
EGFR Signaling in Liver Diseases

Abstract
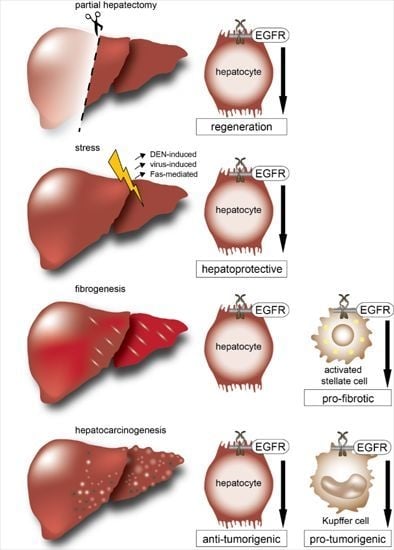
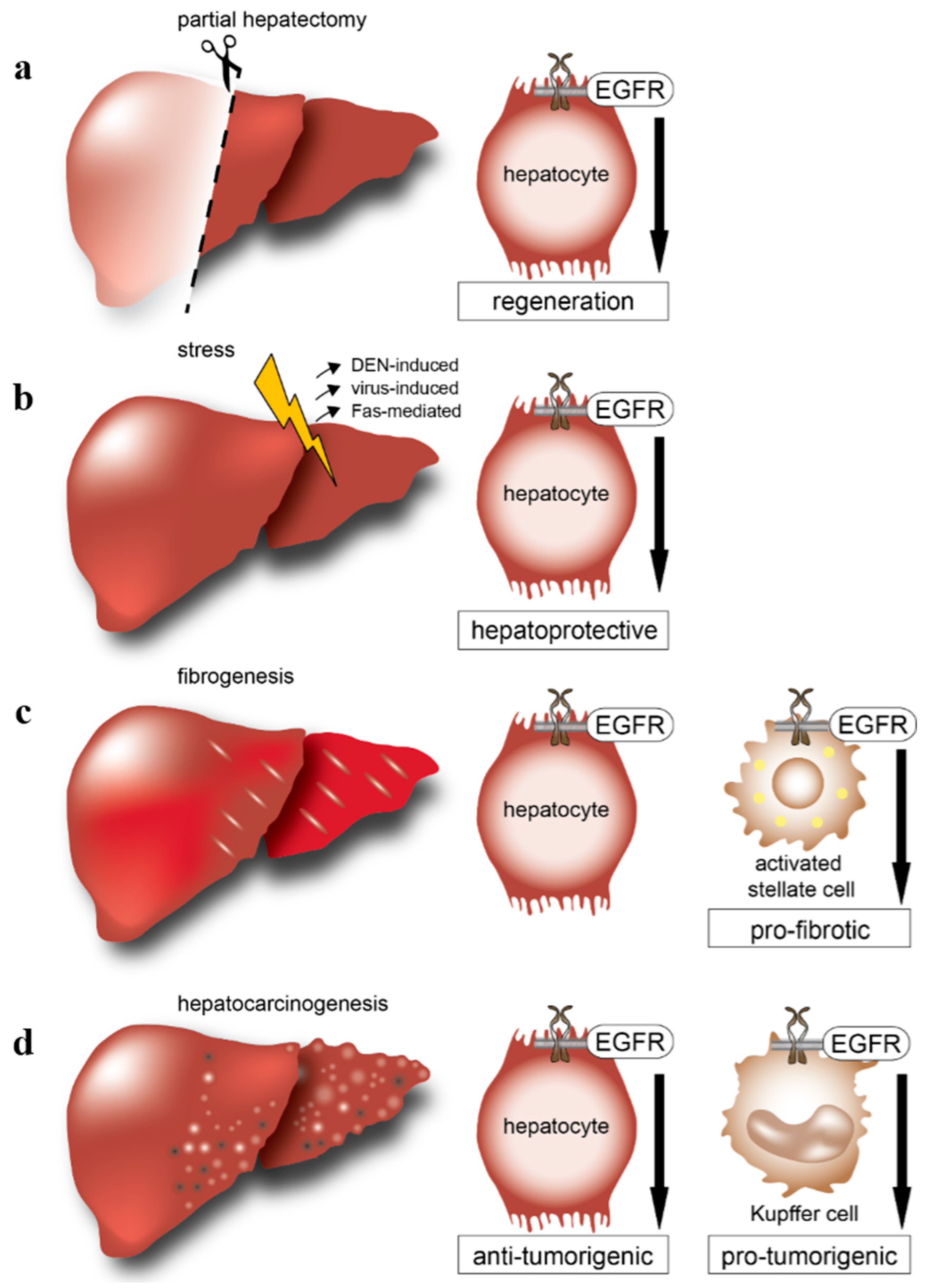
:
{kind=link}
{kind=link}
{kind=link}
1. Epidermal Growth Factor Receptor (EGFR) and Its Ligands
2. EGFR and Its Ligands during Liver Development
3. EGFR and Its Ligands in Liver Regeneration
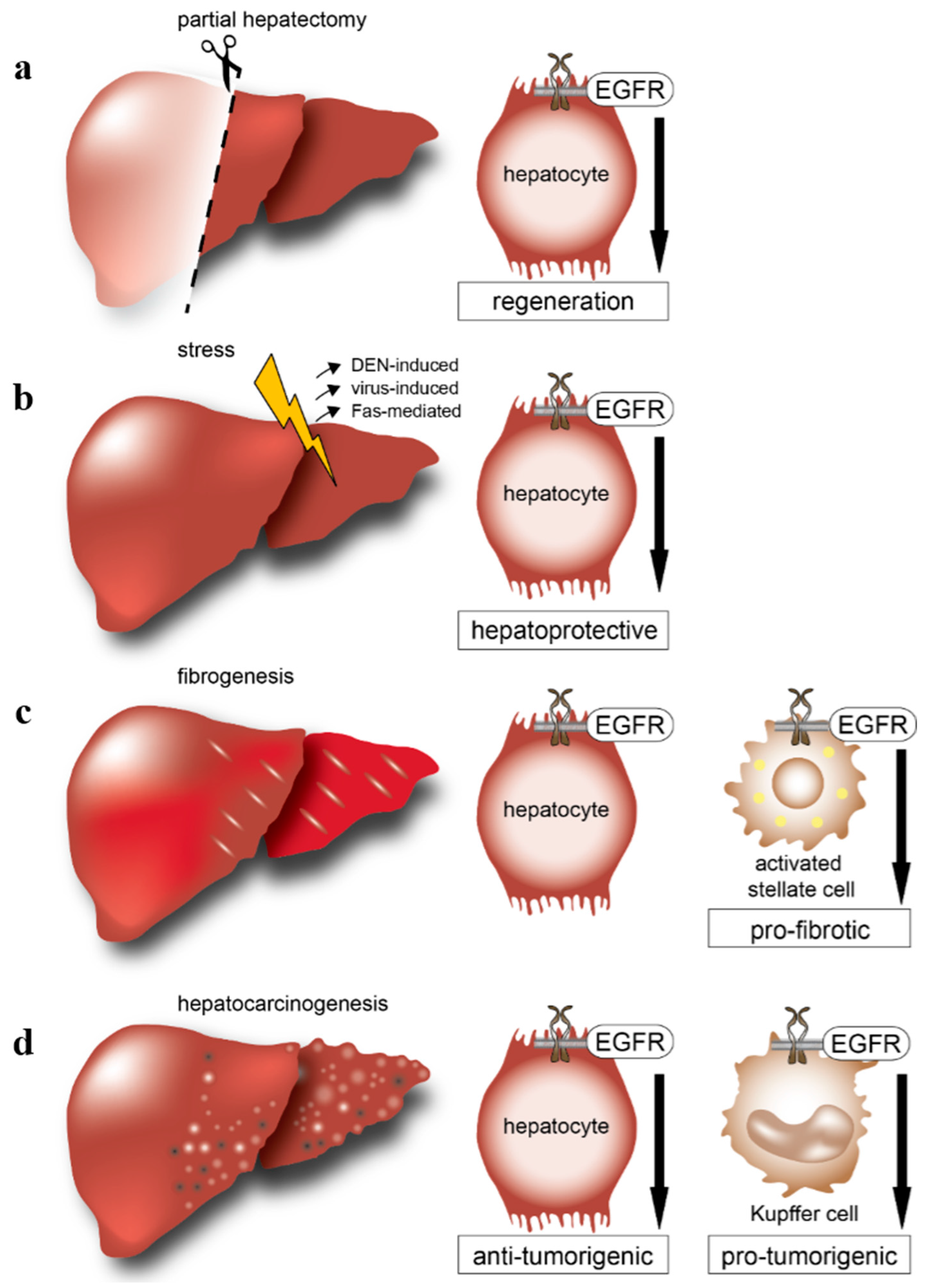
4. EGFR and Its Ligands in Experimental Models of Acute Liver Damage
5. EGFR and Its Ligands in Experimental Models of Chronic Liver Damage
6. EGFR and Hepatocellular Carcinoma
7. EGFR and Its Ligands in Hepatitis B Virus-Induced Hepatocellular Carcinoma
8. EGFR in Hepatitis C Virus-Induced Hepatocellular Carcinoma
9. EGFR and Its Ligands in Genetically Engineered Mouse Models (GEMMs) of Hepatocellular Carcinoma
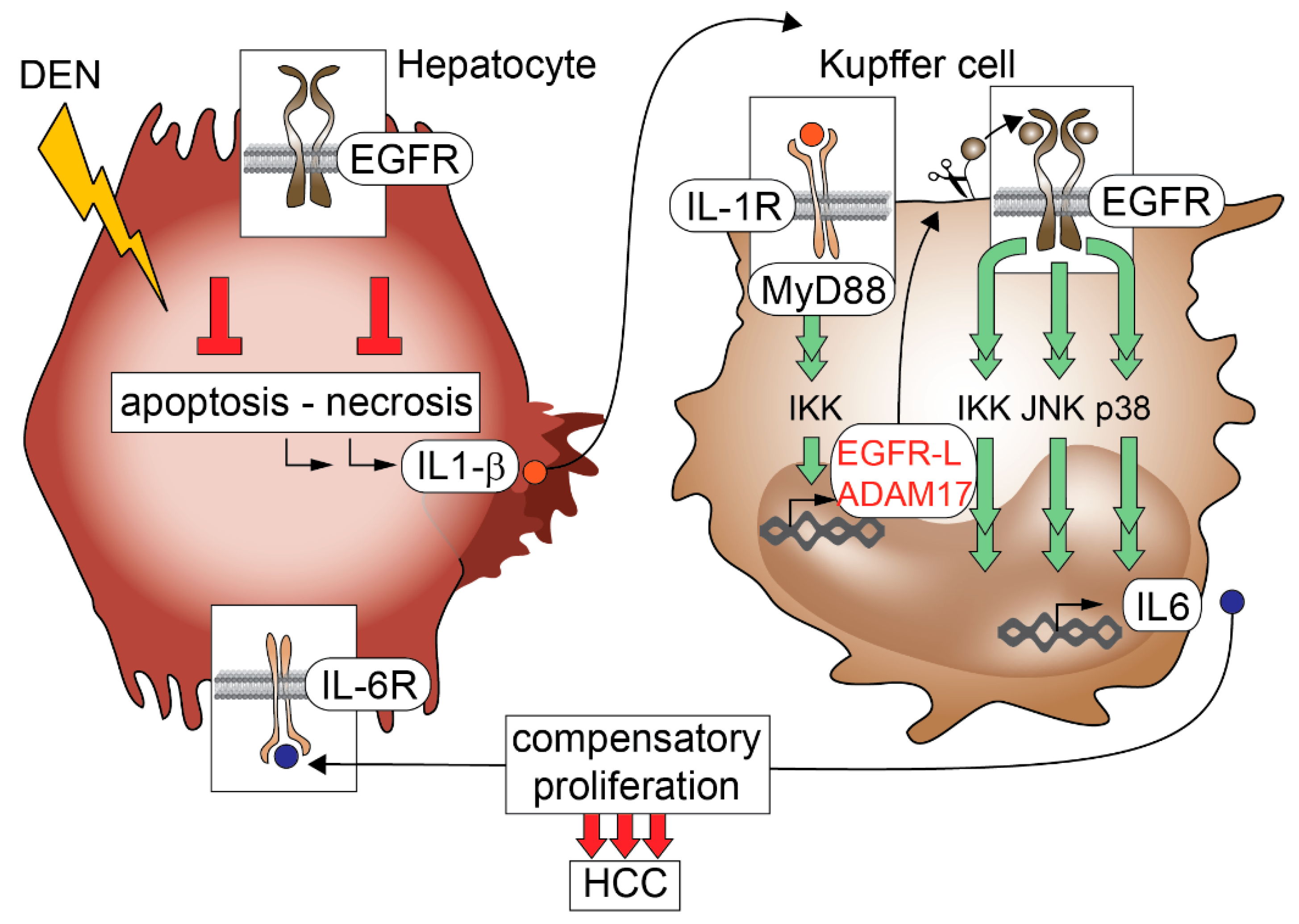
10. EGFR Inhibitors in Experimental Hepatocellular Carcinoma (HCC)
11. EGFR Inhibitors in Human HCC
12. EGFR and Its Ligands in Hepatolithiasis and Cholangiocarcinoma
13. EGFR and Its Ligands in Hepatic Progenitor Cells
14. Conclusions
Acknowledgments
Authors Contributions
Conflicts of Interest
Abbreviations
References
- Schlessinger, J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 2002, 110, 669–672. [Google Scholar] [CrossRef]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signalling. Exp. Cell Res. 2003, 284, 31–53. [Google Scholar] [CrossRef]
- Citri, A.; Yarden, Y. EGF-ErbB signalling: Towards the systems level. Nat. Rev. Mol. Cell Biol. 2006, 7, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.R.; Wolf, E. The epidermal growth factor receptor ligands at a glance. J. Cell. Physiol. 2009, 218, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; de Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; de Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Pandiella, A. Membrane-anchored growth factors. Annu. Rev. Biochem. 1993, 62, 515–541. [Google Scholar] [CrossRef] [PubMed]
- Ohtsu, H.; Dempsey, P.J.; Eguchi, S. ADAMs as mediators of EGF receptor transactivation by g protein-coupled receptors. Am. J. Physiol. Cell Physiol. 2006, 291, C1–C10. [Google Scholar] [CrossRef] [PubMed]
- Blobel, C.P. ADAMs: Key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 2005, 6, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Blobel, C.P. Ectodomain shedding of the EGF-receptor ligand epigen is mediated by ADAM17. FEBS Lett. 2007, 581, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. ErbB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Haendeler, J.; Hojo, Y.; Yamamoto, K.; Berk, B.C. Receptor heterodimerization: Essential mechanism for platelet-derived growth factor-induced epidermal growth factor receptor transactivation. Mol. Cell. Biol. 2001, 21, 6387–6394. [Google Scholar] [CrossRef] [PubMed]
- Habib, A.A.; Hognason, T.; Ren, J.; Stefansson, K.; Ratan, R.R. The epidermal growth factor receptor associates with and recruits phosphatidylinositol 3-kinase to the platelet-derived growth factor β receptor. J. Biol. Chem. 1998, 273, 6885–6891. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Woo, J.K.; Kim, E.S.; Hong, W.K.; Lee, H.Y. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res. 2006, 66, 10100–10111. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.; Stolz, D.B.; Esplen, J.E.; Dorko, K.; Michalopoulos, G.K.; Strom, S.C. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J. Biol. Chem. 2000, 275, 8806–8811. [Google Scholar] [CrossRef] [PubMed]
- Burova, E.; Vassilenko, K.; Dorosh, V.; Gonchar, I.; Nikolsky, N. Interferon γ-dependent transactivation of epidermal growth factor receptor. FEBS Lett. 2007, 581, 1475–1480. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.; Diaz, M.E.; Miquet, J.G.; Sotelo, A.I.; Fernandez, D.; Dominici, F.P.; Bartke, A.; Turyn, D. Gh modulates hepatic epidermal growth factor signaling in the mouse. J. Endocrinol. 2010, 204, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Moro, L.; Dolce, L.; Cabodi, S.; Bergatto, E.; Boeri Erba, E.; Smeriglio, M.; Turco, E.; Retta, S.F.; Giuffrida, M.G.; Venturino, M.; et al. Integrin-induced epidermal growth factor (EGF) receptor activation requires c-Src and p130cas and leads to phosphorylation of specific EGF receptor tyrosines. J. Biol. Chem. 2002, 277, 9405–9414. [Google Scholar] [CrossRef] [PubMed]
- Fischer, O.M.; Hart, S.; Gschwind, A.; Ullrich, A. EGFR signal transactivation in cancer cells. Biochem. Soc. Trans. 2003, 31, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Almendro, V.; Garcia-Recio, S.; Gascon, P. Tyrosine kinase receptor transactivation associated to G protein-coupled receptors. Curr. Drug Targets 2010, 11, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Liebmann, C. EGF receptor activation by GPCRs: An universal pathway reveals different versions. Mol. Cell. Endocrinol. 2011, 331, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Ueki, K.; Tobe, K.; Tamemoto, H.; Sekine, N.; Wada, M.; Honjo, M.; Takahashi, M.; Takahashi, T.; Hirai, H.; et al. Tyrosine phosphorylation of the EGF receptor by the kinase Jak2 is induced by growth hormone. Nature 1997, 390, 91–96. [Google Scholar] [PubMed]
- Werneburg, N.W.; Yoon, J.H.; Higuchi, H.; Gores, G.J. Bile acids activate EGF receptor via a TGF-α-dependent mechanism in human cholangiocyte cell lines. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G31–G36. [Google Scholar] [CrossRef] [PubMed]
- Sibilia, M.; Kroismayr, R.; Lichtenberger, B.M.; Natarajan, A.; Hecking, M.; Holcmann, M. The epidermal growth factor receptor: From development to tumorigenesis. Differentiation 2007, 75, 770–787. [Google Scholar] [CrossRef] [PubMed]
- Luetteke, N.C.; Qiu, T.H.; Fenton, S.E.; Troyer, K.L.; Riedel, R.F.; Chang, A.; Lee, D.C. Targeted inactivation of the EGF and amphiregulin genes reveals distinct roles for EGF receptor ligands in mouse mammary gland development. Development 1999, 126, 2739–2750. [Google Scholar] [PubMed]
- Lee, D.; Pearsall, R.S.; Das, S.; Dey, S.K.; Godfrey, V.L.; Threadgill, D.W. Epiregulin is not essential for development of intestinal tumors but is required for protection from intestinal damage. Mol. Cell. Biol. 2004, 24, 8907–8916. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.F.; Qiu, T.H.; Sunnarborg, S.W.; Chang, A.; Zhang, C.; Patterson, C.; Lee, D.C. Defective valvulogenesis in HB-EGF and TACE-null mice is associated with aberrant bmp signaling. EMBO J. 2003, 22, 2704–2716. [Google Scholar] [CrossRef] [PubMed]
- Dahlhoff, M.; Schafer, M.; Wolf, E.; Schneider, M.R. Genetic deletion of the EGFR ligand epigen does not affect mouse embryonic development and tissue homeostasis. Exp. Cell Res. 2013, 319, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, R.; Yamazaki, S.; Asakura, M.; Takashima, S.; Hasuwa, H.; Miyado, K.; Adachi, S.; Kitakaze, M.; Hashimoto, K.; Raab, G.; et al. Heparin-binding EGF-like growth factor and ErbB signaling is essential for heart function. Proc. Natl. Acad. Sci. USA 2003, 100, 3221–3226. [Google Scholar] [CrossRef] [PubMed]
- Luetteke, N.C.; Qiu, T.H.; Peiffer, R.L.; Oliver, P.; Smithies, O.; Lee, D.C. TGF α deficiency results in hair follicle and eye abnormalities in targeted and waved-1 mice. Cell 1993, 73, 263–278. [Google Scholar] [CrossRef]
- Mann, G.B.; Fowler, K.J.; Gabriel, A.; Nice, E.C.; Williams, R.L.; Dunn, A.R. Mice with a null mutation of the TGF α gene have abnormal skin architecture, wavy hair, and curly whiskers and often develop corneal inflammation. Cell 1993, 73, 249–261. [Google Scholar] [CrossRef]
- Miettinen, P.J.; Berger, J.E.; Meneses, J.; Phung, Y.; Pedersen, R.A.; Werb, Z.; Derynck, R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 1995, 376, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Sibilia, M.; Wagner, E.F. Strain-dependent epithelial defects in mice lacking the EGF receptor. Science 1995, 269, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Threadgill, D.W.; Dlugosz, A.A.; Hansen, L.A.; Tennenbaum, T.; Lichti, U.; Yee, D.; LaMantia, C.; Mourton, T.; Herrup, K.; Harris, R.C.; et al. Targeted disruption of mouse EGF receptor: Effect of genetic background on mutant phenotype. Science 1995, 269, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, A.; Wagner, B.; Sibilia, M. The EGF receptor is required for efficient liver regeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 17081–17086. [Google Scholar] [CrossRef] [PubMed]
- Taub, R. Liver regeneration: From myth to mechanism. Nat. Rev. Mol. Cell Biol. 2004, 5, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Carver, R.S.; Stevenson, M.C.; Scheving, L.A.; Russell, W.E. Diverse expression of ErbB receptor proteins during rat liver development and regeneration. Gastroenterology 2002, 123, 2017–2027. [Google Scholar] [CrossRef] [PubMed]
- Bucher, N.L.; Patel, U.; Cohen, S. Hormonal factors concerned with liver regeneration. In Hepatotrophic Factors; Elsevier Excerpta Media: New York, NY, USA, 1978; pp. 95–107. [Google Scholar]
