
Endoplasmic Reticulum Stress and Associated ROS


Abstract
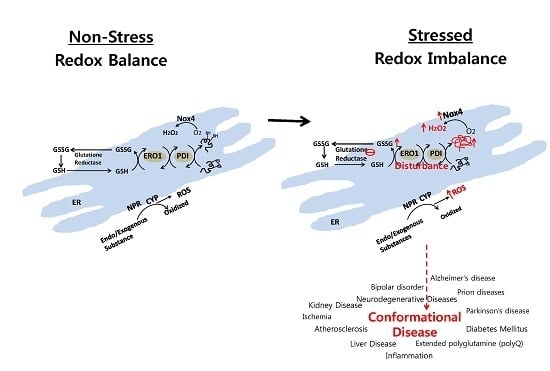
:
{kind=link}
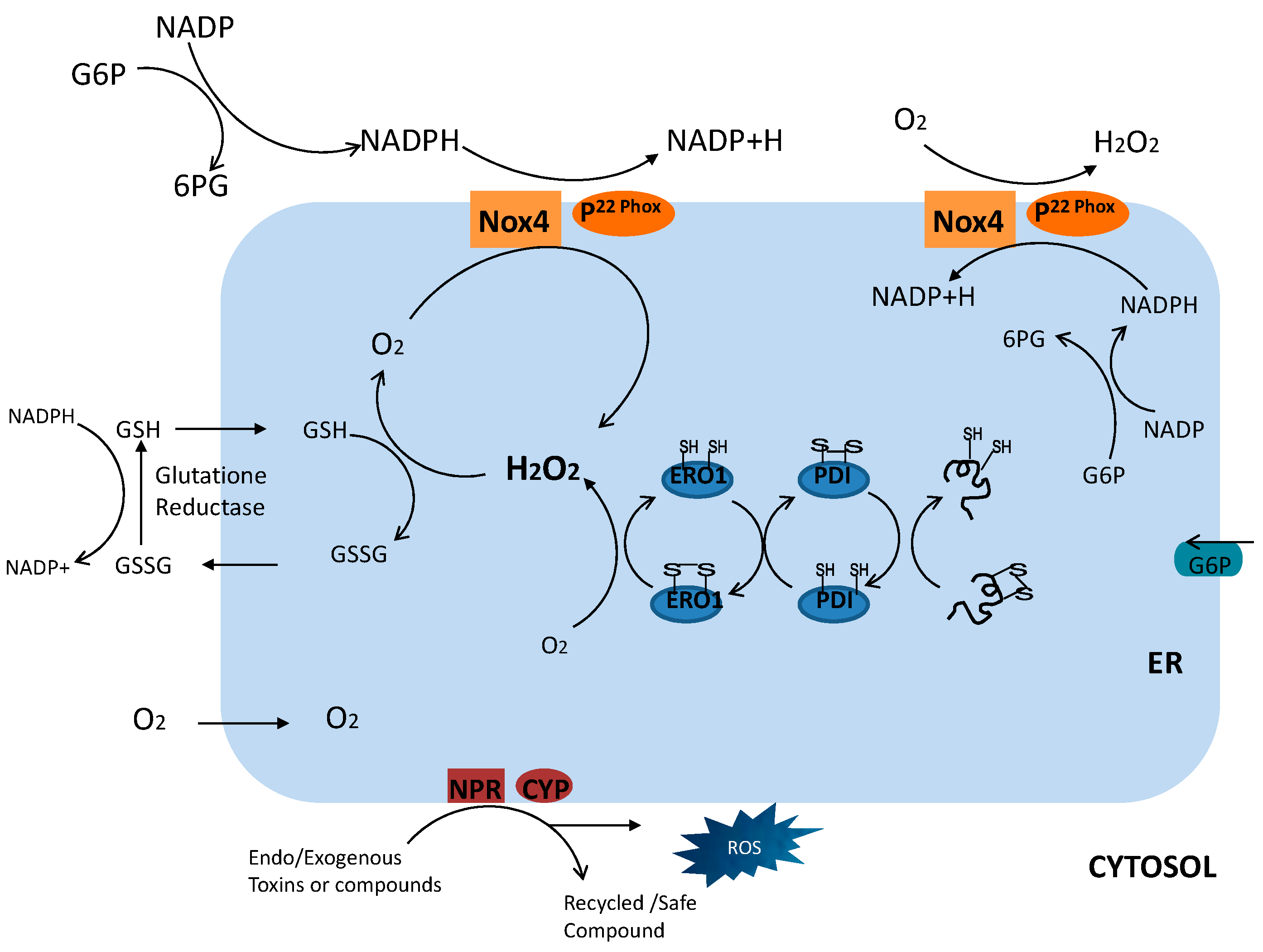{kind=link}
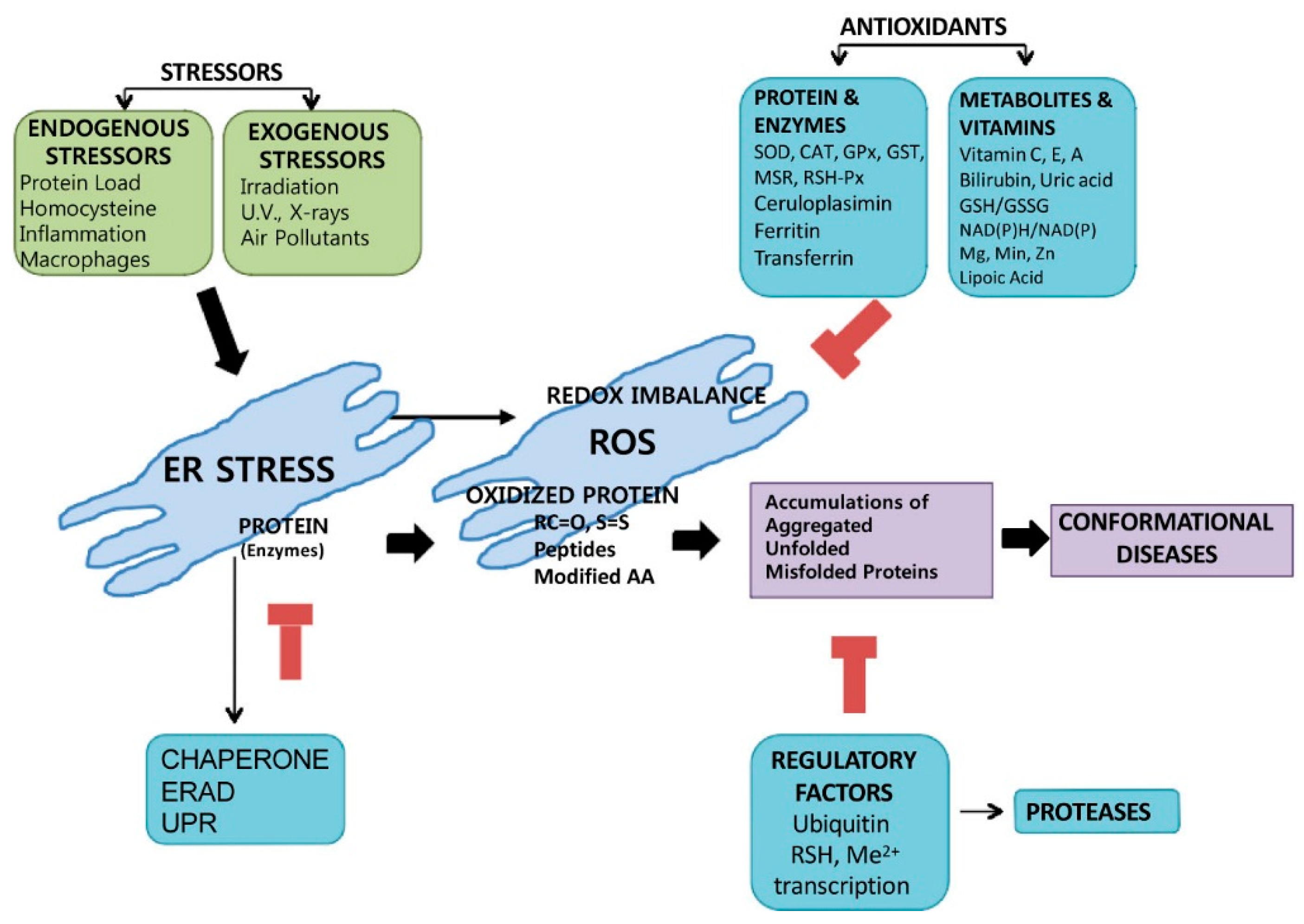{kind=link}
1. Introduction
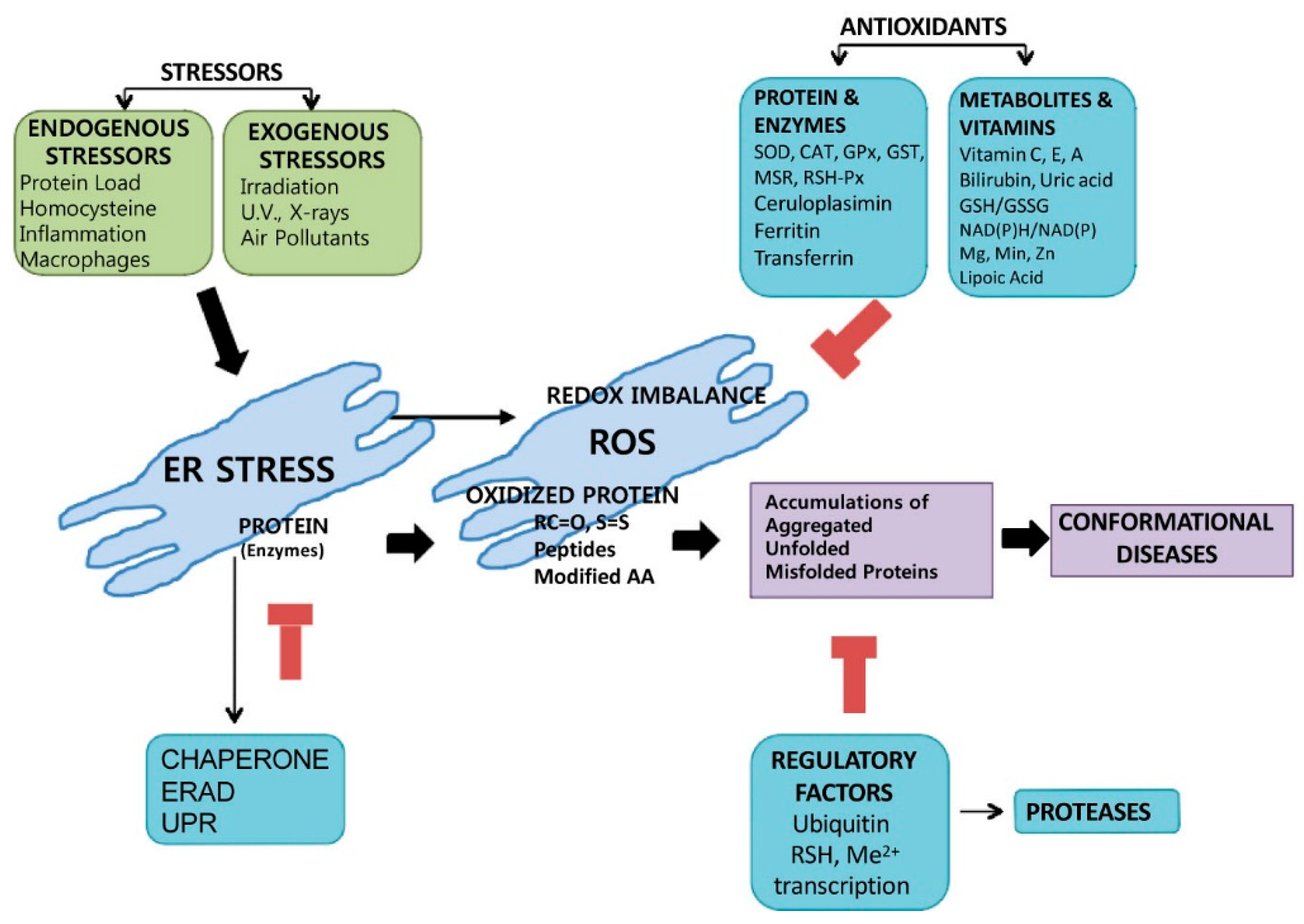
2. How Is Reactive Oxygen Species (ROS) Induced through Endoplasmic Reticulum (ER) Stress?
The Specific Mechanism of ER Stress-Induced ROS during the ER Folding Process
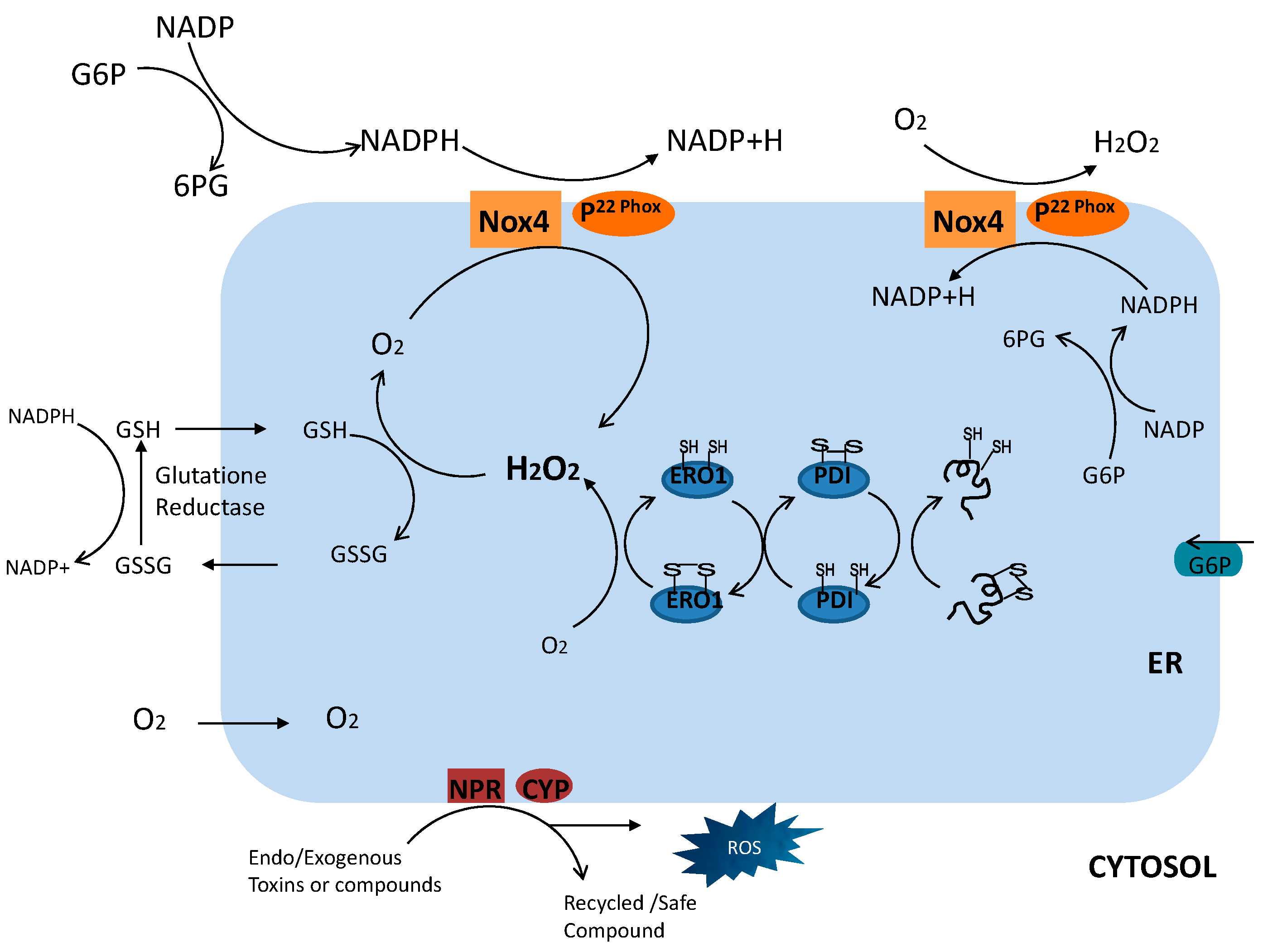
3. Specific Mechanism of ER Stress-Induced ROS: NADPH Oxidase 4 (Nox4)
4. Coupled Glutathione within the ER
5. NADPH-Dependent p450 Reductase and p450 Connection Involvement in ER Stress
6. ER and Mitochondria Connection and Relationship to ROS
7. Disease Application
7.1. ER Stress and Diseases
7.1.1. Neurodegenerative Diseases
7.1.2. Diabetes Mellitus
7.1.3. Atherosclerosis
7.1.4. Inflammation
7.1.5. Liver Disease
7.1.6. Ischemia
7.1.7. Kidney Disease
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.Y.; Chang, C.C.; Ohgami, N.; Yamauchi, Y. Cholesterol sensing, trafficking, and esterification. Annu. Rev. Cell Dev. Biol. 2006, 22, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Hampton, R.Y. ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 2002, 14, 476–482. [Google Scholar] [CrossRef]
- Duncan, M.J.; Stanley, M.; Parkhouse, N.; Cook, K.; Smith, M. Acute caffeine ingestion enhances strength performance and reduces perceived exertion and muscle pain perception during resistance exercise. Eur. J. Sport Sci. 2013, 13, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Nagata, K. Functional in vitro analysis of the ERO1 protein and protein-disulfide isomerase pathway. J. Biol. Chem. 2011, 286, 32705–32712. [Google Scholar] [CrossRef] [PubMed]
- Scriven, P.; Brown, N.J.; Pockley, A.G.; Wyld, L. The unfolded protein response and cancer: A brighter future unfolding? J. Mol. Med. 2007, 85, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Gardner, B.M.; Walter, P. Unfolded proteins are IRE1-activating ligands that directly induce the unfolded protein response. Science 2011, 333, 1891–1894. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.; Verchot, J.; Dickman, M.B. When supply does not meet demand-ER stress and plant programmed cell death. Front. Plant Sci. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Shamu, C.E.; Walter, P. Oligomerization and phosphorylation of the IRE1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 1996, 15, 3028–3039. [Google Scholar] [PubMed]
- Hassler, J.; Cao, S.S.; Kaufman, R.J. IRE1, a double-edged sword in pre-miRNA slicing and cell death. Dev. Cell 2012, 23, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated IRE1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Lei, K.; Davis, R.J. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 2432–2437. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Xiao, L.; Lang, W.; Gao, F.; Ruvolo, P.; May, W.S., Jr. Novel role for JNK as a stress-activated Bcl2 kinase. J. Biol. Chem. 2001, 276, 23681–23688. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Schuck, S.; Prinz, W.A.; Thorn, K.S.; Voss, C.; Walter, P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol. 2009, 187, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.; O’Hare, P. Transmembrane bZIP transcription factors in ER stress signaling and the unfolded protein response. Antioxid. Redox Signal. 2007, 9, 2305–2321. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.; Xie, J.; Zhang, D.; Han, Y.; Wang, C. Polypeptide from Chlamys farreri suppresses ultraviolet-B irradiation-induced apoptosis through restoring ER redox homeostasis, scavenging ROS generation, and suppressing the PERK-eIF2α-CHOP pathway in HaCaT cells. J. Photochem. Photobiol. B 2015, 151, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Ellgaard, L.; Ruddock, L.W. The human protein disulphide isomerase family: Substrate interactions and functional properties. EMBO Rep. 2005, 6, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef] [PubMed]
- Kramer, B.; Ferrari, D.M.; Klappa, P.; Pohlmann, N.; Soling, H.D. Functional roles and efficiencies of the thioredoxin boxes of calcium-binding proteins 1 and 2 in protein folding. Biochem. J. 2001, 357, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Carelli, S.; Ceriotti, A.; Cabibbo, A.; Fassina, G.; Ruvo, M.; Sitia, R. Cysteine and glutathione secretion in response to protein disulfide bond formation in the ER. Science 1997, 277, 1681–1684. [Google Scholar] [CrossRef] [PubMed]
- Frand, A.R.; Kaiser, C.A. The ERO1 gene of yeast is required for oxidation of protein dithiols in the endoplasmic reticulum. Mol. Cell 1998, 1, 161–170. [Google Scholar] [CrossRef]
- Pollard, M.G.; Travers, K.J.; Weissman, J.S. ERO1p: A novel and ubiquitous protein with an essential role in oxidative protein folding in the endoplasmic reticulum. Mol. Cell 1998, 1, 171–182. [Google Scholar] [CrossRef]
