
Streamlining the Pipeline for Generation of Recombinant Affinity Reagents by Integrating the Affinity Maturation Step

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
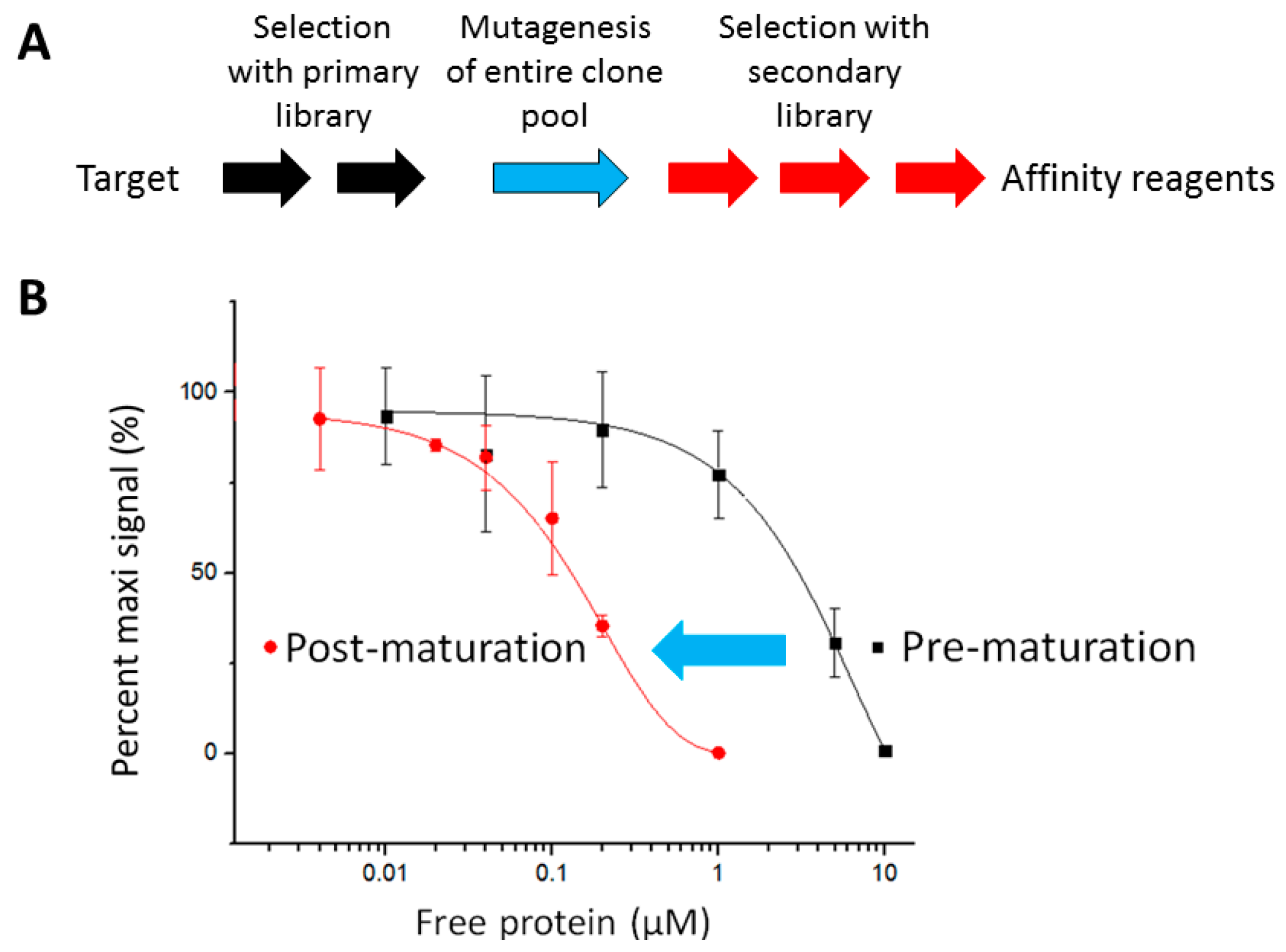
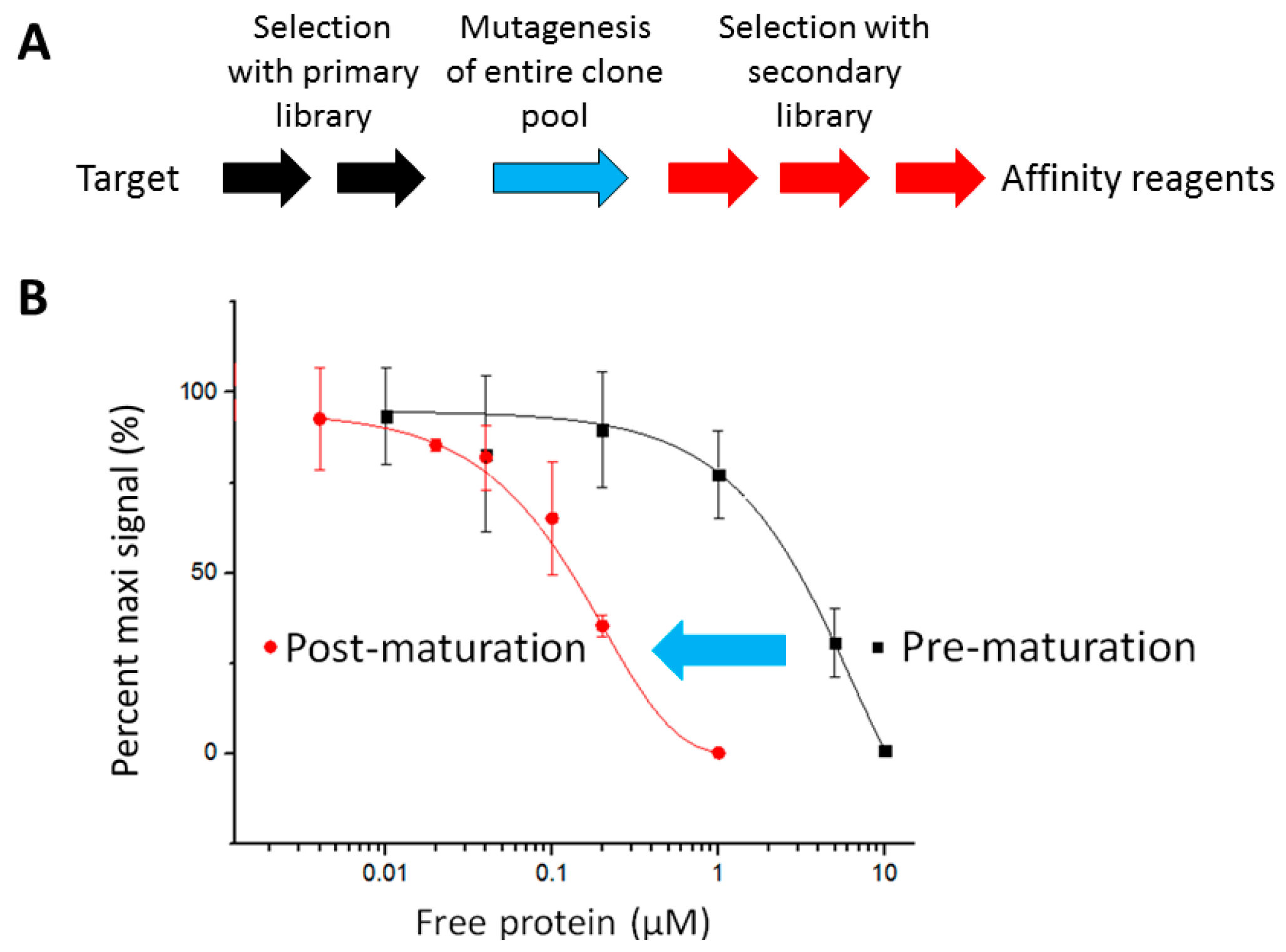
2.1. The Pipeline
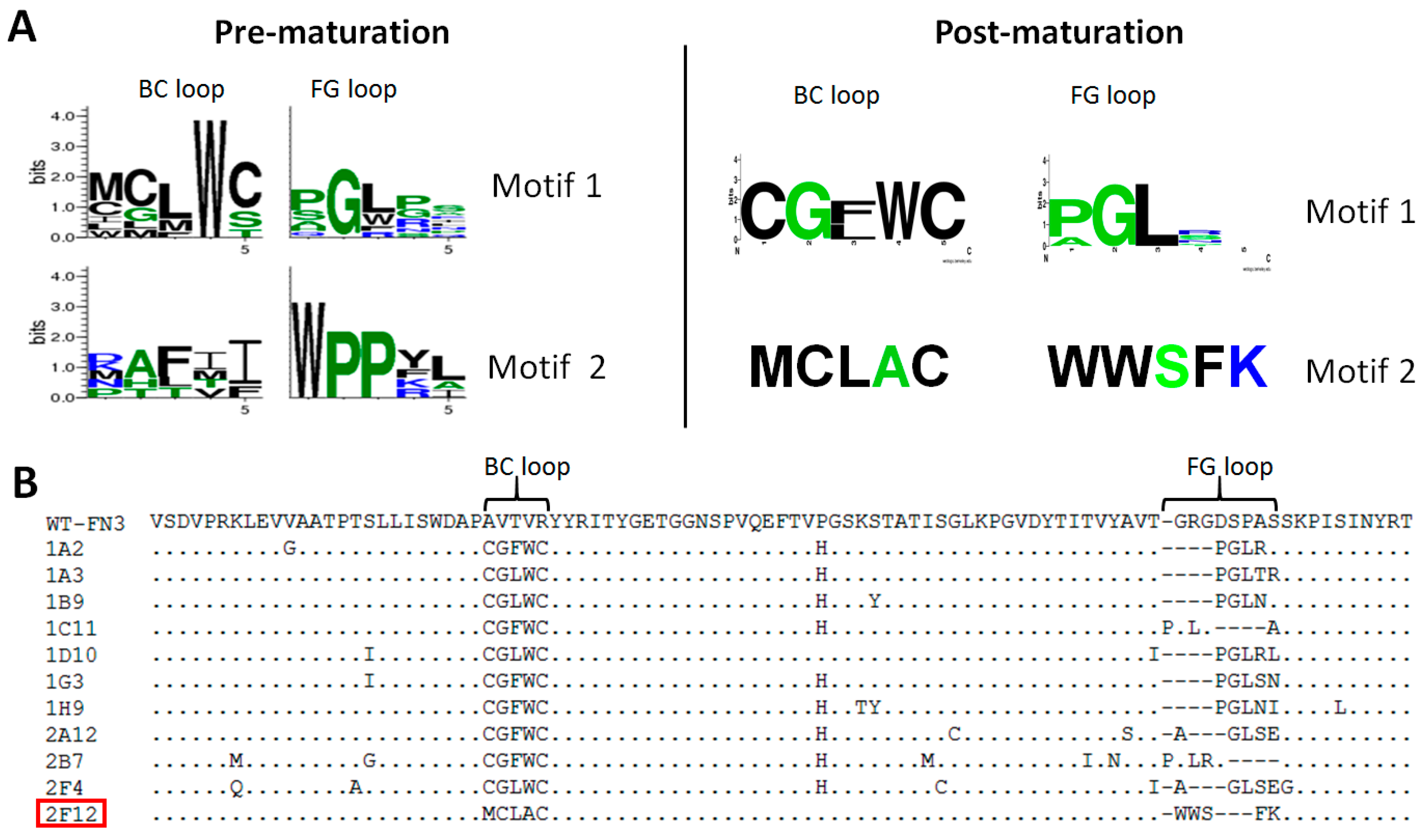
2.2. Affinity Selection and Mutagenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targets | Full Names | Uniprot ID | Biological Processes | Antigens (Amino Acids #) |
|---|---|---|---|---|
| CDC34 | ubiquitin-conjugating enzyme E2 R1 | P49427 | ubiquitin ligase activity | 7–184 |
| CDK2 | cyclin-dependent kinase 2 | P24941 | cell-cycle control | 3–286 |
| COPS5 | COP9 signalosome complex subunit 5 | Q92905 | deubiquitination, JNK signaling, secretion | 9–309 |
| CTBP1 | c-terminal-binding protein 1 | Q13363 | corepressor of transcriptional regulators | 20–440 |
| MAP2K5 | mitogen-activated protein kinase kinase 5 | Q13163 | scaffold for the formation of a signaling process | 5–108 |
| PAK1 | p-21 protein activated kinase 1 | Q13153 | regulation of cell-proliferation, apoptosis | 258–544 |
| PLAA | phospholipase A-2-activating protein | Q9Y263 | maintenance of ubiquitin levels | 338–795 |
| RAB6B | Ras-related protein-6B | Q9NRW1 | retrograde membrane trafficking via Golgi | 6–182 |
| SF3A1 | Splicing factor 3A subunit 1 | Q15459 | mRNA processing, mRNA splicing | 423–790 |
| TDP43 | TAR DNA-binding protein 43 | Q13148 | regulation of transcription and splicing | 1–106 |
| USP11 | Ubiquitin carboxyl-terminal hydrolase 11 | P51784 | deubiquitination, regulator of NF-kappa-B activation | 61–285 |
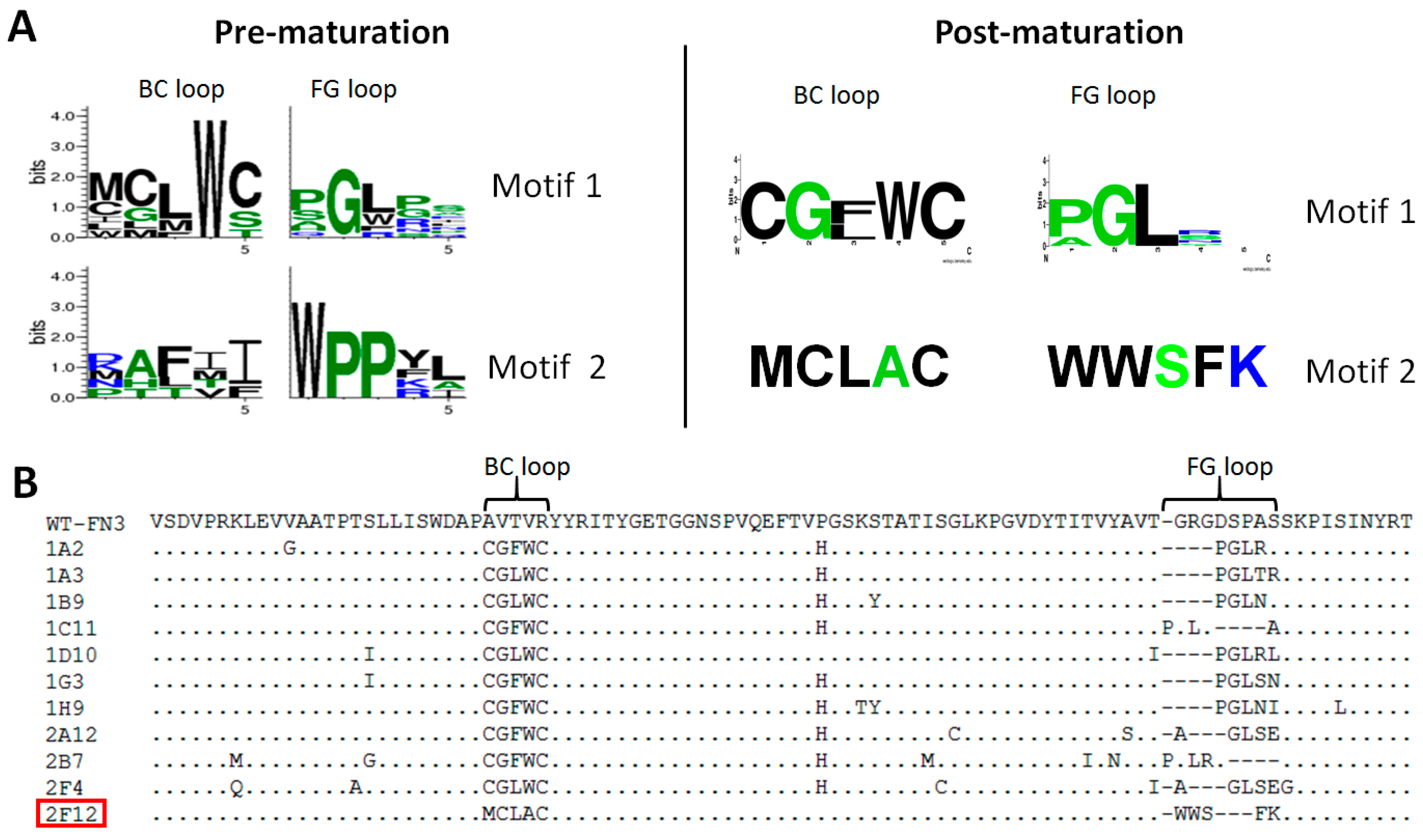
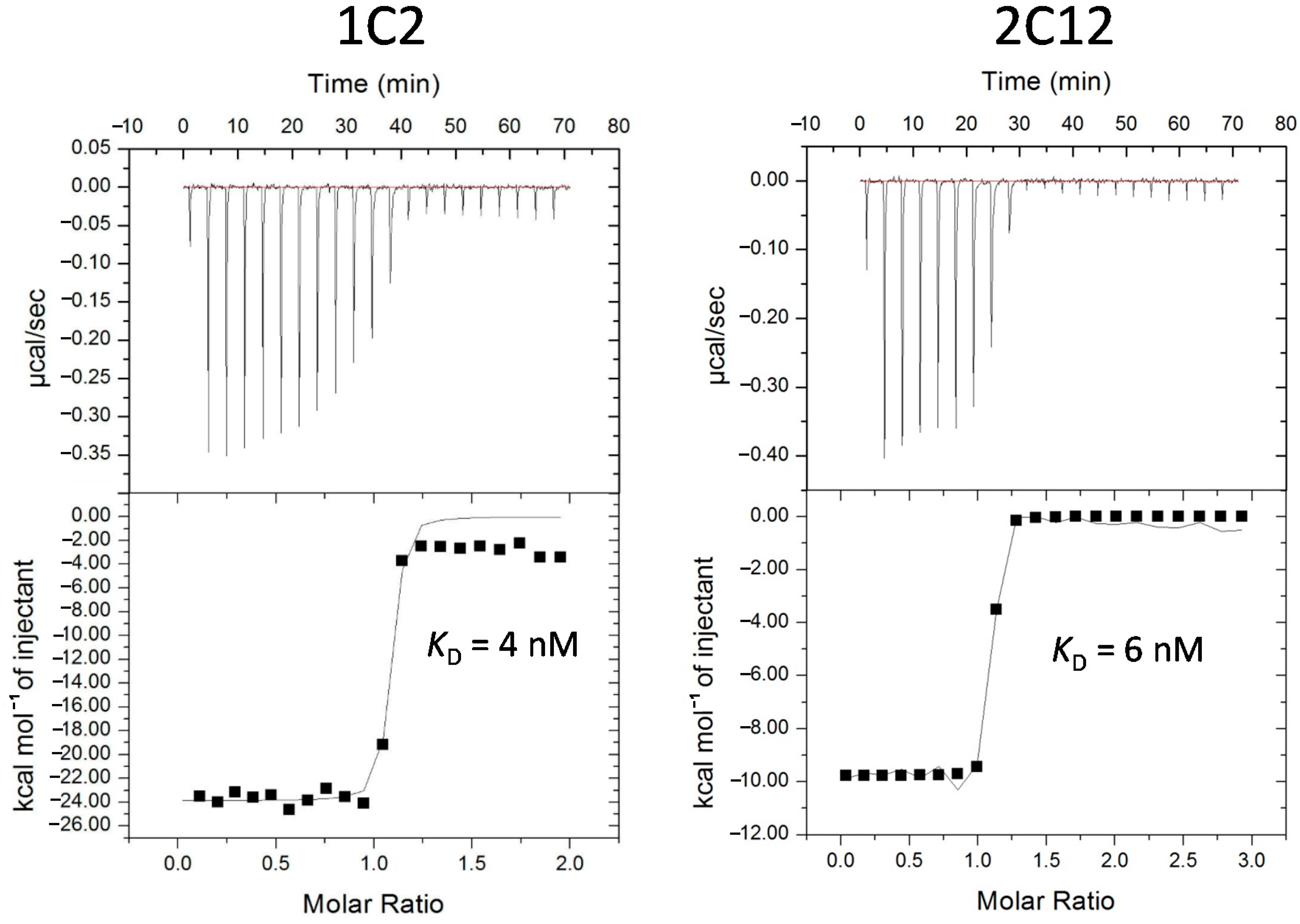
2.3. Characterization of Affinity Matured Monobodies
| Targets | Clones | BC Loop (26–30) | FG Loop (77–81) | Framework Mutations | Affinity (nM) | |
|---|---|---|---|---|---|---|
| ELISA | ITC | |||||
| CDC34 | 1D10 | CGLWC | PGLRL | S17I, T76I | <50 | N/D |
| COPS5 | 1D7 | RRWDV | WGIII | None | <10 | N/D |
| MAP2K5 | 1C4 | CRKCL | RLEWL | P51H, K83N | 6 | 11 |
| 2C12 | CRKCL | RLEFL | None | 17 | 6 | |
| SF3A1 | 1E2 | ALPVY | VWWYE | None | <50 | N/D |
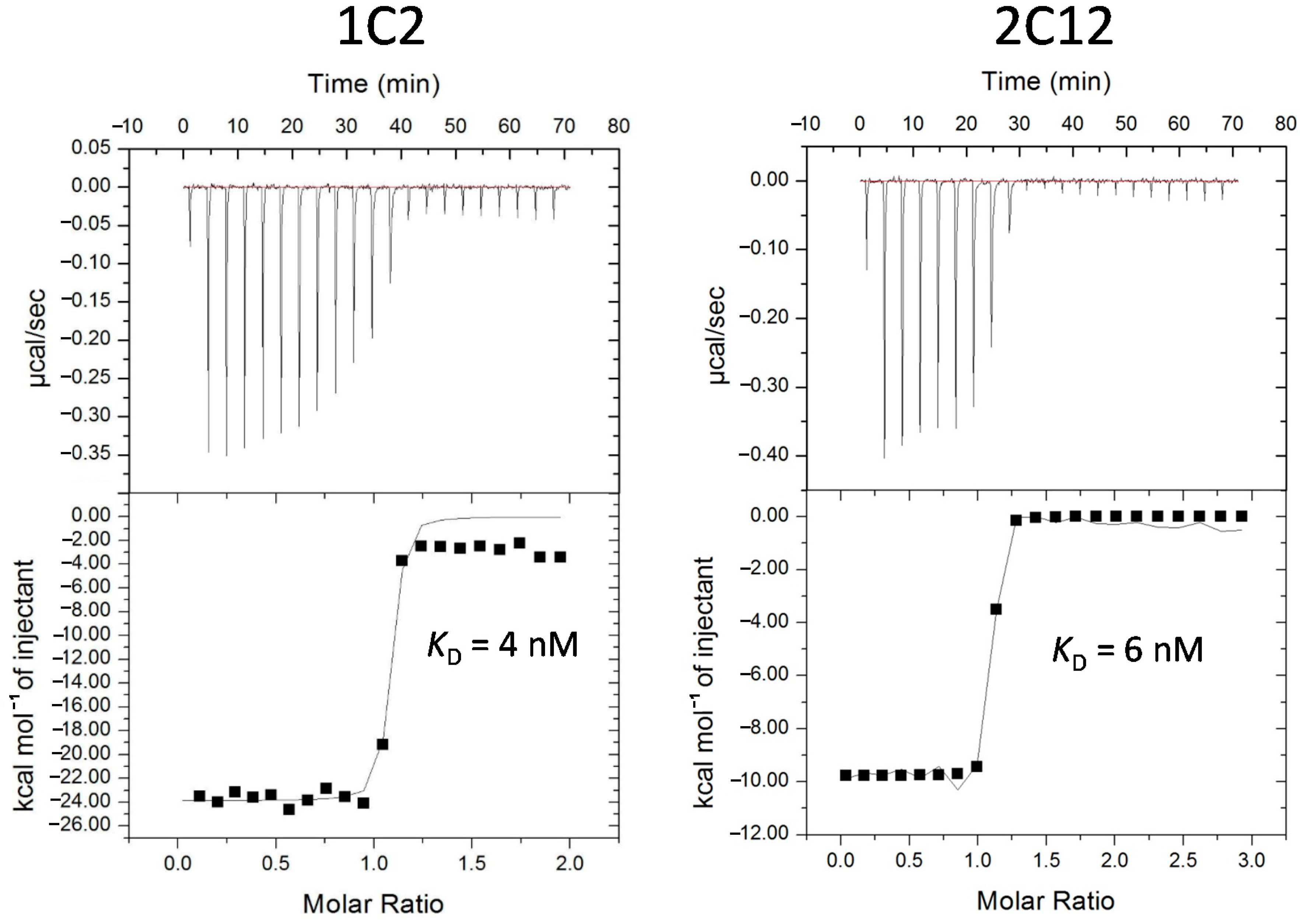
| USP11 | 1C2 | WWVPQ | PGIYQ | L18M, G61C, G65D, S82I | N/D | 4 |
| 1A9 | WWSVP | PGIYA | D67V, S82I, Y92C | 52 | N/D | |
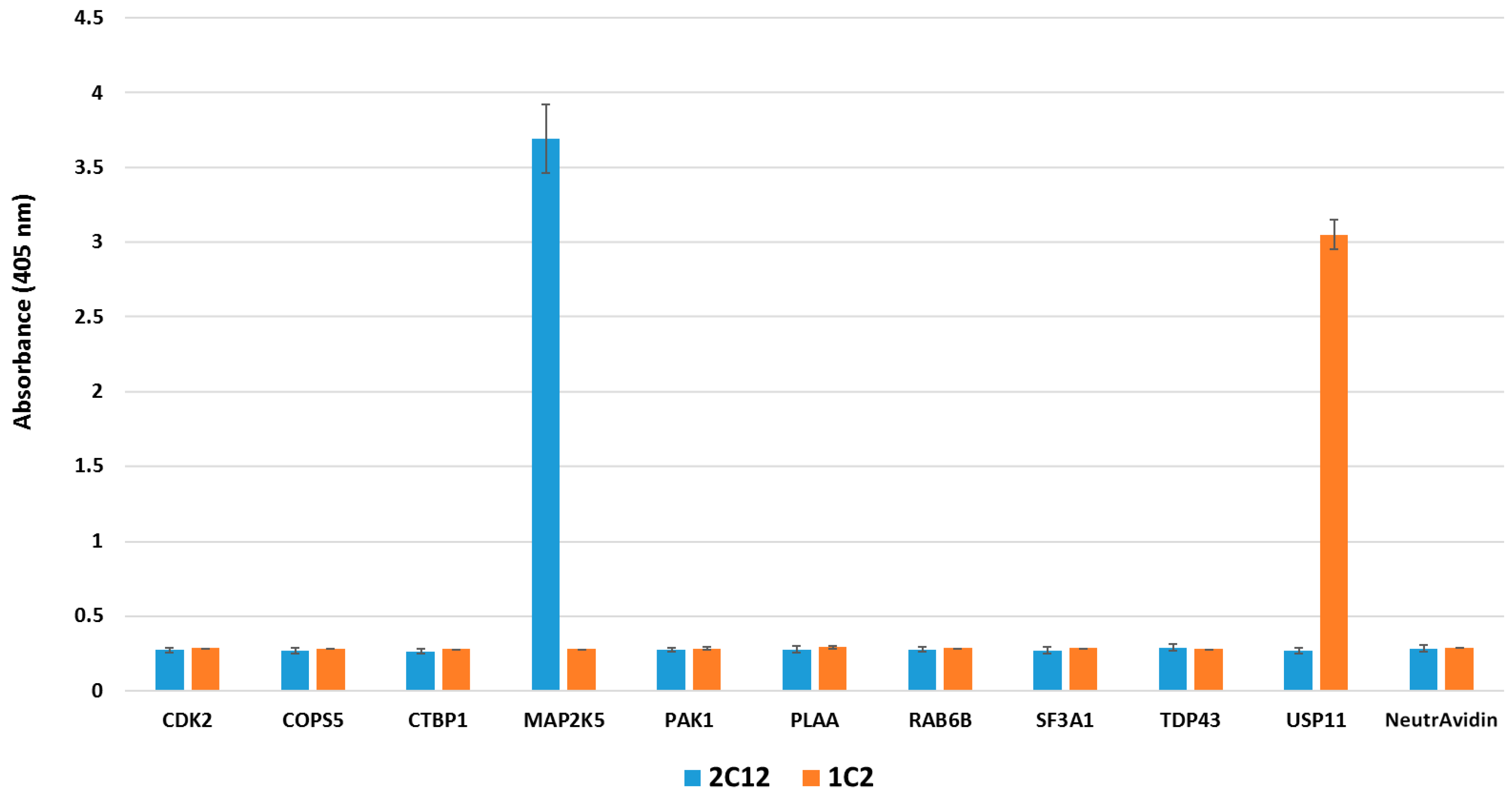