- Hashimoto, M.; Kothary, P.C.; Eckhauser, F.E.; Raper, S.E. Treatment of cirrhotic rats with epidermal growth factor and insulin accelerates liver DNA synthesis after partial hepatectomy. J. Gastroenterol. Hepatol. 1998, 13, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Kawata, S.; Tamura, S.; Kiso, S.; Tsushima, H.; Damm, D.; Abraham, J.A.; Higashiyama, S.; Taniguchi, N.; Matsuzawa, Y. Heparin-binding EGF-like growth factor is a potent mitogen for rat hepatocytes. Biochem. Biophys. Res. Commun. 1994, 198, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Block, G.D.; Locker, J.; Bowen, W.C.; Petersen, B.E.; Katyal, S.; Strom, S.C.; Riley, T.; Howard, T.A.; Michalopoulos, G.K. Population expansion, clonal growth, and specific differentiation patterns in primary cultures of hepatocytes induced by HGF/SF, EGF and TGF α in a chemically defined (HGM) medium. J. Cell Biol. 1996, 132, 1133–1149. [Google Scholar] [CrossRef] [PubMed]
- Fausto, N.; Laird, A.D.; Webber, E.M. Liver regeneration. 2. Role of growth factors and cytokines in hepatic regeneration. FASEB J. 1995, 9, 1527–1536. [Google Scholar] [PubMed]
- Berasain, C.; Garcia-Trevijano, E.R.; Castillo, J.; Erroba, E.; Lee, D.C.; Prieto, J.; Avila, M.A. Amphiregulin: An early trigger of liver regeneration in mice. Gastroenterology 2005, 128, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Draghi, E.; Armato, U.; Andreis, P.G.; Mengato, L. The stimulation by epidermal growth factor (urogastrone) of the growth of neonatal rat hepatocytes in primary tissue culture and its modulation by serum and associated pancreatic hormones. J. Cell. Physiol. 1980, 103, 129–147. [Google Scholar] [CrossRef] [PubMed]
- De Juan, C.; Benito, M.; Alvarez, A.; Fabregat, I. Differential proliferative response of cultured fetal and regenerating hepatocytes to growth factors and hormones. Exp. Cell Res. 1992, 202, 495–500. [Google Scholar] [CrossRef]
- McGowan, J.A.; Strain, A.J.; Bucher, N.L. DNA synthesis in primary cultures of adult rat hepatocytes in a defined medium: Effects of epidermal growth factor, insulin, glucagon, and cyclic-AMP. J. Cell. Physiol. 1981, 108, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Komurasaki, T.; Toyoda, H.; Uchida, D.; Nemoto, N. Mechanism of growth promoting activity of epiregulin in primary cultures of rat hepatocytes. Growth Factors 2002, 20, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, H.; Komurasaki, T.; Uchida, D.; Takayama, Y.; Isobe, T.; Okuyama, T.; Hanada, K. Epiregulin. A novel epidermal growth factor with mitogenic activity for rat primary hepatocytes. J. Biol. Chem. 1995, 270, 7495–7500. [Google Scholar] [PubMed]
- Webber, E.M.; FitzGerald, M.J.; Brown, P.I.; Bartlett, M.H.; Fausto, N. Transforming growth factor-α expression during liver regeneration after partial hepatectomy and toxic injury, and potential interactions between transforming growth factor-α and hepatocyte growth factor. Hepatology 1993, 18, 1422–1431. [Google Scholar] [CrossRef] [PubMed]
- Kiso, S.; Kawata, S.; Tamura, S.; Higashiyama, S.; Ito, N.; Tsushima, H.; Taniguchi, N.; Matsuzawa, Y. Role of heparin-binding epidermal growth factor-like growth factor as a hepatotrophic factor in rat liver regeneration after partial hepatectomy. Hepatology 1995, 22, 1584–1590. [Google Scholar] [CrossRef]
- Lin, X.M.; Liu, Y.B.; Zhou, F.; Wu, Y.L.; Chen, L.; Fang, H.Q. Expression of tumor necrosis factor-α converting enzyme in liver regeneration after partial hepatectomy. World J. Gastroenterol. 2008, 14, 1353–1357. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, C.; Nivison, M.; Jackson, L.F.; Fox, R.; Lee, D.C.; Campbell, J.S.; Fausto, N. Heparin-binding epidermal growth factor-like growth factor links hepatocyte priming with cell cycle progression during liver regeneration. J. Biol. Chem. 2005, 280, 2562–2568. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.E.; Kaufmann, W.K.; Sitaric, S.; Luetteke, N.C.; Lee, D.C. Liver regeneration and hepatocarcinogenesis in transforming growth factor-α-targeted mice. Mol. Carcinog. 1996, 15, 183–189. [Google Scholar] [CrossRef]
- Kokudo, N.; Kothary, P.C.; Eckhauser, F.E.; Raper, S.E. Transforming growth factor-α (TGF-α) improves hepatic DNA synthesis after hepatectomy in cirrhotic rats. J. Surg. Res. 1992, 52, 648–655. [Google Scholar] [CrossRef]
- Liu, Q.; Rehman, H.; Krishnasamy, Y.; Haque, K.; Schnellmann, R.G.; Lemasters, J.J.; Zhong, Z. Amphiregulin stimulates liver regeneration after small-for-size mouse liver transplantation. Am. J. Transpl. 2012, 12, 2052–2061. [Google Scholar] [CrossRef] [PubMed]
- Kosone, T.; Takagi, H.; Horiguchi, N.; Kakizaki, S.; Sato, K.; Watanabe, Y.; Mori, M. Transforming growth factor-α accelerates hepatocyte repopulation after hepatocyte transplantation. J. Gastroenterol. Hepatol. 2008, 23, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Kiso, S.; Kawata, S.; Tamura, S.; Inui, Y.; Yoshida, Y.; Sawai, Y.; Umeki, S.; Ito, N.; Yamada, A.; Miyagawa, J.; et al. Liver regeneration in heparin-binding EGF-like growth factor transgenic mice after partial hepatectomy. Gastroenterology 2003, 124, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Ohba, Y.; Oka, T. Influence of epidermal growth factor on liver regeneration after partial hepatectomy in mice. J. Endocrinol. 1991, 128, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.E., Jr.; Tran-Patterson, R.; Cui, D.M.; Davin, D.; Estell, K.P.; Miller, D.M. Epidermal growth factor secreted from the salivary gland is necessary for liver regeneration. Am. J. Physiol. 1995, 268, G872–G878. [Google Scholar] [PubMed]
- Lambotte, L.; Saliez, A.; Triest, S.; Maiter, D.; Baranski, A.; Barker, A.; Li, B. Effect of sialoadenectomy and epidermal growth factor administration on liver regeneration after partial hepatectomy. Hepatology 1997, 25, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Ohba, Y.; Oka, T. The role of transcription and messenger RNA stability in the regulation of epidermal growth factor receptor gene expression in regenerating mouse liver. Hepatology 1992, 15, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Scheving, L.A.; Zhang, X.; Stevenson, M.C.; Threadgill, D.W.; Russell, W.E. Loss of hepatocyte EGFR has no effect alone but exacerbates carbon tetrachloride-induced liver injury and impairs regeneration in hepatocyte Met-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G364–G377. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Luque, J.; Caballero-Diaz, D.; Martinez-Palacian, A.; Roncero, C.; Moreno-Caceres, J.; Garcia-Bravo, M.; Grueso, E.; Fernandez, A.; Crosas-Molist, E.; Garcia-Alvaro, M.; et al. Dissecting the role of the epidermal growth factor receptor catalytic activity during liver regeneration and hepatocarcinogenesis. Hepatology 2015. [Google Scholar] [CrossRef] [PubMed]
- Paranjpe, S.; Bowen, W.C.; Tseng, G.C.; Luo, J.H.; Orr, A.; Michalopoulos, G.K. RNA interference against hepatic epidermal growth factor receptor has suppressive effects on liver regeneration in rats. Am. J. Pathol. 2010, 176, 2669–2681. [Google Scholar] [CrossRef] [PubMed]
- Van Buren, G., 2nd; Yang, A.D.; Dallas, N.A.; Gray, M.J.; Lim, S.J.; Xia, L.; Fan, F.; Somcio, R.; Wu, Y.; Hicklin, D.J.; et al. Effect of molecular therapeutics on liver regeneration in a murine model. J. Clin. Oncol. 2008, 26, 1836–1842. [Google Scholar] [CrossRef] [PubMed]
- Zerrad-Saadi, A.; Lambert-Blot, M.; Mitchell, C.; Bretes, H.; Collin de l’Hortet, A.; Baud, V.; Chereau, F.; Sotiropoulos, A.; Kopchick, J.J.; Liao, L.; et al. GH receptor plays a major role in liver regeneration through the control of EGFR and ERK1/2 activation. Endocrinology 2011, 152, 2731–2741. [Google Scholar] [CrossRef] [PubMed]
- Collin de l’Hortet, A.; Zerrad-Saadi, A.; Prip-Buus, C.; Fauveau, V.; Helmy, N.; Ziol, M.; Vons, C.; Billot, K.; Baud, V.; Gilgenkrantz, H.; et al. GH administration rescues fatty liver regeneration impairment by restoring GH/EGFR pathway deficiency. Endocrinology 2014, 155, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Behari, J.; Cieply, B.; Michalopoulos, G.K.; Monga, S.P. Conditional deletion of β-catenin reveals its role in liver growth and regeneration. Gastroenterology 2006, 131, 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Fan, Y.; Rao, J.; Xu, Z.; Liu, Y.; Lu, L.; Li, G. Matrix Metalloproteinases-9 deficiency impairs liver regeneration through epidermal growth factor receptor signaling in partial hepatectomy mice. J. Surg. Res. 2015, 197, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Berasain, C.; Garcia-Trevijano, E.R.; Castillo, J.; Erroba, E.; Santamaria, M.; Lee, D.C.; Prieto, J.; Avila, M.A. Novel role for amphiregulin in protection from liver injury. J. Biol. Chem. 2005, 280, 19012–19020. [Google Scholar] [CrossRef] [PubMed]
- Pardo-Saganta, A.; Latasa, M.U.; Castillo, J.; Alvarez-Asiain, L.; Perugorria, M.J.; Sarobe, P.; Rodriguez-Ortigosa, C.M.; Prieto, J.; Berasain, C.; Santamaria, M.; et al. The epidermal growth factor receptor ligand amphiregulin is a negative regulator of hepatic acute-phase gene expression. J. Hepatol. 2009, 51, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