- Hatahet, F.; Ruddock, L.W. Substrate recognition by the protein disulfide isomerases. FEBS J. 2007, 274, 5223–5234. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, W.; Ren, J.; Fang, J.; Ke, H.; Gong, W.; Feng, W.; Wang, C.C. Structural insights into the redox-regulated dynamic conformations of human protein disulfide isomerase. Antioxid. Redox Signal. 2013, 19, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yu, J.; Huo, L.; Wang, L.; Feng, W.; Wang, C.C. Human protein-disulfide isomerase is a redox-regulated chaperone activated by oxidation of domain a’. J. Biol. Chem. 2012, 287, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Wilkinson, B.; Solovyov, A.; Winther, J.R.; Holmgren, A.; Lundstrom-Ljung, J.; Gilbert, H.F. The contributions of protein disulfide isomerase and its homologues to oxidative protein folding in the yeast endoplasmic reticulum. J. Biol. Chem. 2004, 279, 49780–49786. [Google Scholar] [CrossRef] [PubMed]
- Laurindo, F.R.; Pescatore, L.A.; Fernandes Dde, C. Protein disulfide isomerase in redox cell signaling and homeostasis. Free Radic. Biol. Med. 2012, 52, 1954–1969. [Google Scholar] [CrossRef] [PubMed]
- Tu, B.P.; Weissman, J.S. Oxidative protein folding in eukaryotes: Mechanisms and consequences. J. Cell Biol. 2004, 164, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Kleniewska, P.; Piechota, A.; Skibska, B.; Goraca, A. The NADPH oxidase family and its inhibitors. Arch. Immunol. Ther. Exp. 2012, 60, 277–294. [Google Scholar] [CrossRef] [PubMed]
- Anilkumar, N.; San Jose, G.; Sawyer, I.; Santos, C.X.; Sand, C.; Brewer, A.C.; Warren, D.; Shah, A.M. A 28-kDa splice variant of NADPH oxidase-4 is nuclear-localized and involved in redox signaling in vascular cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, e104–e112. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Lardy, B.; Krause, K.H. Nox family NADPH oxidases: Not just in mammals. Biochimie 2007, 89, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y.; Jackson, H.M.; Ogawa, H.; Kawahara, T.; Lambeth, J.D. Constitutive NADPH-dependent electron transferase activity of the Nox4 dehydrogenase domain. Biochemistry 2010, 49, 2433–2442. [Google Scholar] [CrossRef] [PubMed]
- Janiszewski, M.; Lopes, L.R.; Carmo, A.O.; Pedro, M.A.; Brandes, R.P.; Santos, C.X.; Laurindo, F.R. Regulation of NAD(P)H oxidase by associated protein disulfide isomerase in vascular smooth muscle cells. J. Biol. Chem. 2005, 280, 40813–40819. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.X.; Stolf, B.S.; Takemoto, P.V.; Amanso, A.M.; Lopes, L.R.; Souza, E.B.; Goto, H.; Laurindo, F.R. Protein disulfide isomerase (PDI) associates with NADPH oxidase and is required for phagocytosis of leishmania chagasi promastigotes by macrophages. J. Leukoc. Biol. 2009, 86, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.X.; Tanaka, L.Y.; Wosniak, J.; Laurindo, F.R. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: Roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid. Redox Signal. 2009, 11, 2409–2427. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.X.; Nabeebaccus, A.A.; Shah, A.M.; Camargo, L.L.; Filho, S.V.; Lopes, L.R. Endoplasmic reticulum stress and Nox-mediated reactive oxygen species signaling in the peripheral vasculature: Potential role in hypertension. Antioxid. Redox Signal. 2014, 20, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Merksamer, P.I.; Trusina, A.; Papa, F.R. Real-time redox measurements during endoplasmic reticulum stress reveal interlinked protein folding functions. Cell 2008, 135, 933–947. [Google Scholar] [CrossRef] [PubMed]
- Pedruzzi, E.; Guichard, C.; Ollivier, V.; Driss, F.; Fay, M.; Prunet, C.; Marie, J.C.; Pouzet, C.; Samadi, M.; Elbim, C.; et al. NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol. Cell. Biol. 2004, 24, 10703–10717. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.F.; Ma, Z.; Liu, Z.; Terada, L.S. Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local Ras activation. Mol. Cell. Biol. 2010, 30, 3553–3568. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Titus, E.A.; Cooper, A.A. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 2004, 15, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Miao, H.; Zhang, K.; Wolfson, A.; Pennathur, S.; Pipe, S.W.; Kaufman, R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA 2008, 105, 18525–18530. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, D.A.; Forman, H.J. Glutathione in defense and signaling: Lessons from a small thiol. Ann. N. Y. Acad. Sci. 2002, 973, 488–504. [Google Scholar] [CrossRef] [PubMed]
- Tu, B.P.; Weissman, J.S. The FAD- and O2-dependent reaction cycle of ERO1-mediated oxidative protein folding in the endoplasmic reticulum. Mol. Cell 2002, 10, 983–994. [Google Scholar] [CrossRef]