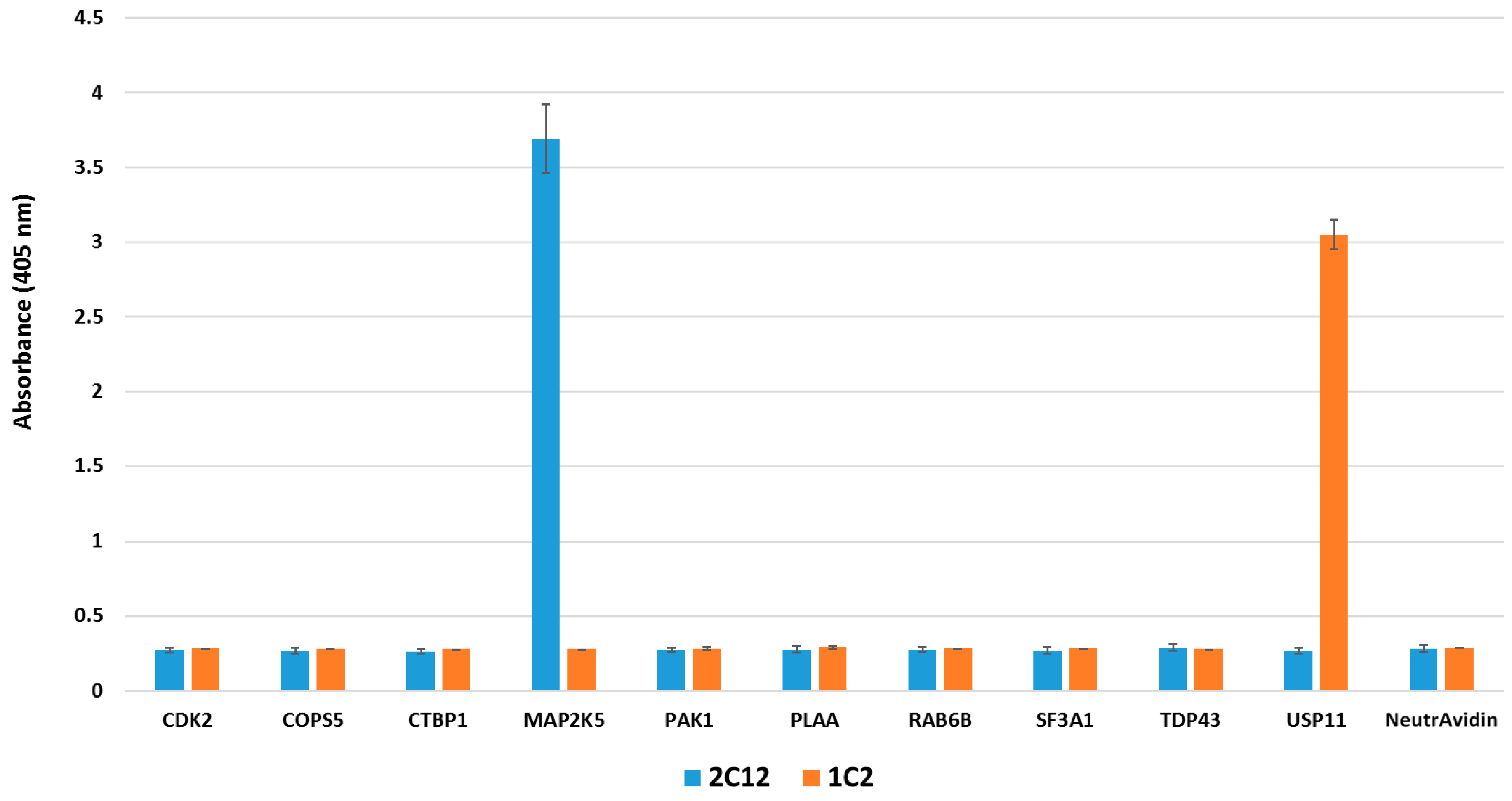
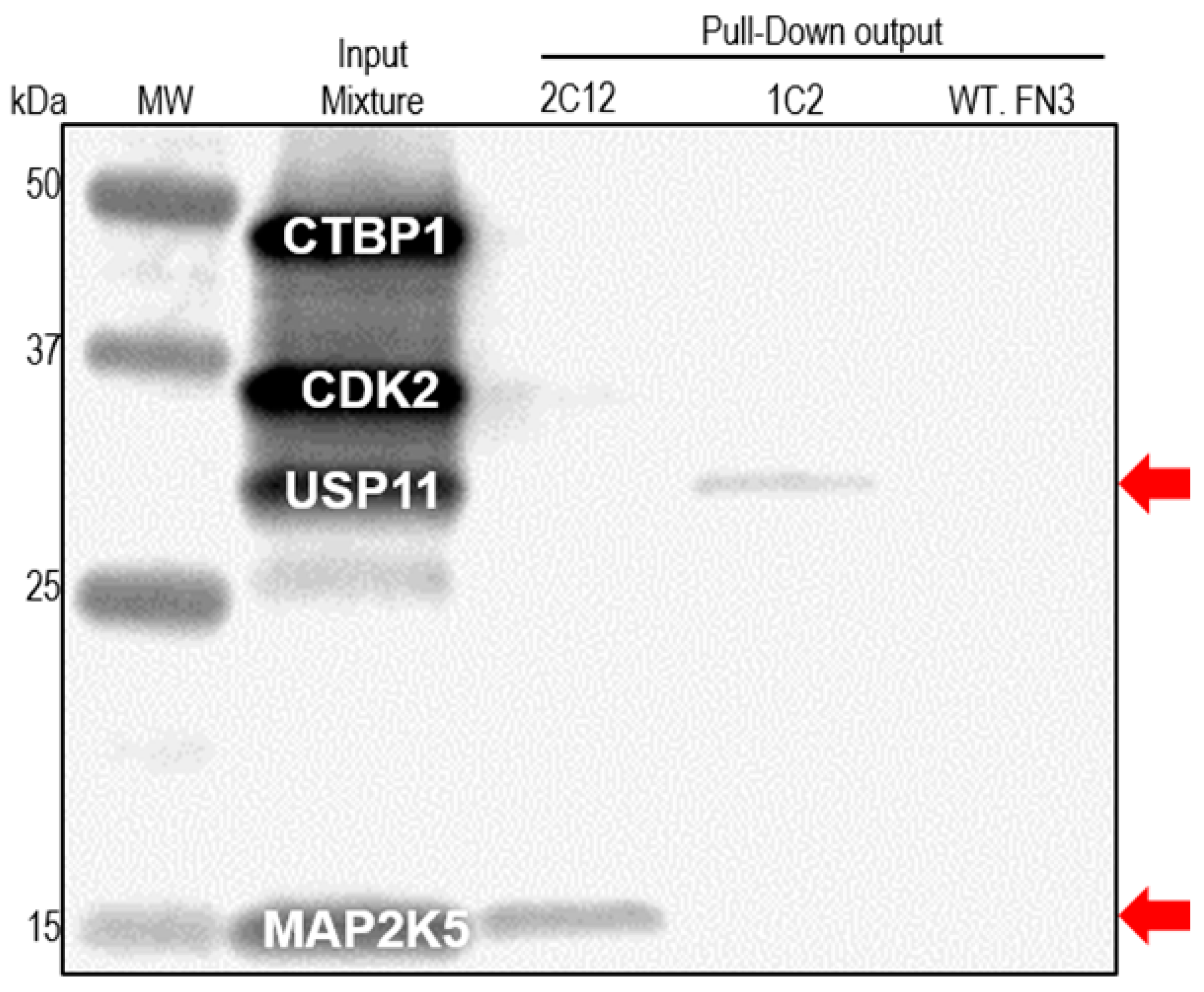
2.4. Pull-down Experiments with a Spiked HeLa Cell Lysate
3. Experimental Section
3.1. Subcloning, Overexpression and Purification of Antigens and Monobodies
3.2. Affinity Selection of the Primary Library
3.3. Secondary Library Construction and Affinity Selection
3.4. Competition ELISA to Estimate Binding Strength
3.5. Isothermal Titration Calorimetry (ITC)
3.6. Pull-down Assays
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bradbury, A.R.; Sidhu, S.; Dubel, S.; McCafferty, J. Beyond natural antibodies: The power of in vitro display technologies. Nat. Biotechnol. 2011, 29, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, A.; Pluckthun, A. Reproducibility: Standardize antibodies used in research. Nature 2015, 518, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Hosse, R.J.; Rothe, A.; Power, B.E. A new generation of protein display scaffolds for molecular recognition. Protein Sci. 2006, 15, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Wahlberg, E.; Lendel, C.; Helgstrand, M.; Allard, P.; Dincbas-Renqvist, V.; Hedqvist, A.; Berglund, H.; Nygren, P.A.; Hard, T. An affibody in complex with a target protein: Structure and coupled folding. Proc. Natl. Acad. Sci. USA 2003, 100, 3185–3190. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, F.Y.; Tolmachev, V. Affibody molecules: New protein domains for molecular imaging and targeted tumor therapy. Curr. Opin. Drug Discov. Dev. 2007, 10, 167–175. [Google Scholar]
- Skerra, A. Lipocalins as a scaffold. Biochim. Biophys. Acta 2000, 1482, 337–350. [Google Scholar] [CrossRef]
- Skerra, A. Alternative binding proteins: Anticalins-harnessing the structural plasticity of the lipocalin ligand pocket to engineer novel binding activities. FEBS J. 2008, 275, 2677–2683. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.; Lu, Q.; Bakker, A.; To, W.; Duguay, A.; Alba, B.M.; Smith, R.; Rivas, A.; Li, P.; Le, H.; et al. Multivalent avimer proteins evolved by exon shuffling of a family of human receptor domains. Nat. Biotechnol. 2005, 23, 1556–1561. [Google Scholar] [CrossRef] [PubMed]
- Binz, H.K.; Stumpp, M.T.; Forrer, P.; Amstutz, P.; Pluckthun, A. Designing repeat proteins: Well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J. Mol. Biol. 2003, 332, 489–503. [Google Scholar] [CrossRef]