- Kanda, D.; Takagi, H.; Toyoda, M.; Horiguchi, N.; Nakajima, H.; Otsuka, T.; Mori, M. Transforming growth factor α protects against Fas-mediated liver apoptosis in mice. FEBS Lett. 2002, 519, 11–15. [Google Scholar] [CrossRef]
- Khai, N.C.; Takahashi, T.; Ushikoshi, H.; Nagano, S.; Yuge, K.; Esaki, M.; Kawai, T.; Goto, K.; Murofushi, Y.; Fujiwara, T.; et al. In vivo hepatic HB-EGF gene transduction inhibits Fas-induced liver injury and induces liver regeneration in mice: A comparative study to HGF. J. Hepatol. 2006, 44, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Murthy, A.; Defamie, V.; Smookler, D.S.; di Grappa, M.A.; Horiuchi, K.; Federici, M.; Sibilia, M.; Blobel, C.P.; Khokha, R. Ectodomain shedding of EGFR ligands and TNFR1 dictates hepatocyte apoptosis during fulminant hepatitis in mice. J. Clin. Investig. 2010, 120, 2731–2744. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Han, C.; Xu, L.; Lim, K.; Isse, K.; Wu, T. Cyclooxygenase-2 prevents Fas-induced liver injury through up-regulation of epidermal growth factor receptor. Hepatology 2009, 50, 834–843. [Google Scholar] [CrossRef] [PubMed]
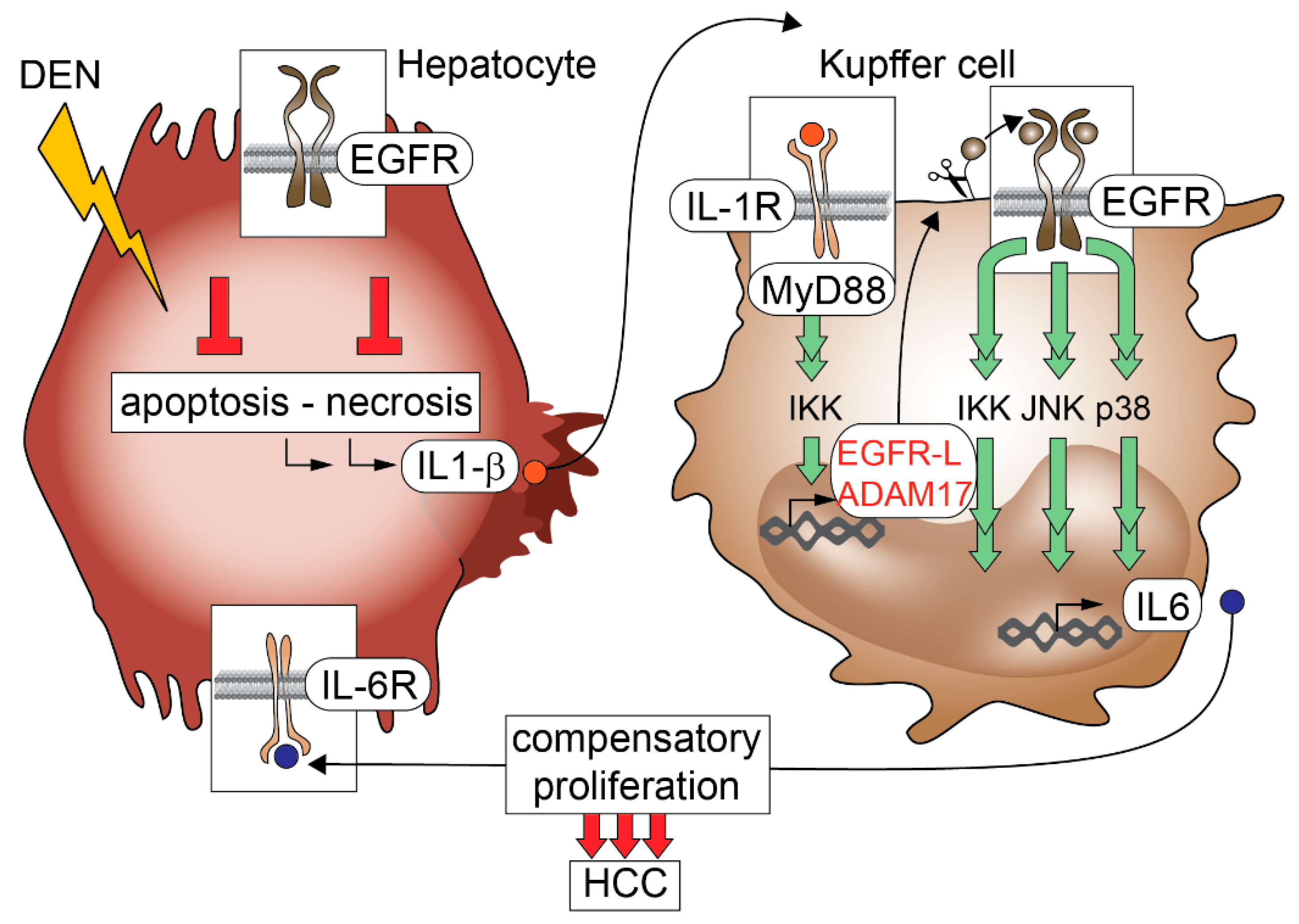
- Lanaya, H.; Natarajan, A.; Komposch, K.; Li, L.; Amberg, N.; Chen, L.; Wculek, S.K.; Hammer, M.; Zenz, R.; Peck-Radosavljevic, M.; et al. EGFR has a tumour-promoting role in liver macrophages during hepatocellular carcinoma formation. Nat. Cell Biol. 2014, 16, 972–981, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Vuppalanchi, R.; Chalasani, N. Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: Selected practical issues in their evaluation and management. Hepatology 2009, 49, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Louvet, A.; Mathurin, P. Alcoholic liver disease: Mechanisms of injury and targeted treatment. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Fuchs, B.C.; Yamada, S.; Lauwers, G.Y.; Kulu, Y.; Goodwin, J.M.; Lanuti, M.; Tanabe, K.K. Mouse model of carbon tetrachloride induced liver fibrosis: Histopathological changes and expression of CD133 and epidermal growth factor. BMC Gastroenterol. 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Kiso, S.; Kawata, S.; Tamura, S.; Ito, N.; Tsushima, H.; Yamada, A.; Higashiyama, S.; Taniguchi, N.; Matsuzawa, Y. Expression of heparin-binding EGF-like growth factor in rat liver injured by carbon tetrachloride or d-galactosamine. Biochem. Biophys. Res. Commun. 1996, 220, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Kiso, S.; Kawata, S.; Tamura, S.; Miyagawa, J.; Ito, N.; Tsushima, H.; Yamada, A.; Umeki, S.; Higashiyama, S.; Taniguchi, N.; et al. Expression of heparin-binding epidermal growth factor-like growth factor in the hepatocytes of fibrotic rat liver during hepatocarcinogenesis. J. Gastroenterol. Hepatol. 1999, 14, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Dalu, A.; Cronin, G.M.; Lyn-Cook, B.D.; Mehendale, H.M. Age-related differences in TGF-α and proto-oncogenes expression in rat liver after a low dose of carbon tetrachloride. J. Biochem. Toxicol. 1995, 10, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Perugorria, M.J.; Latasa, M.U.; Nicou, A.; Cartagena-Lirola, H.; Castillo, J.; Goni, S.; Vespasiani-Gentilucci, U.; Zagami, M.G.; Lotersztajn, S.; Prieto, J.; et al. The epidermal growth factor receptor ligand amphiregulin participates in the development of mouse liver fibrosis. Hepatology 2008, 48, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Takemura, T.; Yoshida, Y.; Kiso, S.; Saji, Y.; Ezaki, H.; Hamano, M.; Kizu, T.; Egawa, M.; Chatani, N.; Furuta, K.; et al. Conditional knockout of heparin-binding epidermal growth factor-like growth factor in the liver accelerates carbon tetrachloride-induced liver injury in mice. Hepatol. Res. 2013, 43, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Besner, G.E.; Brigstock, D.R. Heparin-binding epidermal growth factor-like growth factor suppresses experimental liver fibrosis in mice. Lab. Investig. 2012, 92, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, J.; Jiang, X.; Li, X.; Wang, Y.; Ma, J.; Jiang, H. Heparin-binding epidermal growth factor-like growth factor: A hepatic stellate cell proliferation inducer via ErbB receptors. J. Gastroenterol. Hepatol. 2014, 29, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Wang, C.W.; Xu, J.R.; Zhu, J.Q. Effect of epidermal growth factor on cultured rat hepatocytes poisoned by CCl4. Zhongguo Yao Li Xue Bao 1997, 18, 176–179. [Google Scholar] [PubMed]
- Berlanga, J.; Caballero, M.E.; Ramirez, D.; Torres, A.; Valenzuela, C.; Lodos, J.; Playford, R.J. Epidermal growth factor protects against carbon tetrachloride-induced hepatic injury. Clin. Sci. (Lond.) 1998, 94, 219–223. [Google Scholar] [CrossRef] [PubMed]
- El Taghdouini, A.; Najimi, M.; Sancho-Bru, P.; Sokal, E.; van Grunsven, L.A. In vitro reversion of activated primary human hepatic stellate cells. Fibrogenes. Tissue Repair 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, B.C.; Hoshida, Y.; Fujii, T.; Wei, L.; Yamada, S.; Lauwers, G.Y.; McGinn, C.M.; DePeralta, D.K.; Chen, X.; Kuroda, T.; et al. Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology 2014, 59, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Berasain, C.; Perugorria, M.J.; Latasa, M.U.; Castillo, J.; Goni, S.; Santamaria, M.; Prieto, J.; Avila, M.A. The epidermal growth factor receptor: A link between inflammation and liver cancer. Exp. Biol. Med. (Maywood) 2009, 234, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Arabpour, M.; Poelstra, K.; Helfrich, W.; Bremer, E.; Haisma, H.J. Targeted elimination of activated hepatic stellate cells by an anti-epidermal growth factor-receptor single chain fragment variable antibody-tumor necrosis factor-related apoptosis-inducing ligand (scFv425-sTRAIL). J. Gene Med. 2014, 16, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.P.; Dong, J.Z.; Xiong, L.J.; Shi, K.Q.; Zou, Z.L.; Zhang, S.N.; Cao, S.T.; Lin, Z.; Chen, Y.P. BMP-7 attenuates liver fibrosis via regulation of epidermal growth factor receptor. Int. J. Clin. Exp. Pathol. 2014, 7, 3537–3547. [Google Scholar] [PubMed]
- Svegliati-Baroni, G.; Ridolfi, F.; Hannivoort, R.; Saccomanno, S.; Homan, M.; de Minicis, S.; Jansen, P.L.; Candelaresi, C.; Benedetti, A.; Moshage, H. Bile acids induce hepatic stellate cell proliferation via activation of the epidermal growth factor receptor. Gastroenterology 2005, 128, 1042–1055. [Google Scholar] [CrossRef] [PubMed]
- Sommerfeld, A.; Reinehr, R.; Haussinger, D. Bile acid-induced epidermal growth factor receptor activation in quiescent rat hepatic stellate cells can trigger both proliferation and apoptosis. J. Biol. Chem. 2009, 284, 22173–22183. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, H.; Maesawa, C.; Tatemichi, Y.; Nishinari, Y.; Nishiya, M.; Mizugai, H.; Ikeda, A.; Oikawa, K.; Takikawa, Y.; Masuda, T. A disintegrin and metalloproteinase 17 (ADAM17) mediates epidermal growth factor receptor transactivation by angiotensin ii on hepatic stellate cells. Life Sci. 2014, 97, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Takemura, T.; Yoshida, Y.; Kiso, S.; Kizu, T.; Furuta, K.; Ezaki, H.; Hamano, M.; Egawa, M.; Chatani, N.; Kamada, Y.; et al. Conditional loss of heparin-binding EGF-like growth factor results in enhanced liver fibrosis after bile duct ligation in mice. Biochem. Biophys. Res. Commun. 2013, 437, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Haga, H.; Mizuno, K.; Katsumi, T.; Sato, C.; Okumoto, K.; Nishise, Y.; Watanabe, H.; Saito, T.; Ueno, Y. Epiregulin promotes the emergence and proliferation of adult liver progenitor cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G50–G57. [Google Scholar] [CrossRef] [PubMed]
- McKee, C.; Sigala, B.; Soeda, J.; Mouralidarane, A.; Morgan, M.; Mazzoccoli, G.; Rappa, F.; Cappello, F.; Cabibi, D.; Pazienza, V.; et al. Amphiregulin activates human hepatic stellate cells and is upregulated in non alcoholic steatohepatitis. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, T.; Yamazaki, Y.; Sato, K.; Horiguchi, N.; Ichikawa, T.; Kakizaki, S.; Takagi, H.; Mori, M. Transforming growth factor-α attenuates hepatic fibrosis: Possible involvement of matrix metalloproteinase-1. Liver Int. 2011, 31, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Kato, J.; Sato, Y.; Inui, N.; Nakano, Y.; Takimoto, R.; Takada, K.; Kobune, M.; Kuroiwa, G.; Miyake, S.; Kohgo, Y.; et al. Ethanol induces transforming growth factor-α expression in hepatocytes, leading to stimulation of collagen synthesis by hepatic stellate cells. Alcohol. Clin. Exp. Res. 2003, 27, 58S–63S. [Google Scholar] [CrossRef] [PubMed]
- Deaciuc, I.V.; D’Souza, N.B.; Burikhanov, R.; Lee, E.Y.; Tarba, C.N.; McClain, C.J.; de Villiers, W.J. Epidermal growth factor protects the liver against alcohol-induced injury and sensitization to bacterial lipopolysaccharide. Alcohol. Clin. Exp. Res. 2002, 26, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; El-Serag, H.B. Epidemiology of hepatocellular carcinoma: Consider the population. J. Clin. Gastroenterol. 2013, 47 (Suppl.), S2–S6. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- De Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Farazi, P.A.; DePinho, R.A. Hepatocellular carcinoma pathogenesis: From genes to environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Q.; Zhang, J.; Wang, W.; Li, Q. Over expression of transforming growth factor-α and epidermal growth factor receptor in human hepatic cirrhosis tissues. Hepatogastroenterology 2008, 55, 169–172. [Google Scholar] [PubMed]
