results from a Phase I Safety Trial of Haadc Gene Therapy for Parkinson Disease. Neurology 2008, 70, 1980–1983. [Google Scholar] [CrossRef] [PubMed]
- Jarraya, B.; Boulet, S.; Ralph, G.S.; Jan, C.; Bonvento, G.; Azzouz, M.; Miskin, J.E.; Shin, M.; Delzescaux, T.; Drouot, X.; et al. Dopamine Gene Therapy for Parkinson’s Disease in a Nonhuman Primate without Associated Dyskinesia. Sci. Transl. Med. 2009, 1, 2ra4. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.E.; Mikulis, D. A New Sensitive Imaging Biomarker for Parkinson Disease? Neurology 2009, 72, 1374–1375. [Google Scholar] [CrossRef] [PubMed]
- Rascol, O. “Disease-Modification” Trials in Parkinson Disease: Target Populations, Endpoints and Study Design. Neurology 2009, 72, S51–S58. [Google Scholar] [CrossRef] [PubMed]
- Ansari, K.A.; Johnson, A. Olfactory Function in Patients with Parkinson’s Disease. J. Chronic Dis. 1975, 28, 493–497. [Google Scholar] [CrossRef]
- Haehner, A.; Hummel, T.; Hummel, C.; Sommer, U.; Junghanns, S.; Reichmann, H. Olfactory Loss May Be a First Sign of Idiopathic Parkinson’s Disease. Mov. Disord. 2007, 22, 839–842. [Google Scholar] [CrossRef] [PubMed]
- Haehner, A.; Hummel, T.; Reichmann, H. Olfactory Dysfunction As a Diagnostic Marker for Parkinson’s Disease. Expert Rev. Neurother. 2009, 9, 1773–1779. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.F.; Duda, J.E. Olfaction As a Biomarker in Parkinson’s Disease. Biomark. Med. 2010, 4, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Doty, R.L.; Deems, D.A.; Stellar, S. Olfactory Dysfunction in Parkinsonism: A General Deficit Unrelated to Neurologic Signs, Disease Stage, or Disease Duration. Neurology 1988, 38, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.; Shah, M.; Findley, L. Olfactory Function in Essential Tremor: A Deficit Unrelated to Disease Duration or Severity. Neurology 2003, 61, 871–872. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.H.; Shephard, B.C.; Daniel, S.E. Olfactory Dysfunction in Parkinson’s Disease. J. Neurol. Neurosurg. Psychiatry 1997, 62, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Stern, M.B. The Preclinical Detection of Parkinson’s Disease: Ready for Prime Time? Ann. Neurol. 2004, 56, 169–171. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.; Mungersdorf, M.; Reichmann, H.; Strehle, G.; Hummel, T. Olfactory Function in Parkinsonian Syndromes. J. Clin. Neurosci. 2002, 9, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; You, H.; Liu, J.F.; Ni, D.F.; Zhang, Z.X.; Guan, J. Association of Olfactory Bulb Volume and Olfactory Sulcus Depth with Olfactory Function in Patients with Parkinson Disease. AJNR Am. J. Neuroradiol. 2011, 32, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Witt, M.; Bormann, K.; Gudziol, V.; Pehlke, K.; Barth, K.; Minovi, A.; Hahner, A.; Reichmann, H.; Hummel, T. Biopsies of Olfactory Epithelium in Patients with Parkinson’s Disease. Mov. Disord. 2009, 24, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Huisman, E.; Uylings, H.B.; Hoogland, P.V. A 100% Increase of Dopaminergic Cells in the Olfactory Bulb May Explain Hyposmia in Parkinson’s Disease. Mov. Disord. 2004, 19, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Ross, G.W.; Abbott, R.D.; Petrovitch, H.; White, L.R.; Tanner, C.M. Relationship between Caffeine Intake and Parkinson Disease. JAMA 2000, 284, 1378–1379. [Google Scholar] [PubMed]