- Boersma, Y.L.; Pluckthun, A. Darpins and other repeat protein scaffolds: Advances in engineering and applications. Curr. Opin. Biotechnol. 2011, 22, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Koide, A.; Bailey, C.W.; Huang, X.; Koide, S. The fibronectin type III domain as a scaffold for novel binding proteins. J. Mol. Biol. 1998, 284, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Karatan, E.; Merguerian, M.; Han, Z.; Scholle, M.D.; Koide, S.; Kay, B.K. Molecular recognition properties of FN3 monobodies that bind the Src SH3 domain. Chem. Biol. 2004, 11, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Koide, A.; Koide, S. Monobodies: Antibody mimics based on the scaffold of the fibronectin type III domain. Methods Mol. Biol. 2007, 352, 95–109. [Google Scholar] [PubMed]
- Lipovsek, D. Adnectins: Engineered target-binding protein therapeutics. Protein Eng. Des. Sel. 2011, 24, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Fellouse, F.A.; Wiesmann, C.; Sidhu, S.S. Synthetic antibodies from a four-amino-acid code: A dominant role for tyrosine in antigen recognition. Proc. Natl. Acad. Sci. USA 2004, 101, 12467–12472. [Google Scholar] [CrossRef] [PubMed]
- Birtalan, S.; Zhang, Y.; Fellouse, F.A.; Shao, L.; Schaefer, G.; Sidhu, S.S. The intrinsic contributions of tyrosine, serine, glycine and arginine to the affinity and specificity of antibodies. J. Mol. Biol. 2008, 377, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Miersch, S.; Sidhu, S.S. Synthetic antibodies: Concepts, potential and practical considerations. Methods 2012, 57, 486–498. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.L. Antibody fragments: Hope and hype. MAbs 2010, 2, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Z.A.; Yeap, S.K.; Ali, A.M.; Ho, W.Y.; Alitheen, N.B.; Hamid, M. scFv antibody: Principles and clinical application. Clin. Dev. Immunol. 2012, 2012, 980250. [Google Scholar] [CrossRef] [PubMed]
- De Marco, A. Biotechnological applications of recombinant single-domain antibody fragments. Microb. Cell Fact. 2011, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, T.; Muyldermans, S.; Depicker, A. Nanobody-based products as research and diagnostic tools. Trends Biotechnol. 