- Harada, K.; Shiota, G.; Kawasaki, H. Transforming growth factor-α and epidermal growth factor receptor in chronic liver disease and hepatocellular carcinoma. Liver 1999, 19, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Kira, S.; Nakanishi, T.; Suemori, S.; Kitamoto, M.; Watanabe, Y.; Kajiyama, G. Expression of transforming growth factor α and epidermal growth factor receptor in human hepatocellular carcinoma. Liver 1997, 17, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Takeda, T.; Sakon, M.; Tsujimoto, M.; Higashiyama, S.; Noda, K.; Miyoshi, E.; Monden, M.; Matsuura, N. Expression and clinical significance of ErbB receptor family in hepatocellular carcinoma. Br. J. Cancer 2001, 84, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Daveau, M.; Scotte, M.; Francois, A.; Coulouarn, C.; Ros, G.; Tallet, Y.; Hiron, M.; Hellot, M.F.; Salier, J.P. Hepatocyte growth factor, transforming growth factor α, and their receptors as combined markers of prognosis in hepatocellular carcinoma. Mol. Carcinogenes. 2003, 36, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.H.; Kim, J.A.; Song, B.C.; Lee, G.C.; Koh, M.S.; Lee, Y.S.; Lee, S.G.; Suh, D.J. Expression of transforming growth factor-α mRNA in livers of patients with chronic viral hepatitis and hepatocellular carcinoma. Cancer 2000, 89, 977–982. [Google Scholar] [CrossRef]
- Moon, W.S.; Park, H.S.; Yu, K.H.; Park, M.Y.; Kim, K.R.; Jang, K.Y.; Kim, J.S.; Cho, B.H. Expression of β cellulin and epidermal growth factor receptor in hepatocellular carcinoma: Implications for angiogenesis. Hum. Pathol. 2006, 37, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Inui, Y.; Higashiyama, S.; Kawata, S.; Tamura, S.; Miyagawa, J.; Taniguchi, N.; Matsuzawa, Y. Expression of heparin-binding epidermal growth factor in human hepatocellular carcinoma. Gastroenterology 1994, 107, 1799–1804. [Google Scholar] [PubMed]
- Castillo, J.; Erroba, E.; Perugorria, M.J.; Santamaria, M.; Lee, D.C.; Prieto, J.; Avila, M.A.; Berasain, C. Amphiregulin contributes to the transformed phenotype of human hepatocellular carcinoma cells. Cancer Res. 2006, 66, 6129–6138. [Google Scholar] [CrossRef] [PubMed]
- Buckley, A.F.; Burgart, L.J.; Sahai, V.; Kakar, S. Epidermal growth factor receptor expression and gene copy number in conventional hepatocellular carcinoma. Am. J. Clin. Pathol. 2008, 129, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Abu Dayyeh, B.K.; Yang, M.; Fuchs, B.C.; Karl, D.L.; Yamada, S.; Sninsky, J.J.; O’Brien, T.R.; Dienstag, J.L.; Tanabe, K.K.; Chung, R.T. A functional polymorphism in the epidermal growth factor gene is associated with risk for hepatocellular carcinoma. Gastroenterology 2011, 141, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K.K.; Lemoine, A.; Finkelstein, D.M.; Kawasaki, H.; Fujii, T.; Chung, R.T.; Lauwers, G.Y.; Kulu, Y.; Muzikansky, A.; Kuruppu, D.; et al. Epidermal growth factor gene functional polymorphism and the risk of hepatocellular carcinoma in patients with cirrhosis. JAMA 2008, 299, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Keng, V.W.; Sia, D.; Sarver, A.L.; Tschida, B.R.; Fan, D.; Alsinet, C.; Sole, M.; Lee, W.L.; Kuka, T.P.; Moriarity, B.S.; et al. Sex bias occurrence of hepatocellular carcinoma in poly7 molecular subclass is associated with EGFR. Hepatology 2013, 57, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Liu, Y. Expression of c-ErbB-2 protein and EGF receptor in hepatitis b, cirrhosis and hepatocellular carcinoma. Zhonghua Bing Li Xue Za Zhi Chin. J. Pathol. 1995, 24, 93–95. [Google Scholar]
- Kew, M.C. Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2011, 26 (Suppl. 1), 144–152. [Google Scholar] [CrossRef] [PubMed]
- Menzo, S.; Clementi, M.; Alfani, E.; Bagnarelli, P.; Iacovacci, S.; Manzin, A.; Dandri, M.; Natoli, G.; Levrero, M.; Carloni, G. Trans-activation of epidermal growth factor receptor gene by the hepatitis B virus x-gene product. Virology 1993, 196, 878–882. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, M.; Sato, C.; Sakai, K.; Konishi, M.; Tanaka, K.; Muraoka, M.; Kikuchi-Yanoshita, R.; Nadaoka, Y.; Kanda, H.; Kitagawa, T. Malignant transformation and EGFR activation of immortalized mouse liver epithelial cells caused by HBV enhancer-X from a human hepatocellular carcinoma. Int. J. Cancer 2000, 85, 518–522. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, Y.; Chen, J.; Cheng, G.; Xue, J. Transgenic mice expressing hepatitis B virus x protein are more susceptible to carcinogen induced hepatocarcinogenesis. Exp. Mol. Pathol. 2004, 76, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Chien, P.H.; Chen, W.S.; Chien, Y.F.; Hsu, Y.Y.; Wang, L.Y.; Chen, J.Y.; Lin, C.W.; Huang, T.C.; Yu, Y.L.; et al. Hepatitis B virus-encoded x protein downregulates EGFR expression via inducing microRNA-7 in hepatocellular carcinoma cells. Evid. Based Complement. Altern. Med. 2013, 2013, 682380. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, W.L.; Li, Q.; Qiao, Q. Expression of transforming growth factor-α and hepatitis B surface antigen in human hepatocellular carcinoma tissues and its significance. World J. Gastroenterol. 2004, 10, 830–833. [Google Scholar] [PubMed]
- Dai, K.; Huang, L.; Chen, J.; Yang, L.; Gong, Z. Amphiregulin promotes the immunosuppressive activity of intrahepatic CD4+ regulatory T cells to impair CD8+ T cell immunity against hepatitis B virus infection. Immunology 2014, 144, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A.; El-Hindawi, A.; Hammam, O.; Moussa, M.; Gabal, S.; Said, N. Impact of epidermal growth factor receptor and transforming growth factor-α on hepatitis C virus-induced hepatocarcinogenesis. APMIS 2015, 123, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Shehata, F.; Abdel Monem, N.; Sakr, M.; Kasem, S.; Balbaa, M. Epidermal growth factor, its receptor and transforming growth factor-β1 in the diagnosis of HCV-induced hepatocellular carcinoma. Med. Oncol. 2013, 30, 673. [Google Scholar] [CrossRef] [PubMed]
- Divella, R.; Daniele, A.; Gadaleta, C.; Tufaro, A.; Venneri, M.T.; Paradiso, A.; Quaranta, M. Circulating transforming growth factor-β and epidermal growth factor receptor as related to virus infection in liver carcinogenesis. Anticancer Res. 2012, 32, 141–145. [Google Scholar] [PubMed]
- Pei, R.; Chen, H.; Lu, L.; Zhu, W.; Beckebaum, S.; Cicinnati, V.; Lu, M.; Chen, X. Hepatitis c virus infection induces the expression of amphiregulin, a factor related to the activation of cellular survival pathways and required for efficient viral assembly. J. Gen. Virol. 2011, 92, 2237–2248. [Google Scholar] [CrossRef] [PubMed]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis c virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Kato, J.; Takimoto, R.; Takada, K.; Kawano, Y.; Miyanishi, K.; Kobune, M.; Sato, Y.; Takayama, T.; Matunaga, T.; et al. Hepatitis c virus core protein promotes proliferation of human hepatoma cells through enhancement of transforming growth factor α expression via activation of nuclear factor-κB. Gut 2006, 55, 1801–1808. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Pantua, H.; Ngu, H.; Komuves, L.; Diehl, L.; Schaefer, G.; Kapadia, S.B. Hepatitis C virus induces epidermal growth factor receptor activation via CD81 binding for viral internalization and entry. J. Virol. 2012, 86, 10935–10949. [Google Scholar] [CrossRef] [PubMed]
- Lupberger, J.; Zeisel, M.B.; Xiao, F.; Thumann, C.; Fofana, I.; Zona, L.; Davis, C.; Mee, C.J.; Turek, M.; Gorke, S.; et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011, 17, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Zona, L.; Lupberger, J.; Sidahmed-Adrar, N.; Thumann, C.; Harris, H.J.; Barnes, A.; Florentin, J.; Tawar, R.G.; Xiao, F.; Turek, M.; et al. HRAs signal transduction promotes hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell Host Microbe 2013, 13, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Ishida, H.; Yamane, D.; Yi, M.; Swinney, D.C.; Foung, S.; Lemon, S.M. Contrasting roles of mitogen-activated protein kinases in cellular entry and replication of hepatitis c virus: Mknk1 facilitates cell entry. J. Virol. 2013, 87, 4214–4224. [Google Scholar] [CrossRef] [PubMed]
