Metaphylogenomic and Potential Functionality of the Limpet Patella pellucida’s Gastrointestinal Tract Microbiome

Abstract
:1. Introduction

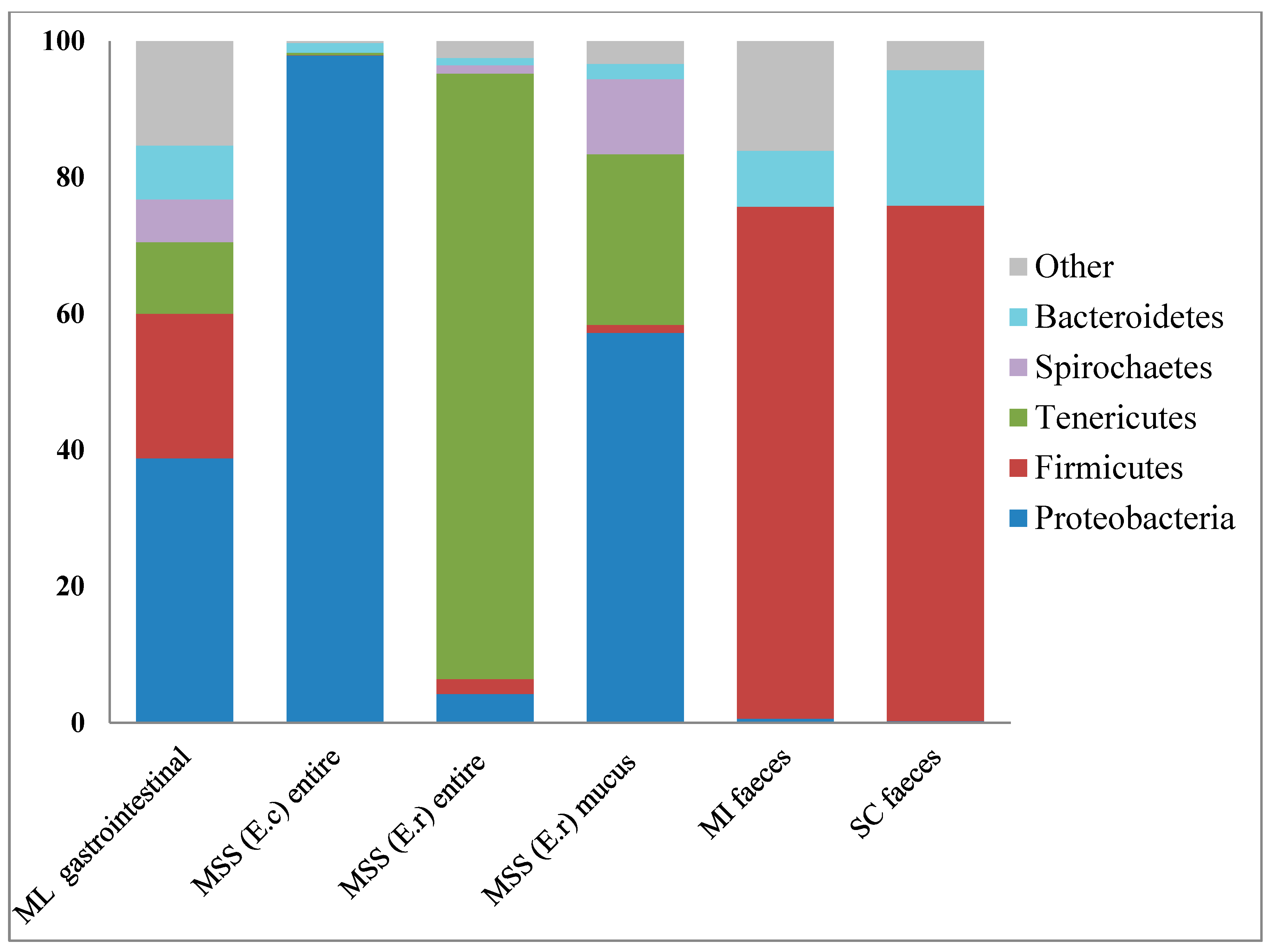
2. Results and Discussion
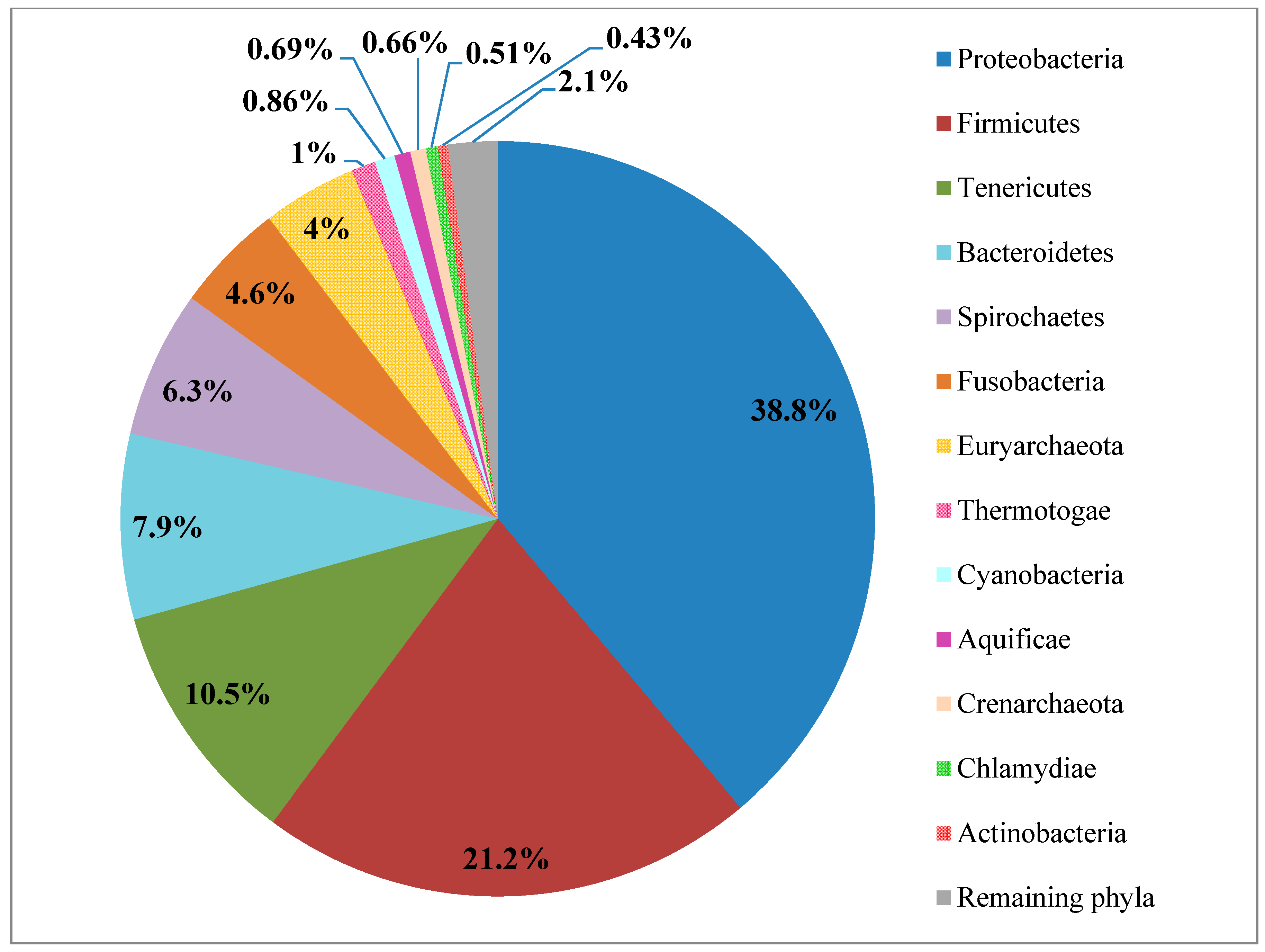