2014, 32, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, R. Advantages of mRNA display selections over other selection techniques for investigation of protein-protein interactions. Expert Rev. Proteome 2011, 8, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, J.W.; Kay, B.K. Filamentous phage display in the new millennium. Chem. Rev. 2005, 105, 4056–4072. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.M.; Weiss, G.A. Optimizing the affinity and specificity of proteins with molecular display. Mol. Biosyst. 2006, 2, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Vodnik, M.; Zager, U.; Strukelj, B.; Lunder, M. Phage display: Selecting straws instead of a needle from a haystack. Molecules 2011, 16, 790–817. [Google Scholar] [CrossRef] [PubMed]
- Pluckthun, A. Ribosome display: A perspective. Methods Mol. Biol. 2012, 805, 3–28. [Google Scholar] [PubMed]
- Feldhaus, M.J.; Siegel, R.W. Yeast display of antibody fragments: A discovery and characterization platform. J. Immunol. Methods 2004, 290, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Pepper, L.R.; Cho, Y.K.; Boder, E.T.; Shusta, E.V. A decade of yeast surface display technology: Where are we now? Comb. Chem. High Throughput Screen. 2008, 11, 127–134. [Google Scholar] [PubMed]
- Gera, N.; Hussain, M.; Rao, B.M. Protein selection using yeast surface display. Methods 2013, 60, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, R.C.; Joyce, G.F. Mutagenic PCR. Genome Res. 1994, 3, S136–S140. [Google Scholar] [CrossRef]
- Zaccolo, M.; Williams, D.M.; Brown, D.M.; Gherardi, E. An approach to random mutagenesis of DNA using mixtures of triphosphate derivatives of nucleoside analogues. J. Mol. Biol. 1996, 255, 589–603. [Google Scholar] [CrossRef] [PubMed]
- McCullum, E.O.; Williams, B.A.; Zhang, J.; Chaput, J.C. Random mutagenesis by error-prone PCR. Methods Mol. Biol. 2010, 634, 103–109. [Google Scholar] [PubMed]
- Stemmer, W.P. DNA shuffling by random fragmentation and reassembly: In vitro recombination for molecular evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 10747–10751. [Google Scholar] [CrossRef] [PubMed]
- Graddis, T.J.; Remmele, R.L., Jr.; McGrew, J.T. Designing proteins that work using recombinant technologies. Curr. Pharm. Biotechnol. 2002, 3, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Binkowski, B.F.; Richmond, K.E.; Kaysen, J.; Sussman, M.R.; Belshaw, P.J. Correcting errors in synthetic DNA through consensus shuffling. Nucleic Acids Res. 2005, 33, e55. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, I.; Kojoh, K.; Tabata, N.; Doi, N.; Takashima, H.; Miyamoto-Sato, E.; Yanagawa, H. In vitro evolution of single-chain antibodies using mRNA display. Nucleic Acids Res. 2006, 34, e127. [Google Scholar] [CrossRef] [PubMed]