- Mankouri, J.; Griffin, S.; Harris, M. The hepatitis c virus non-structural protein NS5A alters the trafficking profile of the epidermal growth factor receptor. Traffic 2008, 9, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Igloi, Z.; Kazlauskas, A.; Saksela, K.; Macdonald, A.; Mankouri, J.; Harris, M. Hepatitis c virus NS5A protein blocks epidermal growth factor receptor degradation via a proline motif-dependent interaction. J. Gen. Virol. 2015, 96, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Patton, J.B.; George, D.; Chang, K.O. Bile acids promote HCV replication through the EGFR/ERK pathway in replicon-harboring cells. Intervirology 2011, 54, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.; Kwon, Y.C.; Liu, S.; Hagedorn, C.H.; Ray, R.B.; Ray, R. Interferon-α inducible protein 6 impairs EGFR activation by CD81 and inhibits hepatitis c virus infection. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Lupberger, J.; Duong, F.H.; Fofana, I.; Zona, L.; Xiao, F.; Thumann, C.; Durand, S.C.; Pessaux, P.; Zeisel, M.B.; Heim, M.H.; et al. Epidermal growth factor receptor signaling impairs the antiviral activity of interferon-α. Hepatology 2013, 58, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.W.; Kwan, R.W.; Mak, P.H.; Mak, K.K.; Sham, M.H.; Chan, S.Y. Overexpression of epidermal growth factor induced hypospermatogenesis in transgenic mice. J. Biol. Chem. 2000, 275, 18297–18301. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.Y.; Wong, R.W. Expression of epidermal growth factor in transgenic mice causes growth retardation. J. Biol. Chem. 2000, 275, 38693–38698. [Google Scholar] [CrossRef] [PubMed]
- Jhappan, C.; Stahle, C.; Harkins, R.N.; Fausto, N.; Smith, G.H.; Merlino, G.T. TGF α overexpression in transgenic mice induces liver neoplasia and abnormal development of the mammary gland and pancreas. Cell 1990, 61, 1137–1146. [Google Scholar] [CrossRef]
- Sandgren, E.P.; Luetteke, N.C.; Palmiter, R.D.; Brinster, R.L.; Lee, D.C. Overexpression of TGF α in transgenic mice: Induction of epithelial hyperplasia, pancreatic metaplasia, and carcinoma of the breast. Cell 1990, 61, 1121–1135. [Google Scholar] [CrossRef]
- Cook, P.W.; Piepkorn, M.; Clegg, C.H.; Plowman, G.D.; DeMay, J.M.; Brown, J.R.; Pittelkow, M.R. Transgenic expression of the human amphiregulin gene induces a psoriasis-like phenotype. J. Clin. Investig. 1997, 100, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.R.; Dahlhoff, M.; Herbach, N.; Renner-Mueller, I.; Dalke, C.; Puk, O.; Graw, J.; Wanke, R.; Wolf, E. Βcellulin overexpression in transgenic mice causes disproportionate growth, pulmonary hemorrhage syndrome, and complex eye pathology. Endocrinology 2005, 146, 5237–5246. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, A.P.; Besner, G.E.; James, P.F.; Harding, P.A. Heparin-binding EGF-like growth factor (HB-EGF) overexpression in transgenic mice downregulates insulin-like growth factor binding protein (IGFBP)-3 and -4 mRNA. Growth Factors 2005, 23, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Dahlhoff, M.; Muller, A.K.; Wolf, E.; Werner, S.; Schneider, M.R. Epigen transgenic mice develop enlarged sebaceous glands. J. Investig. Dermatol. 2010, 130, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Webber, E.M.; Wu, J.C.; Wang, L.; Merlino, G.; Fausto, N. Overexpression of transforming growth factor-α causes liver enlargement and increased hepatocyte proliferation in transgenic mice. Am. J. Pathol. 1994, 145, 398–408. [Google Scholar] [PubMed]
- Pires, P.W.; Furtado, K.S.; Justullin, L.A., Jr.; Rodrigues, M.A.; Felisbino, S.L.; Barbisan, L.F. Chronic ethanol intake promotes double gluthatione S-transferase/transforming growth factor-α-positive hepatocellular lesions in male wistar rats. Cancer Sci. 2008, 99, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Thorgeirsson, S.S.; Santoni-Rugiu, E. Transgenic mouse models in carcinogenesis: Interaction of c-myc with transforming growth factor α and hepatocyte growth factor in hepatocarcinogenesis. Br. J. Clin. Pharmacol. 1996, 42, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Thorgeirsson, S.S.; Santoni-Rugiu, E.; Davis, C.D.; Snyderwine, E.G. Hepatic tumor induction in c-myc mono-transgenic and TGF-α/c-myc double-transgenic mice. Arch. Toxicol. Suppl. 1997, 19, 359–366. [Google Scholar] [PubMed]
- Komuves, L.G.; Feren, A.; Jones, A.L.; Fodor, E. Expression of epidermal growth factor and its receptor in cirrhotic liver disease. J. Histochem. Cytochem. 2000, 48, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Borlak, J.; Meier, T.; Halter, R.; Spanel, R.; Spanel-Borowski, K. Epidermal growth factor-induced hepatocellular carcinoma: Gene expression profiles in precursor lesions, early stage and solitary tumours. Oncogene 2005, 24, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, Y.; Villanueva, A.; Kobayashi, M.; Peix, J.; Chiang, D.Y.; Camargo, A.; Gupta, S.; Moore, J.; Wrobel, M.J.; Lerner, J.; et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 1995–2004. [Google Scholar] [CrossRef] [PubMed]
- Sagmeister, S.; Drucker, C.; Losert, A.; Grusch, M.; Daryabeigi, A.; Parzefall, W.; Rohr-Udilova, N.; Bichler, C.; Smedsrod, B.; Kandioler, D.; et al. HB-EGF is a paracrine growth stimulator for early tumor prestages in inflammation-associated hepatocarcinogenesis. J. Hepatol. 2008, 49, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Berasain, C.; Nicou, A.; Garcia-Irigoyen, O.; Latasa, M.U.; Urtasun, R.; Elizalde, M.; Salis, F.; Perugorria, M.J.; Prieto, J.; Recio, J.A.; et al. Epidermal growth factor receptor signaling in hepatocellular carcinoma: Inflammatory activation and a new intracellular regulatory mechanism. Dig. Dis. 2012, 30, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Latasa, M.U.; Salis, F.; Urtasun, R.; Garcia-Irigoyen, O.; Elizalde, M.; Uriarte, I.; Santamaria, M.; Feo, F.; Pascale, R.M.; Prieto, J.; et al. Regulation of amphiregulin gene expression by β-catenin signaling in human hepatocellular carcinoma cells: A novel crosstalk between FGF19 and the EGFR system. PLoS ONE 2012, 7, e52711. [Google Scholar] [CrossRef] [PubMed]
- Urtasun, R.; Latasa, M.U.; Demartis, M.I.; Balzani, S.; Goni, S.; Garcia-Irigoyen, O.; Elizalde, M.; Azcona, M.; Pascale, R.M.; Feo, F.; et al. Connective tissue growth factor autocriny in human hepatocellular carcinoma: Oncogenic role and regulation by epidermal growth factor receptor/yes-associated protein-mediated activation. Hepatology 2011, 54, 2149–2158. [Google Scholar] [CrossRef] [PubMed]
- Mazzocca, A.; Fransvea, E.; Dituri, F.; Lupo, L.; Antonaci, S.; Giannelli, G. Down-regulation of connective tissue growth factor by inhibition of transforming growth factor β blocks the tumor-stroma cross-talk and tumor progression in hepatocellular carcinoma. Hepatology 2010, 51, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Fabregat, I. NADPH oxidase nox1 controls autocrine growth of liver tumor cells through up-regulation of the epidermal growth factor receptor pathway. J. Biol. Chem. 2010, 285, 24815–24824. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B.; Joe, A.K. Mechanisms of disease: Oncogene addiction—A rationale for molecular targeting in cancer therapy. Nat. Clin. Pract. Oncol. 2006, 3, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Schafer, B.; Gschwind, A.; Ullrich, A. Multiple G-protein-coupled receptor signals converge on the epidermal growth factor receptor to promote migration and invasion. Oncogene 2004, 23, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Reschke, M.; Ferby, I.; Stepniak, E.; Seitzer, N.; Horst, D.; Wagner, E.F.; Ullrich, A. Mitogen-inducible gene-6 is a negative regulator of epidermal growth factor receptor signaling in hepatocytes and human hepatocellular carcinoma. Hepatology 2010, 51, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.Y.; Chen, L.; Chen, H.Y.; Hu, L.; Li, L.; Sun, H.Y.; Jiang, F.; Zhao, J.; Liu, G.M.; Tang, J.; et al. Muc15 inhibits dimerization of EGFR and PI3K-Akt signaling and is associated with aggressive hepatocellular carcinomas in patients. Gastroenterology 2013, 145, 1436–1448, e1431–e1412. [Google Scholar] [CrossRef] [PubMed]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in myd88-dependent IL-6 production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Huether, A.; Hopfner, M.; Sutter, A.P.; Schuppan, D.; Scherubl, H. Erlotinib induces cell cycle arrest and apoptosis in hepatocellular cancer cells and enhances chemosensitivity towards cytostatics. J. Hepatol. 2005, 43, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, M.; Sutter, A.P.; Huether, A.; Schuppan, D.; Zeitz, M.; Scherubl, H. Targeting the epidermal growth factor receptor by Gefitinib for treatment of hepatocellular carcinoma. J. Hepatol. 2004, 41, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Okano, J.; Matsumoto, K.; Nagahara, T.; Murawaki, Y. Gefitinib and the modulation of the signaling pathways downstream of epidermal growth factor receptor in human liver cancer cells. J. Gastroenterol. 2006, 41, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Huether, A.; Hopfner, M.; Sutter, A.P.; Baradari, V.; Schuppan, D.; Scherubl, H. Signaling pathways involved in the inhibition of epidermal growth factor receptor by erlotinib in hepatocellular cancer. World J. Gastroenterol. 2006, 12, 5160–5167. [Google Scholar] [PubMed]