- Chakravarthi, S.; Bulleid, N.J. Glutathione is required to regulate the formation of native disulfide bonds within proteins entering the secretory pathway. J. Biol. Chem. 2004, 279, 39872–39879. [Google Scholar] [CrossRef] [PubMed]
- Sitia, R.; Molteni, S.N. Stress, protein (mis)folding, and signaling: The redox connection. Sci. STKE 2004, 2004. [Google Scholar] [CrossRef] [PubMed]
- Reimann, D.; Dachs, D.; Meye, C.; Gross, P. Amino acid-based peritoneal dialysis solution stimulates mesothelial nitric oxide production. Perit. Dial. Int. 2004, 24, 378–384. [Google Scholar] [PubMed]
- Zhang, K.; Kaufman, R.J. Signaling the unfolded protein response from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 25935–25938. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [PubMed]
- Enoch, H.G.; Strittmatter, P. Cytochrome b5 reduction by NADPH-cytochrome P-450 reductase. J. Biol. Chem. 1979, 254, 8976–8981. [Google Scholar] [PubMed]
- Strittmatter, P.; Spatz, L.; Corcoran, D.; Rogers, M.J.; Setlow, B.; Redline, R. Purification and properties of rat liver microsomal stearyl coenzyme a desaturase. Proc. Natl. Acad. Sci. USA 1974, 71, 4565–4569. [Google Scholar] [CrossRef] [PubMed]
- Davydov, D.R. Microsomal monooxygenase in apoptosis: Another target for cytochrome c signaling? Trends Biochem. Sci. 2001, 26, 155–160. [Google Scholar] [CrossRef]
- Nieto, N.; Friedman, S.L.; Cederbaum, A.I. Stimulation and proliferation of primary rat hepatic stellate cells by cytochrome p450 2E1-derived reactive oxygen species. Hepatology 2002, 35, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Jensen, C.B.; Martin-Gronert, M.S.; Storgaard, H.; Madsbad, S.; Vaag, A.; Ozanne, S.E. Altered PI3-kinase/Akt signalling in skeletal muscle of young men with low birth weight. PLoS ONE 2008, 3, e3738. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Moreau, R.; Pessayre, D.; Epperson, T.K.; Krause, K.H. Nox family NADPH oxidases in liver and in pancreatic islets: A role in the metabolic syndrome and diabetes? Biochem. Soc. Trans. 2008, 36, 920–929. [Google Scholar] [CrossRef] [PubMed]
- Gouillon, Z.; Lucas, D.; Li, J.; Hagbjork, A.L.; French, B.A.; Fu, P.; Fang, C.; Ingelman-Sundberg, M.; Donohue, T.M., Jr.; French, S.W. Inhibition of ethanol-induced liver disease in the intragastric feeding rat model by chlormethiazole. Proc. Soc. Exp. Biol. Med. 2000, 224, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Chtioui, H.; Semela, D.; Ledermann, M.; Zimmermann, A.; Dufour, J.F. Expression and activity of the cytochrome p450 2E1 in patients with nonalcoholic steatosis and steatohepatitis. Liver Int. 2007, 27, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Cederbaum, A.I. Nrf2 is increased by CYP2E1 in rodent liver and HepG2 cells and protects against oxidative stress caused by CYP2E1. Hepatology 2006, 43, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Lee, G.H.; Cho, E.Y.; Chae, S.W.; Ahn, T.; Chae, H.J. Bax inhibitor 1 regulates ER-stress-induced ROS accumulation through the regulation of cytochrome p450 2E1. J. Cell Sci. 2009, 122, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Gerasimenko, J.V.; Gerasimenko, O.V.; Palejwala, A.; Tepikin, A.V.; Petersen, O.H.; Watson, A.J. Menadione-induced apoptosis: Roles of cytosolic Ca2+ elevations and the mitochondrial permeability transition pore. J. Cell Sci. 2002, 115, 485–497. [Google Scholar] [PubMed]
- Li, G.; Mongillo, M.; Chin, K.T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1-α-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, D.L. Redox regulation of endothelial canonical transient receptor potential channels. Antioxid. Redox Signal. 2011, 15, 1567–1582. [Google Scholar] [CrossRef] [PubMed]
- Luciani, D.S.; Gwiazda, K.S.; Yang, T.L.; Kalynyak, T.B.; Bychkivska, Y.; Frey, M.H.; Jeffrey, K.D.; Sampaio, A.V.; Underhill, T.M.; Johnson, J.D. Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and β-cell death. Diabetes 2009, 58, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Lock, J.T.; Sinkins, W.G.; Schilling, W.P. Protein S-glutathionylation enhances Ca2+-induced Ca2+ release via the IP3 receptor in cultured aortic endothelial cells. J. Physiol. 2012, 590, 3431–3447. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki-Mann, M.; Demuro, A.; Parker, I. Modulation of endoplasmic reticulum Ca2+ store filling by cyclic ADP-ribose promotes inositol trisphosphate (IP3)-evoked Ca2+ signals. J. Biol. Chem. 2010, 285, 25053–25061. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, J.; Duchen, M.R. Mitochondrial oxidative stress and cell death in astrocytes—Requirement for stored Ca2+ and sustained opening of the permeability transition pore. J. Cell Sci. 2002, 115, 1175–1188. [Google Scholar] [PubMed]