- Hummel, T.; Sekinger, B.; Wolf, S.R.; Pauli, E.; Kobal, G. ‘Sniffin’ Sticks’: Olfactory Performance Assessed by the Combined Testing of Odor Identification, Odor Discrimination and Olfactory Threshold. Chem. Senses 1997, 22, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Wenning, G.K.; Shephard, B.; Hawkes, C.; Petruckevitch, A.; Lees, A.; Quinn, N. Olfactory Function in Atypical Parkinsonian Syndromes. Acta Neurol. Scand. 1995, 91, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Mckinnon, J.H.; Demaerschalk, B.M.; Caviness, J.N.; Wellik, K.E.; Adler, C.H.; Wingerchuk, D.M. Sniffing out Parkinson Disease: Can Olfactory Testing Differentiate Parkinsonian Disorders? Neurologist 2007, 13, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S.; Sewell, L. Olfactory Dysfunction in Pure Autonomic Failure: Implications for the Pathogenesis of Lewy Body Diseases. Parkinsonism Relat. Disord. 2009, 15, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C. Olfaction in Neurodegenerative Disorder. Mov. Disord. 2003, 18, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Liberini, P.; Parola, S.; Spano, P.F.; Antonini, L. Olfaction in Parkinson’s Disease: Methods of Assessment and Clinical Relevance. J. Neurol. 2000, 247, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.S.; Williams, J.; Combrinck, M.; Christie, S.; Smith, A.D.; Mcshane, R. Olfactory Impairment Is More Marked in Patients with Mild Dementia with Lewy Bodies Than Those with Mild Alzheimer Disease. J. Neurol. Neurosurg. Psychiatry 2009, 80, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Marek, K.; Jennings, D. Can We Image Premotor Parkinson Disease? Neurology 2009, 72, S21–S26. [Google Scholar] [CrossRef] [PubMed]
- Piccini, P.; Whone, A. Functional Brain Imaging in the Differential Diagnosis of Parkinson’s Disease. Lancet Neurol. 2004, 3, 284–290. [Google Scholar] [CrossRef]
- Kagi, G.; Bhatia, K.P.; Tolosa, E. The Role of Dat-SPECT in Movement Disorders. J. Neurol. Neurosurg. Psychiatry 2010, 81, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W. The Parkinson’s Complex: Parkinsonism Is Just the Tip of the Iceberg. Ann. Neurol. 2006, 59, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, E. Movement Disorders: Advances on Many Fronts. Lancet Neurol. 2007, 6, 7–8. [Google Scholar] [CrossRef]
- Tolosa, E.; Compta, Y.; Gaig, C. The Premotor Phase of Parkinson’s Disease. Parkinsonism Relat. Disord. 2007, 13, S2–S7. [Google Scholar] [CrossRef] [PubMed]
- Ponsen, M.M.; Stoffers, D.; Booij, J.; van Eck-Smit, B.L.; Wolters, E.; Berendse, H.W. Idiopathic Hyposmia As a Preclinical Sign of Parkinson’s Disease. Ann. Neurol. 2004, 56, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Stiasny-Kolster, K.; Doerr, Y.; Moller, J.C.; Hoffken, H.; Behr, T.M.; Oertel, W.H.; Mayer, G. Combination Of ‘Idiopathic’ Rem Sleep Behaviour Disorder and Olfactory Dysfunction As Possible Indicator for Alpha-Synucleinopathy Demonstrated by Dopamine Transporter Fp-Cit-SPECT. Brain 2005, 128, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In Vivo Imaging of Microglial Activation with [11c](R)-Pk11195 PET in Idiopathic Parkinson’s Disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s Disease: A Target for Neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial Activation and Dopamine Terminal Loss in Early Parkinson’s Disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.A.; Barker, R.A. The Search for Biomarkers in Parkinson’s Disease: A Critical Review. Expert Rev. Neurother. 2008, 8, 1841–1852. [Google Scholar] [CrossRef] [PubMed]
- Caudle, W.M.; Bammler, T.K.; Lin, Y.; Pan, S.; Zhang, J. Using ‘Omics’ to Define Pathogenesis and Biomarkers of Parkinson’s Disease. Expert Rev. Neurother. 2010, 10, 925–942. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. ‘Metabonomics’: Understanding the Metabolic Responses of Living Systems to Pathophysiological Stimuli Via Multivariate Statistical Analysis of Biological Nmr Spectroscopic Data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Wilson, I.D. Opinion: Understanding ‘Global’ Systems Biology: Metabonomics and the Continuum of Metabolism. Nat. Rev. Drug Discov. 