- Schiffer, E.; Housset, C.; Cacheux, W.; Wendum, D.; Desbois-Mouthon, C.; Rey, C.; Clergue, F.; Poupon, R.; Barbu, V.; Rosmorduc, O. Gefitinib, an EGFR inhibitor, prevents hepatocellular carcinoma development in the rat liver with cirrhosis. Hepatology 2005, 41, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, M.; Sakurai, H.; Saiki, I. ZD1839, a selective epidermal growth factor receptor tyrosine kinase inhibitor, shows antimetastatic activity using a hepatocellular carcinoma model. Mol. Cancer Ther. 2003, 2, 557–561. [Google Scholar] [PubMed]
- Zhu, B.D.; Yuan, S.J.; Zhao, Q.C.; Li, X.; Li, Y.; Lu, Q.Y. Antitumor effect of Gefitinib, an epidermal growth factor receptor tyrosine kinase inhibitor, combined with cytotoxic agent on murine hepatocellular carcinoma. World J. Gastroenterol. 2005, 11, 1382–1386. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Basaki, Y.; Yoshie, M.; Ogawa, K.; Sakisaka, S.; Kuwano, M.; Ono, M. PTEN/Akt signaling through epidermal growth factor receptor is prerequisite for angiogenesis by hepatocellular carcinoma cells that is susceptible to inhibition by Gefitinib. Cancer Res. 2006, 66, 5346–5353. [Google Scholar] [CrossRef] [PubMed]
- Ueno, Y.; Sakurai, H.; Matsuo, M.; Choo, M.K.; Koizumi, K.; Saiki, I. Selective inhibition of TNF-α-induced activation of mitogen-activated protein kinases and metastatic activities by Gefitinib. Br. J. Cancer 2005, 92, 1690–1695. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Arteaga, C.L. Critical update and emerging trends in epidermal growth factor receptor targeting in cancer. J. Clin. Oncol. 2005, 23, 2445–2459. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J. Clinical implications of the mechanism of epidermal growth factor receptor inhibitors. Cancer 2006, 107, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G. Targeting HER1/EGFR in cancer therapy: Experience with erlotinib. Future Oncol. 2005, 1, 449–460. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, P.J.; Giantonio, B.J.; Levy, D.E.; Kauh, J.S.; Fitzgerald, D.B.; Benson, A.B. Gefitinib in advanced unresectable hepatocellular carcinoma: Results from the Eastern Cooperative Oncology Group’s Study E1203. J. Clin. Oncol. 2006, 24, 213S. [Google Scholar]
- Llovet, J.M.; Bruix, J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008, 48, 1312–1327. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.K.; Belani, C.P.; Singh, D.A.; Tanaka, M.; Lenz, H.J.; Yen, Y.; Kindler, H.L.; Iqbal, S.; Longmate, J.; Mack, P.C.; et al. A phase II study of lapatinib in patients with advanced biliary tree and hepatocellular cancer. Cancer Chemother. Pharmacol. 2009, 64, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Philip, P.A.; Mahoney, M.R.; Allmer, C.; Thomas, J.; Pitot, H.C.; Kim, G.; Donehower, R.C.; Fitch, T.; Picus, J.; Erlichman, C. Phase II study of erlotinib (OSI-774) in patients with advanced hepatocellular cancer. J. Clin. Oncol. 2005, 23, 6657–6663. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.B.; Chadha, R.; Glover, K.; Wang, X.; Morris, J.; Brown, T.; Rashid, A.; Dancey, J.; Abbruzzese, J.L. Phase 2 study of erlotinib in patients with unresectable hepatocellular carcinoma. Cancer 2007, 110, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Stuart, K.; Blaszkowsky, L.S.; Muzikansky, A.; Reitberg, D.P.; Clark, J.W.; Enzinger, P.C.; Bhargava, P.; Meyerhardt, J.A.; Horgan, K.; et al. Phase 2 study of cetuximab in patients with advanced hepatocellular carcinoma. Cancer 2007, 110, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Gruenwald, V.; Wilkens, L.; Gebel, M.; Greten, T.F.; Kubicka, S.; Ganser, A.; Manns, M.P.; Malek, N.P. A phase II open-label study of cetuximab in unresectable hepatocellular carcinoma: Final results. J. Clin. Oncol. 2007, 25, 4598. [Google Scholar]
- Asnacios, A.; Fartoux, L.; Romano, O.; Tesmoingt, C.; Louafi, S.S.; Mansoubakht, T.; Artru, P.; Poynard, T.; Rosmorduc, O.; Hebbar, M.; et al. Gemcitabine plus oxaliplatin (GEMOX) combined with cetuximab in patients with progressive advanced stage hepatocellular carcinoma: Results of a multicenter phase 2 study. Cancer 2008, 112, 2733–2739. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.H.; Kang, Y.K.; Yang, T.S.; Shun, C.T.; Shao, Y.Y.; Su, W.C.; Sandoval-Tan, J.; Chiou, T.J.; Jin, K.; Hsu, C.; et al. Bevacizumab with erlotinib as first-line therapy in asian patients with advanced hepatocellular carcinoma: A multicenter phase ii study. Oncology 2013, 85, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Philip, P.A.; Mahoney, M.R.; Holen, K.D.; Northfelt, D.W.; Pitot, H.C.; Picus, J.; Flynn, P.J.; Erlichman, C. Phase 2 study of bevacizumab plus erlotinib in patients with advanced hepatocellular cancer. Cancer 2012, 118, 2424–2430. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.B.; Morris, J.S.; Chadha, R.; Iwasaki, M.; Kaur, H.; Lin, E.; Kaseb, A.; Glover, K.; Davila, M.; Abbruzzese, J. Phase II trial of the combination of bevacizumab and erlotinib in patients who have advanced hepatocellular carcinoma. J. Clin. Oncol. 2009, 27, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Blivet-Van Eggelpoel, M.J.; Chettouh, H.; Fartoux, L.; Aoudjehane, L.; Barbu, V.; Rey, C.; Priam, S.; Housset, C.; Rosmorduc, O.; Desbois-Mouthon, C. Epidermal growth factor receptor and HER-3 restrict cell response to sorafenib in hepatocellular carcinoma cells. J. Hepatol. 2012, 57, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Rosmorduc, O.; Evans, T.R.; Ross, P.J.; Santoro, A.; Carrilho, F.J.; Bruix, J.; Qin, S.; Thuluvath, P.J.; Llovet, J.M.; et al. Search: A phase iii, randomized, double-blind, placebo-controlled trial of sorafenib plus erlotinib in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 2015, 33, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Su, M.C.; Lien, H.C.; Jeng, Y.M. Absence of epidermal growth factor receptor exon 18–21 mutation in hepatocellular carcinoma. Cancer Lett. 2005, 224, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Lim, S.G.; Soo, R.; Hsieh, W.S.; Guo, J.Y.; Putti, T.; Tao, Q.; Soong, R.; Goh, B.C. Lack of somatic mutations in EGFR tyrosine kinase domain in hepatocellular and nasopharyngeal carcinoma. Pharmacogenet. Genom. 2006, 16, 73–74. [Google Scholar] [CrossRef]
- Fuchs, B.C.; Fujii, T.; Dorfman, J.D.; Goodwin, J.M.; Zhu, A.X.; Lanuti, M.; Tanabe, K.K. Epithelial-to-mesenchymal transition and integrin-linked kinase mediate sensitivity to epidermal growth factor receptor inhibition in human hepatoma cells. Cancer Res. 2008, 68, 2391–2399. [Google Scholar] [CrossRef] [PubMed]
- Losert, A.; Lotsch, D.; Lackner, A.; Koppensteiner, H.; Peter-Vorosmarty, B.; Steiner, E.; Holzmann, K.; Grunt, T.; Schmid, K.; Marian, B.; et al. The major vault protein mediates resistance to epidermal growth factor receptor inhibition in human hepatoma cells. Cancer Lett. 2012, 319, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Desbois-Mouthon, C.; Baron, A.; Blivet-Van Eggelpoel, M.J.; Fartoux, L.; Venot, C.; Bladt, F.; Housset, C.; Rosmorduc, O. Insulin-like growth factor-1 receptor inhibition induces a resistance mechanism via the epidermal growth factor receptor/HER3/Akt signaling pathway: Rational basis for cotargeting insulin-like growth factor-1 receptor and epidermal growth factor receptor in hepatocellular carcinoma. Clin. Cancer Res. 2009, 15, 5445–5456. [Google Scholar] [PubMed]
- Desbois-Mouthon, C.; Cacheux, W.; Blivet-Van Eggelpoel, M.J.; Barbu, V.; Fartoux, L.; Poupon, R.; Housset, C.; Rosmorduc, O. Impact of IGF-1R/EGFR cross-talks on hepatoma cell sensitivity to gefitinib. Int. J. Cancer 2006, 119, 2557–2566. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K.; DeFrances, M.C. Liver regeneration. Science 1997, 276, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Scheving, L.A.; Stevenson, M.C.; Taylormoore, J.M.; Traxler, P.; Russell, W.E. Integral role of the EGF receptor in HGF-mediated hepatocyte proliferation. Biochem. Biophys. Res. Commun. 2002, 290, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Gusenbauer, S.; Vlaicu, P.; Ullrich, A. HGF induces novel EGFR functions involved in resistance formation to tyrosine kinase inhibitors. Oncogene 2013, 32, 3846–3856. [Google Scholar] [CrossRef] [PubMed]
- Ou, C.; Wu, F.X.; Luo, Y.; Cao, J.; Zhao, Y.N.; Yuan, W.P.; Li, Y.; Su, J.J. Expression and significance of epidermal growth factor receptor variant type III in hepatocellular carcinoma. Ai Zheng 2005, 24, 166–169. [Google Scholar] [PubMed]