- Okabe, E.; Tsujimoto, Y.; Kobayashi, Y. Calmodulin and cyclic ADP-ribose interaction in Ca2+ signaling related to cardiac sarcoplasmic reticulum: Superoxide anion radical-triggered Ca2+ release. Antioxid. Redox Signal. 2000, 2, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, A.; Klappa, P.; Kietzmann, T. The endoplasmic reticulum: Folding, calcium homeostasis, signaling, and redox control. Antioxid. Redox Signal. 2006, 8, 1391–1418. [Google Scholar] [CrossRef] [PubMed]
- Meares, G.P.; Hughes, K.J.; Naatz, A.; Papa, F.R.; Urano, F.; Hansen, P.A.; Benveniste, E.N.; Corbett, J.A. IRE1-dependent activation of AMPK in response to nitric oxide. Mol. Cell. Biol. 2011, 31, 4286–4297. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Liu, L.; Charles, I.G.; Moncada, S. Nitric oxide induces coupling of mitochondrial signalling with the endoplasmic reticulum stress response. Nat. Cell Biol. 2004, 6, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jekabsone, A.; Ivanoviene, L.; Brown, G.C.; Borutaite, V. Nitric oxide and calcium together inactivate mitochondrial complex I and induce cytochrome c release. J. Mol. Cell. Cardiol. 2003, 35, 803–809. [Google Scholar] [CrossRef]
- Bhandary, B.; Marahatta, A.; Kim, H.R.; Chae, H.J. An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int. J. Mol. Sci. 2012, 14, 434–456. [Google Scholar] [CrossRef] [PubMed]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Schaffar, G.; Breuer, P.; Boteva, R.; Behrends, C.; Tzvetkov, N.; Strippel, N.; Sakahira, H.; Siegers, K.; Hayer-Hartl, M.; Hartl, F.U. Cellular toxicity of polyglutamine expansion proteins: Mechanism of transcription factor deactivation. Mol. Cell 2004, 15, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- Behrends, C.; Langer, C.A.; Boteva, R.; Bottcher, U.M.; Stemp, M.J.; Schaffar, G.; Rao, B.V.; Giese, A.; Kretzschmar, H.; Siegers, K.; et al. Chaperonin TRiC promotes the assembly of polyQ expansion proteins into nontoxic oligomers. Mol. Cell 2006, 23, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Di, L.J. BRCA1 and estrogen/estrogen receptor in breast cancer: Where they interact? Int. J. Biol. Sci. 2014, 10, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Credle, J.J.; Forcelli, P.A.; Delannoy, M.; Oaks, A.W.; Permaul, E.; Berry, D.L.; Duka, V.; Wills, J.; Sidhu, A. α-synuclein-mediated inhibition of ATF6 processing into COPII vesicles disrupts UPR signaling in Parkinson’s disease. Neurobiol. Dis. 2015, 76, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Gan, M.; Lin, W.L.; Yen, S.H. Nutrient deprivation induces α-synuclein aggregation through endoplasmic reticulum stress response and SREBP2 pathway. Front. Aging Neurosci. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Valdes, P.; Mercado, G.; Vidal, R.L.; Molina, C.; Parsons, G.; Court, F.A.; Martinez, A.; Galleguillos, D.; Armentano, D.; Schneider, B.L.; et al. Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc. Natl. Acad. Sci. USA 2014, 111, 6804–6809. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Omura, S.; Takahashi, Y. Generation of superoxide anions by a glycation reaction in conventional laboratory media. J. Biosci. Bioeng. 2012, 114, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Hashida, K.; Kitao, Y.; Sudo, H.; Awa, Y.; Maeda, S.; Mori, K.; Takahashi, R.; Iinuma, M.; Hori, O. ATF6α promotes astroglial activation and neuronal survival in a chronic mouse model of Parkinson’s disease. PLoS ONE 2012, 7, e47950. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M. Molecular pharmacological studies on the protection mechanism against endoplasmic reticulum stress-induced neurodegenerative disease. Yakugaku Zasshi 2012, 132, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Nomura, Y. Pharmacological studies on neurodegenerative diseases focusing on refolding and degradation of unfolded proteins in the endoplasmic reticulum. Yakugaku Zasshi 2014, 134, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.; van Haastert, E.S.; Nijholt, D.A.; Rozemuller, A.J.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am. J. Pathol. 2009, 174, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Voet, T. Alzheimer’s disease: A protective mutation. Nature 2012, 488, 38–39. [Google Scholar] [CrossRef] [PubMed]
- Culmsee, C.; Landshamer, S. Molecular insights into mechanisms of the cell death program: Role in the progression of neurodegenerative disorders. Curr. Alzheimer Res. 2006, 3, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in app protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Dasari, B.; Prasanthi, J.R.; Marwarha, G.; Singh, B.B.; Ghribi, O. The oxysterol 27-hydroxycholesterol increases β-amyloid and oxidative stress in retinal pigment epithelial cells. BMC Ophthalmol. 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, E.; Baldeiras, I.; Ferreira, I.L.; Costa, R.O.; Rego, A.C.; Pereira, C.F.; Oliveira, C.R. Mitochondrial- and endoplasmic reticulum-associated oxidative stress in Alzheimer’s disease: From pathogenesis to biomarkers. Int. J. Cell Biol. 2012, 2012, 735206. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; de Groof, A.J.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, K.; Yan, M.; Wang, Y.; Zheng, X. Protective effects of galantamine against Aβ-induced PC12 cell apoptosis by preventing mitochondrial dysfunction and endoplasmic reticulum stress. Neurochem. Int. 2010, 57, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Ernesto, C.; Thomas, R.G.; Klauber, M.R.; Schafer, K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C.W.; Pfeiffer, E.; et al. A controlled trial of selegiline, α-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s disease cooperative study. N. Engl. J. Med. 1997, 336, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Devore, E.E.; Grodstein, F.; van Rooij, F.J.; Hofman, A.; Stampfer, M.J.; Witteman, J.C.; Breteler, M.M. Dietary antioxidants and long-term risk of dementia. Arch. Neurol. 2010, 67, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Yamamoto, K.; Takahashi, K.; Kaku, Y.; Uchiyama, M.; Sugiyama, K.; Yamada, M. Treatment of Alzheimer-type dementia with intravenous mecobalamin. Clin. Ther. 1992, 14, 426–437. [Google Scholar] [PubMed]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- McLennan, N.F.; Brennan, P.M.; McNeill, A.; Davies, I.; Fotheringham, A.; Rennison, K.A.; Ritchie, D.; Brannan, F.; Head, M.W.; Ironside, J.W.; et al. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am. J. Pathol. 2004, 165, 227–235. [Google Scholar] [CrossRef]
- Beraldo, F.H.; Soares, I.N.; Goncalves, D.F.; Fan, J.; Thomas, A.A.; Santos, T.G.; Mohammad, A.H.; Roffe, M.; Calder, M.D.; Nikolova, S.; et al. Stress-inducible phosphoprotein 1 has unique cochaperone activity during development and regulates cellular response to ischemia via the prion protein. FASEB J. 2013, 27, 3594–3607. [Google Scholar] [CrossRef] [PubMed]
- Bertuchi, F.R.; Bourgeon, D.M.; Landemberger, M.C.; Martins, V.R.; Cerchiaro, G. PrpC displays an essential protective role from oxidative stress in an astrocyte cell line derived from PrpC knockout mice. Biochem. Biophys. Res. Commun. 2012, 418, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, U.S.; Sonati, T.; Falsig, J.; Reimann, R.R.; Dametto, P.; O’Connor, T.; Li, B.; Lau, A.; Hornemann, S.; Sorce, S.; et al. Prion infections and anti-PrP antibodies trigger converging neurotoxic pathways. PLoS Pathog. 2015, 11, e1004662. [Google Scholar]
- Hetz, C.; Russelakis-Carneiro, M.; Maundrell, K.; Castilla, J.; Soto, C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 2003, 22, 5435–5445. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, H.Y.; Orr, H.T. Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 2000, 23, 217–247. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.M.; Yuan, J. Mechanisms of cell death in polyglutamine expansion diseases. Curr. Opin. Pharmacol. 2004, 4, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef] [PubMed]
- Kouroku, Y.; Fujita, E.; Jimbo, A.; Kikuchi, T.; Yamagata, T.; Momoi, M.Y.; Kominami, E.; Kuida, K.; Sakamaki, K.; Yonehara, S.; et al. Polyglutamine aggregates stimulate ER stress signals and caspase-12 activation. Hum. Mol. Genet. 2002, 11, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Yu, Z.; Dadgar, N.; Varambally, S.; Yu, J.; Chinnaiyan, A.M.; Lieberman, A.P. The unfolded protein response modulates toxicity of the expanded glutamine androgen receptor. J. Biol. Chem. 2005, 280, 21264–21271. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Tanaka, K.; Inoue, K.; Kakizuka, A. Functional atpase activity of p97/valosin-containing protein (VCP) is required for the quality control of endoplasmic reticulum in neuronally differentiated mammalian PC12 cells. J. Biol. Chem. 2002, 277, 47358–47365. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, C.; Ishigaki, S.; Oslowski, C.M.; Fonseca, S.G.; Kato, T.; Urano, F. Valproate, a mood stabilizer, induces WFS1 expression and modulates its interaction with ER stress protein GRP94. PLoS ONE 2009, 4, e4134. [Google Scholar] [CrossRef] [PubMed]
- Bown, C.D.; Wang, J.F.; Chen, B.; Young, L.T. Regulation of ER stress proteins by valproate: Therapeutic implications. Bipolar Disord. 2002, 4, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Pandya, C.D.; Howell, K.R.; Pillai, A. Antioxidants as potential therapeutics for neuropsychiatric disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 46, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, C.; Ishiwata, M.; Nanko, S.; Kunugi, H.; Minabe, Y.; Nakamura, K.; Mori, N.; Fujii, K.; Umekage, T.; Tochigi, M.; et al. Functional polymorphisms of HSPA5: Possible association with bipolar disorder. Biochem. Biophys. Res. Commun. 2005, 336, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, C.; Ishiwata, M.; Umekage, T.; Tochigi, M.; Kohda, K.; Sasaki, T.; Kato, T. Association of the XBP1–116C/G polymorphism with schizophrenia in the japanese population. Psychiatry Clin. Neurosci. 