2003, 2, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.; Tsang, T.M.; Tabrizi, S.J. The Application of Nmr-Based Metabonomics in Neurological Disorders. Neurorx 2006, 3, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Michell, A.W.; Luheshi, L.M.; Barker, R.A. Skin and Platelet Alpha-Synuclein As Peripheral Biomarkers of Parkinson’s Disease. Neurosci. Lett. 2005, 381, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Michell, A.W.; Mosedale, D.; Grainger, D.J.; Barker, R.A. Metabolomic Analysis of Urine and Serum in Parkinson’s Disease. Metabolomics 2008, 4, 10. [Google Scholar] [CrossRef]
- Bogdanov, M.; Matson, W.R.; Wang, L.; Matson, T.; Saunders-Pullman, R.; Bressman, S.S.; Flint Beal, M. Metabolomic Profiling to Develop Blood Biomarkers for Parkinson’s Disease. Brain 2008, 131, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Brundin, P.; Strecker, R.E.; Lindvall, O.; Isacson, O.; Nilsson, O.G.; Barbin, G.; Prochiantz, A.; Forni, C.; Nieoullon, A.; Widner, H.; et al. Intracerebral Grafting of Dopamine Neurons. Experimental Basis for Clinical Trials in Patients with Parkinson’s Disease. Ann. N. Y. Acad. Sci. 1987, 495, 473–496. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.A. Developing Stem Cell Therapies for Parkinson’s Disease: Waiting Until the Time Is Right. Cell Stem Cell 2014, 15, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Madhavan, L.; Daley, B.F.; Paumier, K.L.; Collier, T.J. Transplantation of Subventricular Zone Neural Precursors Induces an Endogenous Precursor Cell Response in a Rat Model of Parkinson’s Disease. J. Comp. Neurol. 2009, 515, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Choong, C.J.; Yasuda, T. The Promises of Stem Cells: Stem Cell Therapy for Movement Disorders. Parkinsonism Relat. Disord. 2014, 20 (Suppl. S1), S128–S131. [Google Scholar] [CrossRef]
- Chung, S.; Moon, J.I.; Leung, A.; Aldrich, D.; Lukianov, S.; Kitayama, Y.; Park, S.; Li, Y.; Bolshakov, V.Y.; Lamonerie, T.; et al. Es Cell-Derived Renewable and Functional Midbrain Dopaminergic Progenitors. Proc. Natl. Acad. Sci. USA 2011, 108, 9703–9708. [Google Scholar] [CrossRef] [PubMed]
- Kirkeby, A.; Grealish, S.; Wolf, D.A.; Nelander, J.; Wood, J.; Lundblad, M.; Lindvall, O.; Parmar, M. Generation of Regionally Specified Neural Progenitors and Functional Neurons from Human Embryonic Stem Cells under Defined Conditions. Cell Rep. 2012, 1, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Kriks, S.; Shim, J.W.; Piao, J.; Ganat, Y.M.; Wakeman, D.R.; Xie, Z.; Carrillo-Reid, L.; Auyeung, G.; Antonacci, C.; Buch, A.; et al. Dopamine Neurons Derived from Human Es Cells Efficiently Engraft in Animal Models of Parkinson’s Disease. Nature 2011, 480, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Caldwell, M.; Song, B. Development of Stem Cell-Based Therapies for Parkinson’s Disease. Int. J. Neurosci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lindvall, O.; Kokaia, Z. Prospects of Stem Cell Therapy for Replacing Dopamine Neurons in Parkinson’s Disease. Trends Pharmacol. Sci. 2009, 30, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Morales, P.L.; Revilla, A.; Ocana, I.; Gonzalez, C.; Sainz, P.; Mcguire, D.; Liste, I. Progress in Stem Cell Therapy for Major Human Neurological Disorders. Stem Cell Rev. 2013, 9, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Rhee, Y.H.; Ko, J.Y.; Chang, M.Y.; Yi, S.H.; Kim, D.; Kim, C.H.; Shim, J.W.; Jo, A.Y.; Kim, B.W.; Lee, H.; et al. Protein-Based Human Ips Cells Efficiently Generate Functional Dopamine Neurons and Can Treat a Rat Model of Parkinson Disease. J. Clin. Investig. 2011, 121, 2326–2335. [Google Scholar] [CrossRef] [PubMed]