- Thie, H.; Voedisch, B.; Dubel, S.; Hust, M.; Schirrmann, T. Affinity maturation by phage display. Methods Mol. Biol. 2009, 525, 309–322. [Google Scholar] [PubMed]
- Jermutus, L.; Honegger, A.; Schwesinger, F.; Hanes, J.; Pluckthun, A. Tailoring in vitro evolution for protein affinity or stability. Proc. Natl. Acad. Sci. USA 2001, 98, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Zahnd, C.; Sarkar, C.A.; Pluckthun, A. Computational analysis of off-rate selection experiments to optimize affinity maturation by directed evolution. Protein Eng. Des. Sel. 2010, 23, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Detanico, T.; St Clair, J.B.; Aviszus, K.; Kirchenbaum, G.; Guo, W.; Wysocki, L.J. Somatic mutagenesis in autoimmunity. Autoimmunity 2013, 46, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T.A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. USA 1985, 82, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Fang, P.; Kay, B.K. Improvements to the Kunkel mutagenesis protocol for constructing primary and secondary phage-display libraries. Methods 2012, 58, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Holland, E.G.; Buhr, D.L.; Acca, F.E.; Alderman, D.; Bovat, K.; Busygina, V.; Kay, B.K.; Weiner, M.P.; Kiss, M.M. AXM mutagenesis: An efficient means for the production of libraries for directed evolution of proteins. J. Immunol. Methods 2013, 394, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, J.; Hantschel, O.; Grebien, F.; Kaupe, I.; Bennett, K.L.; Barkinge, J.; Jones, R.B.; Koide, A.; Superti-Furga, G.; Koide, S. A potent and highly specific FN3 monobody inhibitor of the abl SH2 domain. Nat. Struct. Mol. Biol. 2010, 17, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Grebien, F.; Hantschel, O.; Wojcik, J.; Kaupe, I.; Kovacic, B.; Wyrzucki, A.M.; Gish, G.D.; Cerny-Reiterer, S.; Koide, A.; Beug, H.; et al. Targeting the SH2-kinase interface in Bcr-Abl inhibits leukemogenesis. Cell 2011, 147, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Wu, J.; Valencia, C.A.; Liu, R. Fibronectin type III domain based monobody with high avidity. Biochemistry 2007, 46, 12656–12664. [Google Scholar] [CrossRef] [PubMed]