- Zhou, M.; Gong, B.; Gu, J.; Li, Z. EGFRVIII mRNA detection in the serum of patients with hepatocellular carcinoma. Liver Int. 2010, 30, 925–927. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jiang, H.; Zhou, M.; Xu, Z.; Liu, S.; Shi, B.; Yao, X.; Yao, M.; Gu, J.; Li, Z. Epidermal growth factor receptor viii enhances tumorigenicity and resistance to 5-fluorouracil in human hepatocellular carcinoma. Cancer Lett. 2009, 279, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wang, H.; Tan, Z.; Hu, S.; Wang, H.; Shi, B.; Yang, L.; Li, P.; Gu, J.; Wang, H.; et al. Growth suppression of human hepatocellular carcinoma xenografts by a monoclonal antibody ch12 directed to epidermal growth factor receptor variant iii. J. Biol. Chem. 2011, 286, 5913–5920. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Dong, Q.; Luo, X.; Shi, B.; Wang, H.; Gao, H.; Kong, J.; Zhang, J.; Li, Z. The monoclonal antibody ch12 augments 5-fluorouracil-induced growth suppression of hepatocellular carcinoma xenografts expressing epidermal growth factor receptor variant iii. Cancer Lett. 2014, 342, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jiang, H.; Gao, H.; Kong, J.; Zhang, P.; Hu, S.; Shi, B.; Zhang, P.; Yao, M.; Li, Z. The monoclonal antibody ch12 enhances the sorafenib-mediated growth inhibition of hepatocellular carcinoma xenografts expressing epidermal growth factor receptor variant iii. Neoplasia 2012, 14, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Alpini, G.; McGill, J.M.; Larusso, N.F. The pathobiology of biliary epithelia. Hepatology 2002, 35, 1256–1268. [Google Scholar] [CrossRef] [PubMed]
- Wehbe, H.; Henson, R.; Meng, F.; Mize-Berge, J.; Patel, T. Interleukin-6 contributes to growth in cholangiocarcinoma cells by aberrant promoter methylation and gene expression. Cancer Res. 2006, 66, 10517–10524. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Takeda, T.; Sasaki, Y.; Sakon, M.; Yamada, T.; Ishiguro, S.; Imaoka, S.; Tsujimoto, M.; Higashiyama, S.; Monden, M.; et al. Expression and clinical significance of the ErbB family in intrahepatic cholangiocellular carcinoma. Pathol. Res. Pract. 2001, 197, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Altimari, A.; Fiorentino, M.; Gabusi, E.; Gruppioni, E.; Corti, B.; D’Errico, A.; Grigioni, W.F. Investigation of ErbB1 and ErbB2 expression for therapeutic targeting in primary liver tumours. Dig. Liver Dis. 2003, 35, 332–338. [Google Scholar] [CrossRef]
- Nakazawa, K.; Dobashi, Y.; Suzuki, S.; Fujii, H.; Takeda, Y.; Ooi, A. Amplification and overexpression of c-ErbB-2, epidermal growth factor receptor, and c-Met in biliary tract cancers. J. Pathol. 2005, 206, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Nonomura, A.; Ohta, G.; Nakanuma, Y.; Izumi, R.; Mizukami, Y.; Matsubara, F.; Hayashi, M.; Watanabe, K.; Takayanagi, N. Simultaneous detection of epidermal growth factor receptor (EGF-R), epidermal growth factor (EGF) and ras p21 in cholangiocarcinoma by an immunocytochemical method. Liver 1988, 8, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Jan, Y.Y.; Yeh, T.S.; Yeh, J.N.; Yang, H.R.; Chen, M.F. Expression of epidermal growth factor receptor, apomucins, matrix metalloproteinases, and p53 in rat and human cholangiocarcinoma: Appraisal of an animal model of cholangiocarcinoma. Ann. Surg. 2004, 240, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, D.; Ojima, H.; Iwasaki, M.; Hiraoka, N.; Kosuge, T.; Kasai, S.; Hirohashi, S.; Shibata, T. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br. J. Cancer 2008, 98, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, W.; Wang, C.; Wang, L.; Yang, M.; Qi, M.; Su, H.; Sun, X.; Liu, Z.; Zhang, J.; et al. Characterization of EGFR family gene aberrations in cholangiocarcinoma. Oncol. Rep. 2014, 32, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Harder, J.; Waiz, O.; Otto, F.; Geissler, M.; Olschewski, M.; Weinhold, B.; Blum, H.E.; Schmitt-Graeff, A.; Opitz, O.G. EGFR and HER2 expression in advanced biliary tract cancer. World J. Gastroenterol. 2009, 15, 4511–4517. [Google Scholar] [CrossRef] [PubMed]
- Bekaii-Saab, T.; Williams, N.; Plass, C.; Calero, M.V.; Eng, C. A novel mutation in the tyrosine kinase domain of ErbB2 in hepatocellular carcinoma. BMC Cancer 2006, 6, 278. [Google Scholar] [CrossRef] [PubMed]
- Leone, F.; Cavalloni, G.; Pignochino, Y.; Sarotto, I.; Ferraris, R.; Piacibello, W.; Venesio, T.; Capussotti, L.; Risio, M.; Aglietta, M. Somatic mutations of epidermal growth factor receptor in bile duct and gallbladder carcinoma. Clin. Cancer Res. 2006, 12, 1680–1685. [Google Scholar] [CrossRef] [PubMed]
- Gwak, G.Y.; Yoon, J.H.; Shin, C.M.; Ahn, Y.J.; Chung, J.K.; Kim, Y.A.; Kim, T.Y.; Lee, H.S. Detection of response-predicting mutations in the kinase domain of the epidermal growth factor receptor gene in cholangiocarcinomas. J. Cancer Res. Clin. Oncol. 2005, 131, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, J.S.; Kang, C.D.; Lee, S.J.; Kim, J.Y.; Yeon, J.E.; Park, J.J.; Shim, J.J.; Byun, K.S.; Bak, Y.T.; et al. Expression of epidermal growth factor receptor, ErbB2 and matrix metalloproteinase-9 in hepatolithiasis and cholangiocarcinoma. Korean J. Gastroenterol. 2005, 45, 52–59. [Google Scholar] [PubMed]
- Zhou, Q.; Gong, Y.; Huang, F.; Lin, Q.; Zeng, B.; Li, Z.; Chen, R. Expression levels and significance of nuclear factor-κB and epidermal growth factor receptor in hepatolithiasis associated with intrahepatic cholangiocarcinoma. Dig. Surg. 2013, 30, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Sung, R.; Lee, S.H.; Ji, M.; Han, J.H.; Kang, M.H.; Kim, J.H.; Choi, J.W.; Kim, Y.C.; Park, S.M. Epithelial-mesenchymal transition-related protein expression in biliary epithelial cells associated with hepatolithiasis. J. Gastroenterol. Hepatol. 2014, 29, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Claperon, A.; Mergey, M.; Nguyen Ho-Bouldoires, T.H.; Vignjevic, D.; Wendum, D.; Chretien, Y.; Merabtene, F.; Frazao, A.; Paradis, V.; Housset, C.; et al. EGF/EGFR axis contributes to the progression of cholangiocarcinoma through the induction of an epithelial-mesenchymal transition. J. Hepatol. 2014, 61, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Claperon, A.; Guedj, N.; Mergey, M.; Vignjevic, D.; Desbois-Mouthon, C.; Boissan, M.; Saubamea, B.; Paradis, V.; Housset, C.; Fouassier, L. Loss of EBP50 stimulates EGFR activity to induce EMT phenotypic features in biliary cancer cells. Oncogene 2012, 31, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- Carraway, K.L.; Ramsauer, V.P.; Haq, B.; Carothers Carraway, C.A. Cell signaling through membrane mucins. BioEssays 2003, 25, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Hollingsworth, M.A. Cell surface-associated mucins in signal transduction. Trends Cell Biol. 2006, 16, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Pochampalli, M.R.; el Bejjani, R.M.; Schroeder, J.A. Muc1 is a novel regulator of ErbB1 receptor trafficking. Oncogene 2007, 26, 1693–1701. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Yu, G.R.; Yoo, H.J.; Kim, J.H.; Yoon, B.I.; Choi, Y.K.; Kim, D.G. Anxa8 down-regulation by EGF-FOXO4 signaling is involved in cell scattering and tumor metastasis of cholangiocarcinoma. Gastroenterology 2009, 137, 1138–1150, 1150 e1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, J.; Kikuchi, K.; Mizuguchi, Y.; Kawahigashi, Y.; Yoshida, H.; Uchida, E.; Takizawa, T. miR-376c down-regulation accelerates EGF-dependent migration by targeting GRB2 in the HuCCT1 human intrahepatic cholangiocarcinoma cell line. PLoS ONE 2013, 8, e69496. [Google Scholar] [CrossRef] [PubMed]
- Gui, A.; Kobayashi, A.; Motoyama, H.; Kitazawa, M.; Takeoka, M.; Miyagawa, S. Impaired degradation followed by enhanced recycling of epidermal growth factor receptor caused by hypo-phosphorylation of tyrosine 1045 in rbe cells. BMC Cancer 2012, 12, 179. [Google Scholar] [CrossRef] [PubMed]
- Claperon, A.; Mergey, M.; Aoudjehane, L.; Ho-Bouldoires, T.H.; Wendum, D.; Prignon, A.; Merabtene, F.; Firrincieli, D.; Desbois-Mouthon, C.; Scatton, O.; et al. Hepatic myofibroblasts promote the progression of human cholangiocarcinoma through activation of epidermal growth factor receptor. Hepatology 2013, 58, 2001–2011. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hausmann, M.; Dietmaier, W.; Kellermeier, S.; Pesch, T.; Stieber-Gunckel, M.; Lippert, E.; Klebl, F.; Rogler, G. Expression of growth factor receptors and targeting of EGFR in cholangiocarcinoma cell lines. BMC Cancer 2010, 10, 302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Li, L.; Yang, X.; Wang, B.; Zhao, J.; Lu, S.; Yu, X. Expression and activation of EGFR and STAT3 during the multistage carcinogenesis of intrahepatic cholangiocarcinoma induced by 3′-methyl-4 dimethylaminoazobenzene in rats. J. Toxicol. Pathol. 2015, 28, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Sirica, A.E. Role of ErbB family receptor tyrosine kinases in intrahepatic cholangiocarcinoma. World J. Gastroenterol. 2008, 14, 7033–7058. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.B.; Spee, B.; Blechacz, B.R.; Avital, I.; Komuta, M.; Barbour, A.; Conner, E.A.; Gillen, M.C.; Roskams, T.; Roberts, L.R.; et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology 2012, 142, 1021–1031 e1015. [Google Scholar] [CrossRef] [PubMed]
- Philip, P.A.; Mahoney, M.R.; Allmer, C.; Thomas, J.; Pitot, H.C.; Kim, G.; Donehower, R.C.; Fitch, T.; Picus, J.; Erlichman, C. Phase ii study of erlotinib in patients with advanced biliary cancer. J. Clin. Oncol. 2006, 24, 3069–3074. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.W.; Wang, C.H.; Hsieh, C.B. Effects of the anti-epidermal growth factor receptor antibody cetuximab on cholangiocarcinoma of the liver. Onkologie 2007, 30, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Sprinzl, M.F.; Schimanski, C.C.; Moehler, M.; Schadmand-Fischer, S.; Galle, P.R.; Kanzler, S. Gemcitabine in combination with EGF-receptor antibody (Cetuximab) as a treatment of cholangiocarcinoma: A case report. BMC Cancer 2006, 6. [Google Scholar] [CrossRef] [PubMed]