2004, 58, 438–440. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, C.; Iwamoto, K.; Ishiwata, M.; Bundo, M.; Kasahara, T.; Kusumi, I.; Tsujita, T.; Okazaki, Y.; Nanko, S.; Kunugi, H.; et al. Impaired feedback regulation of XBP1 as a genetic risk factor for bipolar disorder. Nat. Genet. 2003, 35, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Kusumi, I.; Masui, T.; Kakiuchi, C.; Suzuki, K.; Akimoto, T.; Hashimoto, R.; Kunugi, H.; Kato, T.; Koyama, T. Relationship between XBP1 genotype and personality traits assessed by TCI and NEO-FFI. Neurosci. Lett. 2005, 391, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Cichon, S.; Buervenich, S.; Kirov, G.; Akula, N.; Dimitrova, A.; Green, E.; Schumacher, J.; Klopp, N.; Becker, T.; Ohlraun, S.; et al. Lack of support for a genetic association of the XBP1 promoter polymorphism with bipolar disorder in probands of european origin. Nat. Genet. 2004, 36, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Andreazza, A.C.; Kauer-Sant’Anna, M.; Frey, B.N.; Bond, D.J.; Kapczinski, F.; Young, L.T.; Yatham, L.N. Oxidative stress markers in bipolar disorder: A meta-analysis. J. Affect. Disord. 2008, 111, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Sun, X.; Xu, L.; Young, L.T.; Wang, J.F. Mood stabilizing drug lithium increases expression of endoplasmic reticulum stress proteins in primary cultured rat cerebral cortical cells. Life Sci. 2006, 78, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Rovira-Llopis, S.; Banuls, C.; Apostolova, N.; Morillas, C.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Is glycemic control modulating endoplasmic reticulum stress in leukocytes of type 2 diabetic patients? Antioxid. Redox Signal. 2014, 21, 1759–1765. [Google Scholar] [CrossRef] [PubMed]
- Papa, F.R. Endoplasmic reticulum stress, pancreatic β-cell degeneration, and diabetes. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zeng, H.; Zhang, Y.; Jungries, R.; Chung, P.; Plesken, H.; Sabatini, D.D.; Ron, D. Diabetes mellitus and exocrine pancreatic dysfunction in PERK−/− mice reveals a role for translational control in secretory cell survival. Mol. Cell 2001, 7, 1153–1163. [Google Scholar] [CrossRef]
- Thomas, S.E.; Dalton, L.E.; Daly, M.L.; Malzer, E.; Marciniak, S.J. Diabetes as a disease of endoplasmic reticulum stress. Diabetes Metab. Res. Rev. 2010, 26, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, D.; Song, B.; McEwen, E.; Liu, C.; Laybutt, R.; Gillespie, P.; Saunders, T.; Bonner-Weir, S.; Kaufman, R.J. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 2001, 7, 1165–1176. [Google Scholar] [CrossRef]
- Lim, S.; Rashid, M.A.; Jang, M.; Kim, Y.; Won, H.; Lee, J.; Woo, J.T.; Kim, Y.S.; Murphy, M.P.; Ali, L.; et al. Mitochondria-targeted antioxidants protect pancreatic β-cells against oxidative stress and improve insulin secretion in glucotoxicity and glucolipotoxicity. Cell. Physiol. Biochem. 2011, 28, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Marre, M.L.; James, E.A.; Piganelli, J.D. β cell ER stress and the implications for immunogenicity in type 1 diabetes. Front. Cell Dev. Biol. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Hossain, G.S.; van Thienen, J.V.; Werstuck, G.H.; Zhou, J.; Sood, S.K.; Dickhout, J.G.; de Koning, A.B.; Tang, D.; Wu, D.; Falk, E.; et al. TDAG51 is induced by homocysteine, promotes detachment-mediated programmed cell death, and contributes to the cevelopment of atherosclerosis in hyperhomocysteinemia. J. Biol. Chem. 2003, 278, 30317–30327. [Google Scholar] [CrossRef] [PubMed]
- Werstuck, G.H.; Lentz, S.R.; Dayal, S.; Hossain, G.S.; Sood, S.K.; Shi, Y.Y.; Zhou, J.; Maeda, N.; Krisans, S.K.; Malinow, M.R.; et al. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J. Clin. Investig. 2001, 107, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Werstuck, G.H.; Lhotak, S.; de Koning, A.B.; Sood, S.K.; Hossain, G.S.; Moller, J.; Ritskes-Hoitinga, M.; Falk, E.; Dayal, S.; et al. Association of multiple cellular stress pathways with accelerated atherosclerosis in hyperhomocysteinemic apolipoprotein E-deficient mice. Circulation 2004, 110, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Gargalovic, P.S.; Gharavi, N.M.; Clark, M.J.; Pagnon, J.; Yang, W.P.; He, A.; Truong, A.; Baruch-Oren, T.; Berliner, J.A.; Kirchgessner, T.G.; et al. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2490–2496. [Google Scholar] [CrossRef] [PubMed]