- Hackel, B.J.; Kapila, A.; Wittrup, K.D. Picomolar affinity fibronectin domains engineered utilizing loop length diversity, recursive mutagenesis, and loop shuffling. J. Mol. Biol. 2008, 381, 1238–1252. [Google Scholar] [CrossRef] [PubMed]
- Koide, A.; Gilbreth, R.N.; Esaki, K.; Tereshko, V.; Koide, S. High-affinity single-domain binding proteins with a binary-code interface. Proc. Natl. Acad. Sci. USA 2007, 104, 6632–6637. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.A.; Liao, H.I.; Sun, R.; Roberts, R.W. mRNA display selection of a high-affinity, modification-specific phospho-ikappabalpha-binding fibronectin. ACS Chem. Biol. 2008, 3, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.H.; Chen, Y.; Danehy, F.; Dufu, K.; Ekstrom, J.; Getmanova, E.; Gokemeijer, J.; Xu, L.; Lipovsek, D. Antibody mimics based on human fibronectin type three domain engineered for thermostability and high-affinity binding to vascular endothelial growth factor receptor two. Protein Eng. Des. Sel. 2005, 18, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Bloom, L.; Calabro, V. FN3: A new protein scaffold reaches the clinic. Drug Discov. Today 2009, 14, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Martin-Killias, P.; Wyss-Stoeckle, S.; Honegger, A.; Zangemeister-Wittke, U.; Pluckthun, A. Darpins recognizing the tumor-associated antigen epcam selected by phage and ribosome display and engineered for multivalency. J. Mol. Biol. 2011, 413, 826–843. [Google Scholar] [CrossRef] [PubMed]
- Zahnd, C.; Wyler, E.; Schwenk, J.M.; Steiner, D.; Lawrence, M.C.; McKern, N.M.; Pecorari, F.; Ward, C.W.; Joos, T.O.; Pluckthun, A. A designed ankyrin repeat protein evolved to picomolar affinity to Her2. J. Mol. Biol. 2007, 369, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Parizek, P.; Kummer, L.; Rube, P.; Prinz, A.; Herberg, F.W.; Pluckthun, A. Designed ankyrin repeat proteins (DARPins) as novel isoform-specific intracellular inhibitors of c-Jun N-terminal kinases. ACS Chem. Biol. 2012, 7, 1356–1366. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. Weblogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Klein, F.; Diskin, R.; Scheid, J.F.; Gaebler, C.; Mouquet, H.; Georgiev, I.S.; Pancera, M.; Zhou, T.; Incesu, R.B.; Fu, B.Z.; et al. Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell 2013, 153, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Buhr, D.L.; Acca, F.E.; Holland, E.G.; Johnson, K.; Maksymiuk, G.M.; Vaill, A.; Kay, B.K.; Weitz, D.A.; Weiner, M.P.; Kiss, M.M. Use of micro-emulsion technology for the directed evolution of antibodies. Methods 2012, 58, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Ravn, U.; Didelot, G.; Venet, S.; Ng, K.T.; Gueneau, F.; Rousseau, F.; Calloud, S.; Kosco-Vilbois, M.; Fischer, N. Deep sequencing of phage display libraries to support antibody discovery. Methods 2013, 60, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Campoy, A.; Leavitt, S.A.; Freire, E. Characterization of protein-protein interactions by isothermal titration calorimetry. Methods Mol. Biol. 2015, 1278, 183–204. [Google Scholar] [PubMed]
- Poulsen, T.R.; Jensen, A.; Haurum, J.S.; Andersen, P.S. Limits for antibody affinity maturation and repertoire diversification in hypervaccinated humans. J. Immunol. 2011, 187, 4229–4235. [Google Scholar] [CrossRef] [PubMed]
- Koide, A.; Wojcik, J.; Gilbreth, R.N.; Hoey, R.J.; Koide, S. Teaching an old scaffold new tricks: Monobodies constructed using alternative surfaces of the FN3 scaffold. J. Mol. Biol. 2012, 415, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Willuda, J.; Honegger, A.; Waibel, R.; Schubiger, P.A.; Stahel, R.; Zangemeister-Wittke, U.; Pluckthun, A. High thermal stability is essential for tumor targeting of antibody fragments: Engineering of a humanized anti-epithelial glycoprotein-2 (epithelial cell adhesion molecule) single-chain Fv fragment. Cancer Res. 1999, 59, 5758–5767. [Google Scholar] [PubMed]
- Holland, E.G.; Acca, F.E.; Belanger, K.M.; Bylo, M.E.; Kay, B.K.; Weiner, M.P.; Kiss, M.M. In vivo elimination of parental clones in general and site-directed mutagenesis. J. Immunol. Methods 2015, 417, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Huovinen, T.; Brockmann, E.C.; Akter, S.; Perez-Gamarra, S.; Yla-Pelto, J.; Liu, Y.; Lamminmaki, U. Primer extension mutagenesis powered by selective rolling circle amplification. PLoS ONE 2012, 7, e31817. [Google Scholar] [CrossRef] [PubMed]
- Porath, J. Immobilized metal ion affinity chromatography. Protein Expr. Purif. 1992, 3, 263–281. [Google Scholar] [CrossRef]