- Paule, B.; Herelle, M.O.; Rage, E.; Ducreux, M.; ADAM, R.; Guettier, C.; Bralet, M.P. Cetuximab plus gemcitabine-oxaliplatin (GEMOX) in patients with refractory advanced intrahepatic cholangiocarcinomas. Oncology 2007, 72, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Siegel-Lakhai, W.S.; Beijnen, J.H.; Vervenne, W.L.; Boot, H.; Keessen, M.; Versola, M.; Koch, K.M.; Smith, D.A.; Pandite, L.; Richel, D.J.; et al. Phase I pharmacokinetic study of the safety and tolerability of lapatinib (GW572016) in combination with oxaliplatin/fluorouracil/leucovorin (FOLFOX4) in patients with solid tumors. Clin. Cancer Res. 2007, 13, 4495–4502. [Google Scholar] [CrossRef] [PubMed]
- Changbumrung, S.; Tungtrongchitr, R.; Migasena, P.; Chamroenngan, S. Serum unconjugated primary and secondary bile acids in patients with cholangiocarcinoma and hepatocellular carcinoma. J. Med. Assoc. Thail. 1990, 73, 81–90. [Google Scholar]
- Kinami, Y.; Ashida, Y.; Gotoda, H.; Seto, K.; Kojima, Y.; Takashima, S. Promoting effects of bile acid load on the occurrence of cholangiocarcinoma induced by diisopropanolnitrosamine in hamsters. Oncology 1993, 50, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Kinami, Y.; Miyakoshi, M.; Fujikawa, K. Bile acid load on the DNA distribution pattern of bile ductules and cholangiocarcinoma induced by diisopropanolnitrosamine in hamsters. Oncology 1998, 55, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhao, R.; Zhou, X.; Liang, X.; Campbell, D.J.; Zhang, X.; Zhang, L.; Shi, R.; Wang, G.; Pandak, W.M.; et al. Conjugated bile acids promote cholangiocarcinoma cell invasive growth through activation of sphingosine 1-phosphate receptor 2. Hepatology 2014, 60, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.; Alpini, G.; Francis, H. Bile acid signaling and biliary functions. Acta Pharm. Sin. B 2015, 5, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Higuchi, H.; Werneburg, N.W.; Kaufmann, S.H.; Gores, G.J. Bile acids induce cyclooxygenase-2 expression via the epidermal growth factor receptor in a human cholangiocarcinoma cell line. Gastroenterology 2002, 122, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Werneburg, N.W.; Higuchi, H.; Canbay, A.E.; Kaufmann, S.H.; Akgul, C.; Edwards, S.W.; Gores, G.J. Bile acids inhibit Mcl-1 protein turnover via an epidermal growth factor receptor/Raf-1-dependent mechanism. Cancer Res. 2002, 62, 6500–6505. [Google Scholar] [PubMed]
- Kim, K.M.; Yoon, J.H.; Gwak, G.Y.; Kim, W.; Lee, S.H.; Jang, J.J.; Lee, H.S. Bile acid-mediated induction of cyclooxygenase-2 and Mcl-1 in hepatic stellate cells. Biochem. Biophys. Res. Commun. 2006, 342, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jiang, L.; Sun, Q.; Peng, T.; Lou, K.; Liu, N.; Leng, J. Prostaglandin e2 enhances mitogen-activated protein kinase/ERK pathway in human cholangiocarcinoma cells: Involvement of ep1 receptor, calcium and EGF receptors signaling. Mol. Cell. Biochem. 2007, 305, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Gwak, G.Y.; Lee, H.S.; Bronk, S.F.; Werneburg, N.W.; Gores, G.J. Enhanced epidermal growth factor receptor activation in human cholangiocarcinoma cells. J. Hepatol. 2004, 41, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Bird, T.G.; Lorenzini, S.; Forbes, S.J. Activation of stem cells in hepatic diseases. Cell Tissue Res. 2008, 331, 283–300. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Russo, F.P.; Parola, M. Stem and progenitor cells in liver regeneration and repair. Cytotherapy 2011, 13, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Roskams, T.; Katoonizadeh, A.; Komuta, M. Hepatic progenitor cells: An update. Clin. Liver Dis. 2010, 14, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Rountree, C.B.; Mishra, L.; Willenbring, H. Stem cells in liver diseases and cancer: Recent advances on the path to new therapies. Hepatology 2012, 55, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Bisgaard, H.C.; Santoni-Rugiu, E.; Thorgeirsson, S.S. In vivo infusion of growth factors enhances the mitogenic response of rat hepatic ductal (oval) cells after administration of 2-acetylaminofluorene. Hepatology 1996, 23, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Kitade, M.; Factor, V.M.; Andersen, J.B.; Tomokuni, A.; Kaji, K.; Akita, H.; Holczbauer, A.; Seo, D.; Marquardt, J.U.; Conner, E.A.; et al. Specific fate decisions in adult hepatic progenitor cells driven by Met and EGFR signaling. Genes Dev. 2013, 27, 1706–1717. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Samuelson, L.; Cui, C.B.; Sun, Y.; Gerber, D.A. MAPK/ERK and Wnt/β-catenin pathways are synergistically involved in proliferation of Sca-1 positive hepatic progenitor cells. Biochem. Biophys. Res. Commun. 2011, 409, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Huitfeldt, H.S.; Skarpen, E.; Lindeman, B.; Becher, R.; Thrane, E.V.; Schwarze, P.E. Differential distribution of Met and epidermal growth factor receptor in normal and carcinogen-treated rat liver. J. Histochem. Cytochem. Soc. 1996, 44, 227–233. [Google Scholar] [CrossRef]
- Hu, Z.; Evarts, R.P.; Fujio, K.; Omori, N.; Omori, M.; Marsden, E.R.; Thorgeirsson, S.S. Expression of transforming growth factor α/epidermal growth factor receptor, hepatocyte growth factor/c-Met and acidic fibroblast growth factor/fibroblast growth factor receptors during hepatocarcinogenesis. Carcinogenesis 1996, 17, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Evarts, R.P.; Nakatsukasa, H.; Marsden, E.R.; Hu, Z.; Thorgeirsson, S.S. Expression of transforming growth factor-α in regenerating liver and during hepatic differentiation. Mol. Carcinogenes. 1992, 5, 25–31. [Google Scholar] [CrossRef]
- Evarts, R.P.; Hu, Z.; Fujio, K.; Marsden, E.R.; Thorgeirsson, S.S. Activation of hepatic stem cell compartment in the rat: Role of transforming growth factor α, hepatocyte growth factor, and acidic fibroblast growth factor in early proliferation. Cell Growth Differ. 1993, 4, 555–561. [Google Scholar] [PubMed]
- Isfort, R.J.; Cody, D.B.; Stuard, S.B.; Randall, C.J.; Miller, C.; Ridder, G.M.; Doersen, C.J.; Richards, W.G.; Yoder, B.K.; Wilkinson, J.E.; et al. The combination of epidermal growth factor and transforming growth factor-β induces novel phenotypic changes in mouse liver stem cell lines. J. Cell Sci. 1997, 110 Pt 24, 3117–3129. [Google Scholar] [PubMed]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liu, T.; Cong, M.; Wu, X.; Bai, Y.; Yin, C.; An, W.; Wang, B.; Jia, J.; You, H. Expression of extracellular matrix genes in cultured hepatic oval cells: An origin of hepatic stellate cells through transforming growth factor β? Liver Int. 2009, 29, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Ding, J.; Chen, C.; Sun, W.; Ning, B.F.; Wen, W.; Huang, L.; Han, T.; Yang, W.; Wang, C.; et al. Hepatic transforming growth factor β gives rise to tumor-initiating cells and promotes liver cancer development. Hepatology 2012, 56, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I.; Sanchez, A.; Alvarez, A.M.; Nakamura, T.; Benito, M. Epidermal growth factor, but not hepatocyte growth factor, suppresses the apoptosis induced by transforming growth factor-β in fetal hepatocytes in primary culture. FEBS Lett. 1996, 384, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Palacian, A.; del Castillo, G.; Herrera, B.; Fernandez, M.; Roncero, C.; Fabregat, I.; Sanchez, A. EGFR is dispensable for c-Met-mediated proliferation and survival activities in mouse adult liver oval cells. Cell. Signal. 2012, 24, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Yang, A.T.; Cong, M.; Liu, T.H.; Zhang, D.; Huang, J.; Tong, X.F.; Zhu, S.T.; Xu, Y.; Tang, S.Z.; et al. EGF suppresses the initiation and drives the reversion of TGF-β1-induced transition in hepatic oval cells showing the plasticity of progenitor cells. J. Cell. Physiol. 2015, 230, 2362–2370. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Rimassa, L.; Borbath, I.; Daniele, B.; Salvagni, S.; Van Laethem, J.L.; Van Vlierberghe, H.; Trojan, J.; Kolligs, F.T.; Weiss, A.; et al. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: A randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013, 14, 55–63. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komposch, K.; Sibilia, M. EGFR Signaling in Liver Diseases. Int. J. Mol. Sci. 2016, 17, 30. https://doi.org/10.3390/ijms17010030
Komposch K, Sibilia M. EGFR Signaling in Liver Diseases. International Journal of Molecular Sciences. 2016; 17(1):30. https://doi.org/10.3390/ijms17010030
Chicago/Turabian StyleKomposch, Karin, and Maria Sibilia. 2016. "EGFR Signaling in Liver Diseases" International Journal of Molecular Sciences 17, no. 1: 30. https://doi.org/10.3390/ijms17010030
APA StyleKomposch, K., & Sibilia, M. (2016). EGFR Signaling in Liver Diseases. International Journal of Molecular Sciences, 17(1), 30. https://doi.org/10.3390/ijms17010030