- Devries-Seimon, T.; Li, Y.; Yao, P.M.; Stone, E.; Wang, Y.; Davis, R.J.; Flavell, R.; Tabas, I. Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. J. Cell Biol. 2005, 171, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Yao, P.M.; Li, Y.; Devlin, C.M.; Zhang, D.; Harding, H.P.; Sweeney, M.; Rong, J.X.; Kuriakose, G.; Fisher, E.A.; et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 2003, 5, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Liang, C.P.; DeVries-Seimon, T.; Ranalletta, M.; Welch, C.L.; Collins-Fletcher, K.; Accili, D.; Tabas, I.; Tall, A.R. Macrophage insulin receptor deficiency increases ER stress-induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metab. 2006, 3, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Schwabe, R.F.; DeVries-Seimon, T.; Yao, P.M.; Gerbod-Giannone, M.C.; Tall, A.R.; Davis, R.J.; Flavell, R.; Brenner, D.A.; Tabas, I. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-α and interleukin-6: Model of NF-κB- and map kinase-dependent inflammation in advanced atherosclerosis. J. Biol. Chem. 2005, 280, 21763–21772. [Google Scholar] [CrossRef] [PubMed]
- Horke, S.; Witte, I.; Wilgenbus, P.; Kruger, M.; Strand, D.; Forstermann, U. Paraoxonase-2 reduces oxidative stress in vascular cells and decreases endoplasmic reticulum stress-induced caspase activation. Circulation 2007, 115, 2055–2064. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, A.; Grijalva, V.R.; Bourquard, N.; Meriwether, D., 3rd; Imaizumi, S.; Shin, B.C.; Devaskar, S.U.; Reddy, S.T. Macrophage paraoxonase 2 regulates calcium homeostasis and cell survival under endoplasmic reticulum stress conditions and is sufficient to prevent the development of aggravated atherosclerosis in paraoxonase 2 deficiency/apoE−/− mice on a western diet. Mol. Genet. Metab. 2012, 107, 416–427. [Google Scholar] [PubMed]
- Lenin, R.; Maria, M.S.; Agrawal, M.; Balasubramanyam, J.; Mohan, V.; Balasubramanyam, M. Amelioration of glucolipotoxicity-induced endoplasmic reticulum stress by a “chemical chaperone” in human THP-1 monocytes. Exp. Diabetes Res. 2012, 2012, 356487. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Mori, M.; Akira, S.; Gotoh, T. C/EBP homologous protein (CHOP) is crucial for the induction of caspase-11 and the pathogenesis of lipopolysaccharide-induced inflammation. J. Immunol. 2006, 176, 6245–6253. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jiang, W.; Hu, S.; Song, W.; Ji, L.; Wang, Y.; Cai, L. Fucosylated chondroitin sulphate from Cusumaria frondosa mitigates hepatic endoplasmic reticulum stress and inflammation in insulin resistant mice. Food Funct. 2015, 6, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Muramoto, M.; Oe, T.; Morikawa, N.; Okitsu, O.; Nagashima, T.; Nishimura, S.; Katayama, Y.; Kita, Y. Diclofenac, a non-steroidal anti-inflammatory drug, suppresses apoptosis induced by endoplasmic reticulum stresses by inhibiting caspase signaling. Neuropharmacology 2006, 50, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.H.; Parveen, A.; Ahmad, I.; Ahmad, I.; Verma, A.K.; Arshad, M.; Mahdi, A.A. Aluminum activates PERK-eIF2α signaling and inflammatory proteins in human neuroblastoma SH-SY5Y cells. Biol. Trace Elem. Res. 2015, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Kaczmarek, A.; Krysko, O.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. ER stress-induced inflammation: Does it aid or impede disease progression? Trends Mol. Med. 2012, 18, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Na, B.; Huang, Z.; Wang, Q.; Qi, Z.; Tian, Y.; Lu, C.C.; Yu, J.; Hanes, M.A.; Kakar, S.; Huang, E.J.; et al. Transgenic expression of entire hepatitis B virus in mice induces hepatocarcinogenesis independent of chronic liver injury. PLoS ONE 2011, 6, e26240. [Google Scholar] [CrossRef] [PubMed]
- Kaplowitz, N.; Ji, C. Unfolding new mechanisms of alcoholic liver disease in the endoplasmic reticulum. J. Gastroenterol. Hepatol. 2006, 21 (Suppl. S3), S7–S9. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kuhnisch, J.; Mustafa, A.; Lhotak, S.; Schlachterman, A.; Slifker, M.J.; Klein-Szanto, A.; High, K.A.; Austin, R.C.; Kruger, W.D. Mouse models of cystathionine β-synthase deficiency reveal significant threshold effects of hyperhomocysteinemia. FASEB J. 2009, 23, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.H.; Bhandary, B.; Lee, E.M.; Park, J.K.; Jeong, K.S.; Kim, I.K.; Kim, H.R.; Chae, H.J. The roles of ER stress and p450 2E1 in CCI4-induced steatosis. Int. J. Biochem. Cell Biol. 2011, 43, 1469–1482. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Deng, Q.; Kaplowitz, N. Role of TNF-α in ethanol-induced hyperhomocysteinemia and murine alcoholic liver injury. Hepatology 2004, 40, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Kaplowitz, N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology 2003, 124, 1488–1499. [Google Scholar] [CrossRef]