- Panavas, T.; Sanders, C.; Butt, T.R. Sumo fusion technology for enhanced protein production in prokaryotic and eukaryotic expression systems. Methods Mol. Biol. 2009, 497, 303–317. [Google Scholar] [PubMed]
- Beckett, D.; Kovaleva, E.; Schatz, P.J. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 1999, 8, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Cull, M.G.; Schatz, P.J. Biotinylation of proteins in vivo and in vitro using small peptide tags. Methods Enzymol. 2000, 326, 430–440. [Google Scholar] [PubMed]
- Huang, R.; Fang, P.; Kay, B.K. Isolation of monobodies that bind specifically to the SH3 domain of the Fyn tyrosine protein kinase. New Biotechnol. 2012, 29, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Kay, B.K.; Thai, S.; Volgina, V.V. High-throughput biotinylation of proteins. Methods Mol. Biol. 2009, 498, 185–196. [Google Scholar] [PubMed]
- Ferrara, F.; D’Angelo, S.; Gaiotto, T.; Naranjo, L.; Tian, H.; Graslund, S.; Dobrovetsky, E.; Hraber, P.; Lund-Johansen, F.; Saragozza, S.; et al. Recombinant renewable polyclonal antibodies. MAbs 2015, 7, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. Nih image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, R.; Gorman, K.T.; Vinci, C.R.; Dobrovetsky, E.; Gräslund, S.; Kay, B.K. Streamlining the Pipeline for Generation of Recombinant Affinity Reagents by Integrating the Affinity Maturation Step. Int. J. Mol. Sci. 2015, 16, 23587-23603. https://doi.org/10.3390/ijms161023587
Huang R, Gorman KT, Vinci CR, Dobrovetsky E, Gräslund S, Kay BK. Streamlining the Pipeline for Generation of Recombinant Affinity Reagents by Integrating the Affinity Maturation Step. International Journal of Molecular Sciences. 2015; 16(10):23587-23603. https://doi.org/10.3390/ijms161023587
Chicago/Turabian StyleHuang, Renhua, Kevin T. Gorman, Chris R. Vinci, Elena Dobrovetsky, Susanne Gräslund, and Brian K. Kay. 2015. "Streamlining the Pipeline for Generation of Recombinant Affinity Reagents by Integrating the Affinity Maturation Step" International Journal of Molecular Sciences 16, no. 10: 23587-23603. https://doi.org/10.3390/ijms161023587
APA StyleHuang, R., Gorman, K. T., Vinci, C. R., Dobrovetsky, E., Gräslund, S., & Kay, B. K. (2015). Streamlining the Pipeline for Generation of Recombinant Affinity Reagents by Integrating the Affinity Maturation Step. International Journal of Molecular Sciences, 16(10), 23587-23603. https://doi.org/10.3390/ijms161023587