- Fricker-Gates, R.A.; Gates, M.A. Stem Cell-Derived Dopamine Neurons for Brain Repair in Parkinson’s Disease. Regen. Med. 2010, 5, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Foldes, A.; Kadar, K.; Keremi, B.; Zsembery, A.; Gyires, K.; Zadori, Z.S.; Varga, G. Mesenchymal Stem Cells of Dental Origin—Their Potential for Anti-Inflammatory and Regenerative Actions in Brain and Gut Damage. Curr. Neuropharmacol. 2016, 14. [Google Scholar] [CrossRef]
- Pollock, K.; Dahlenburg, H.; Nelson, H.; Fink, K.D.; Cary, W.; Hendrix, K.; Annett, G.; Torrest, A.; Deng, P.; Gutierrez, J.; et al. Human Mesenchymal Stem Cells Genetically Engineered to Overexpress Brain-Derived Neurotrophic Factor Improve Outcomes in Huntington’s Disease Mouse Models. Mol. Ther. 2016, 24, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Glavaski-Joksimovic, A.; Bohn, M.C. Mesenchymal Stem Cells and Neuroregeneration in Parkinson’s Disease. Exp. Neurol. 2013, 247, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Offen, D.; Barhum, Y.; Levy, Y.S.; Burshtein, A.; Panet, H.; Cherlow, T.; Melamed, E. Intrastriatal Transplantation of Mouse Bone Marrow-Derived Stem Cells Improves Motor Behavior in a Mouse Model of Parkinson’s Disease. J. Neural. Transm. Suppl. 2007, 72, 133–143. [Google Scholar] [PubMed]
- Mathieu, P.; Roca, V.; Gamba, C.; del Pozo, A.; Pitossi, F. Neuroprotective Effects of Human Umbilical Cord Mesenchymal Stromal Cells in an Immunocompetent Animal Model of Parkinson’s Disease. J. Neuroimmunol. 2012, 246, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Xiong, N.; Zhang, Z.; Huang, J.; Chen, C.; Zhang, Z.; Jia, M.; Xiong, J.; Liu, X.; Wang, F.; Cao, X.; et al. Vegf-Expressing Human Umbilical Cord Mesenchymal Stem Cells, an Improved Therapy Strategy for Parkinson’s Disease. Gene Ther. 2011, 18, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.L.; Zhang, C.W. Molecular Events Underlying Parkinson’s Disease—An Interwoven Tapestry. Front. Neurol. 2013, 4, 33. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| PD Pathogenic Mechanism | Targets for Neuroprotection |
|---|---|
| Oxidative stress and mitochondrial dysfunction | Inhibitors of dopamine metabolism (e.g., MAO inhibitors, dopamine receptor agonists) |
| Electron transport enhancers (e.g., CoQ10) | |
| Other Oxidants (e.g., Vitamin E, Uric acid) | |
| Glutathione promoters (e.g., selenium) | |
| Inhibitors of a-synuclein aggregation | |
| Therapeutic agents that reduce α-synuclein protein levels | |
| Enhancers of parkin function | |
| Enhancers of UCH-L1 function | |
| Enhancers of proteosomal or lysosomal pathways | |
| Protein aggregation and misfolding | Anti-inflammatory agents (e.g., NSAID, statins, minocycline) |
| NMDA receptor antagonists, Calcium channel antagonists | |
| Anti-apoptotic agents | |
| Neuroinflammation | Neurotrpohic factors (e.g., GDNF, neurturin) |
| Excitotoxicity | |
| Apoptosis and cell death pathways | |
| Loss of trophic factors |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarkar, S.; Raymick, J.; Imam, S. Neuroprotective and Therapeutic Strategies against Parkinson’s Disease: Recent Perspectives. Int. J. Mol. Sci. 2016, 17, 904. https://doi.org/10.3390/ijms17060904
Sarkar S, Raymick J, Imam S. Neuroprotective and Therapeutic Strategies against Parkinson’s Disease: Recent Perspectives. International Journal of Molecular Sciences. 2016; 17(6):904. https://doi.org/10.3390/ijms17060904
Chicago/Turabian StyleSarkar, Sumit, James Raymick, and Syed Imam. 2016. "Neuroprotective and Therapeutic Strategies against Parkinson’s Disease: Recent Perspectives" International Journal of Molecular Sciences 17, no. 6: 904. https://doi.org/10.3390/ijms17060904
APA StyleSarkar, S., Raymick, J., & Imam, S. (2016). Neuroprotective and Therapeutic Strategies against Parkinson’s Disease: Recent Perspectives. International Journal of Molecular Sciences, 17(6), 904. https://doi.org/10.3390/ijms17060904