- Arai, M.; Kondoh, N.; Imazeki, N.; Hada, A.; Hatsuse, K.; Kimura, F.; Matsubara, O.; Mori, K.; Wakatsuki, T.; Yamamoto, M. Transformation-associated gene regulation by ATF6α during hepatocarcinogenesis. FEBS Lett. 2006, 580, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Chiang, P.C.; Chien, C.L.; Pan, S.L.; Chen, W.P.; Teng, C.M.; Shen, Y.C.; Guh, J.H. Induction of endoplasmic reticulum stress and apoptosis by a marine prostanoid in human hepatocellular carcinoma. J. Hepatol. 2005, 43, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Kondoh, N.; Imazeki, N.; Tanaka, K.; Okada, T.; Mori, K.; Hada, A.; Arai, M.; Wakatsuki, T.; Matsubara, O.; et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: A possible involvement of the er stress pathway in hepatocarcinogenesis. J. Hepatol. 2003, 38, 605–614. [Google Scholar] [CrossRef]
- Galligan, J.J.; Smathers, R.L.; Shearn, C.T.; Fritz, K.S.; Backos, D.S.; Jiang, H.; Franklin, C.C.; Orlicky, D.J.; Maclean, K.N.; Petersen, D.R. Oxidative stress and the ER stress response in a murine model for early-stage alcoholic liver disease. J. Toxicol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Rissanen, A.; Sivenius, J.; Jolkkonen, J. Prolonged bihemispheric alterations in unfolded protein response related gene expression after experimental stroke. Brain Res. 2006, 1087, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Tajiri, S.; Oyadomari, S.; Yano, S.; Morioka, M.; Gotoh, T.; Hamada, J.I.; Ushio, Y.; Mori, M. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 2004, 11, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Azfer, A.; Niu, J.; Rogers, L.M.; Adamski, F.M.; Kolattukudy, P.E. Activation of endoplasmic reticulum stress response during the development of ischemic heart disease. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1411–H1420. [Google Scholar] [CrossRef] [PubMed]
- Thuerauf, D.J.; Marcinko, M.; Gude, N.; Rubio, M.; Sussman, M.A.; Glembotski, C.C. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ. Res. 2006, 99, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, T.; Mori, M. Nitric oxide and endoplasmic reticulum stress. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Shintani-Ishida, K.; Nakajima, M.; Uemura, K.; Yoshida, K. Ischemic preconditioning protects cardiomyocytes against ischemic injury by inducing grp78. Biochem. Biophys. Res. Commun. 2006, 345, 1600–1605. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zong, L.; Wang, X. TGF-β improves myocardial function and prevents apoptosis induced by anoxia-reoxygenation, through the reduction of endoplasmic reticulum stress. Can. J. Physiol. Pharmacol. 2015, 94, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Hu, H.; Chen, B.; Yue, R.; Zhou, Z.; Liu, Y.; Zhang, S.; Xu, L.; Wang, H.; Yu, Z. Lycopene protects against hypoxia/reoxygenation injury by alleviating ER stress induced apoptosis in neonatal mouse cardiomyocytes. PLoS ONE 2015, 10, e0136443. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Okuma, Y.; Hosoi, T.; Nomura, Y. Edaravone protects against hypoxia/ischemia-induced endoplasmic reticulum dysfunction. J. Pharmacol. Exp. Ther. 2004, 311, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Adler, H.T.; Chinery, R.; Wu, D.Y.; Kussick, S.J.; Payne, J.M.; Fornace, A.J., Jr.; Tkachuk, D.C. Leukemic HRX fusion proteins inhibit GADD34-induced apoptosis and associate with the GADD34 and hSNF5/INI1 proteins. Mol. Cell. Biol. 1999, 19, 7050–7060. [Google Scholar] [CrossRef] [PubMed]
- Boussabbeh, M.; Prola, A.; Ben Salem, I.; Guilbert, A.; Bacha, H.; Lemaire, C.; Abis-Essefi, S. Crocin and quercetin prevent PAT-induced apoptosis in mammalian cells: Involvement of ROS-mediated ER stress pathway. Environ. Toxicol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Lorz, C.; Justo, P.; Sanz, A.; Subira, D.; Egido, J.; Ortiz, A. Paracetamol-induced renal tubular injury: A role for ER stress. J. Am. Soc. Nephrol. 2004, 15, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, N. The endoplasmic reticulum stress response and aging. Rev. Neurosci. 2009, 20, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Adam, J.; Bollee, G.; Fougeray, S.; Noel, L.H.; Antignac, C.; Knebelman, B.; Pallet, N. Endoplasmic reticulum stress in UMOD-related kidney disease: A human pathologic study. Am. J. Kidney Dis. 2012, 59, 117–121. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeeshan, H.M.A.; Lee, G.H.; Kim, H.-R.; Chae, H.-J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. https://doi.org/10.3390/ijms17030327
Zeeshan HMA, Lee GH, Kim H-R, Chae H-J. Endoplasmic Reticulum Stress and Associated ROS. International Journal of Molecular Sciences. 2016; 17(3):327. https://doi.org/10.3390/ijms17030327
Chicago/Turabian StyleZeeshan, Hafiz Maher Ali, Geum Hwa Lee, Hyung-Ryong Kim, and Han-Jung Chae. 2016. "Endoplasmic Reticulum Stress and Associated ROS" International Journal of Molecular Sciences 17, no. 3: 327. https://doi.org/10.3390/ijms17030327
APA StyleZeeshan, H. M. A., Lee, G. H., Kim, H.-R., & Chae, H.-J. (2016). Endoplasmic Reticulum Stress and Associated ROS. International Journal of Molecular Sciences, 17(3), 327. https://doi.org/10.3390/ijms17030327