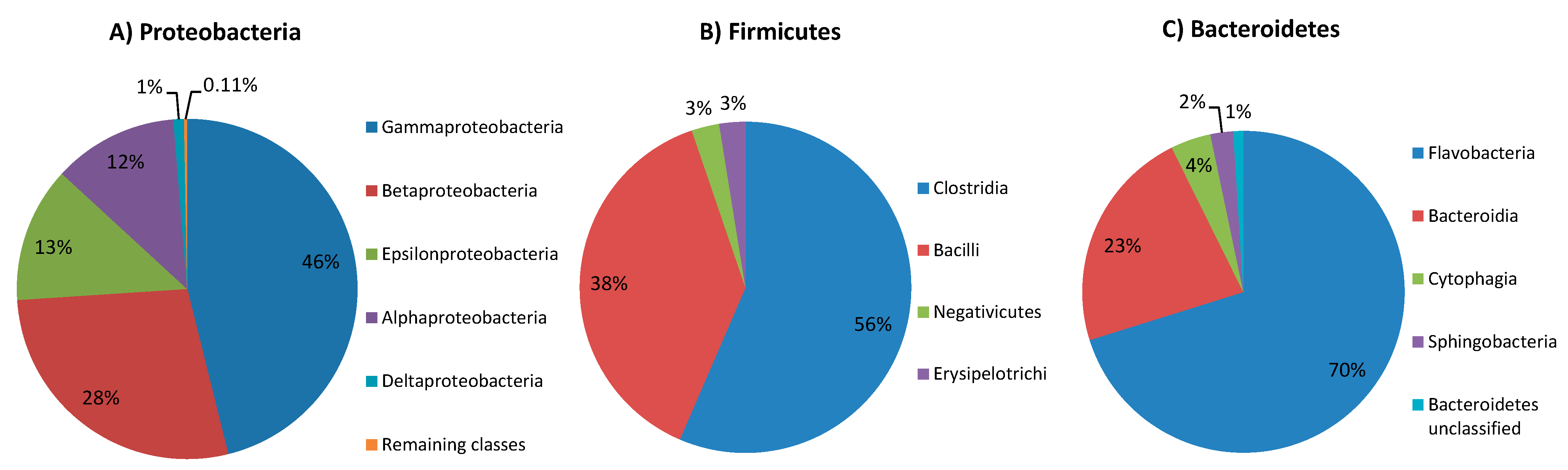
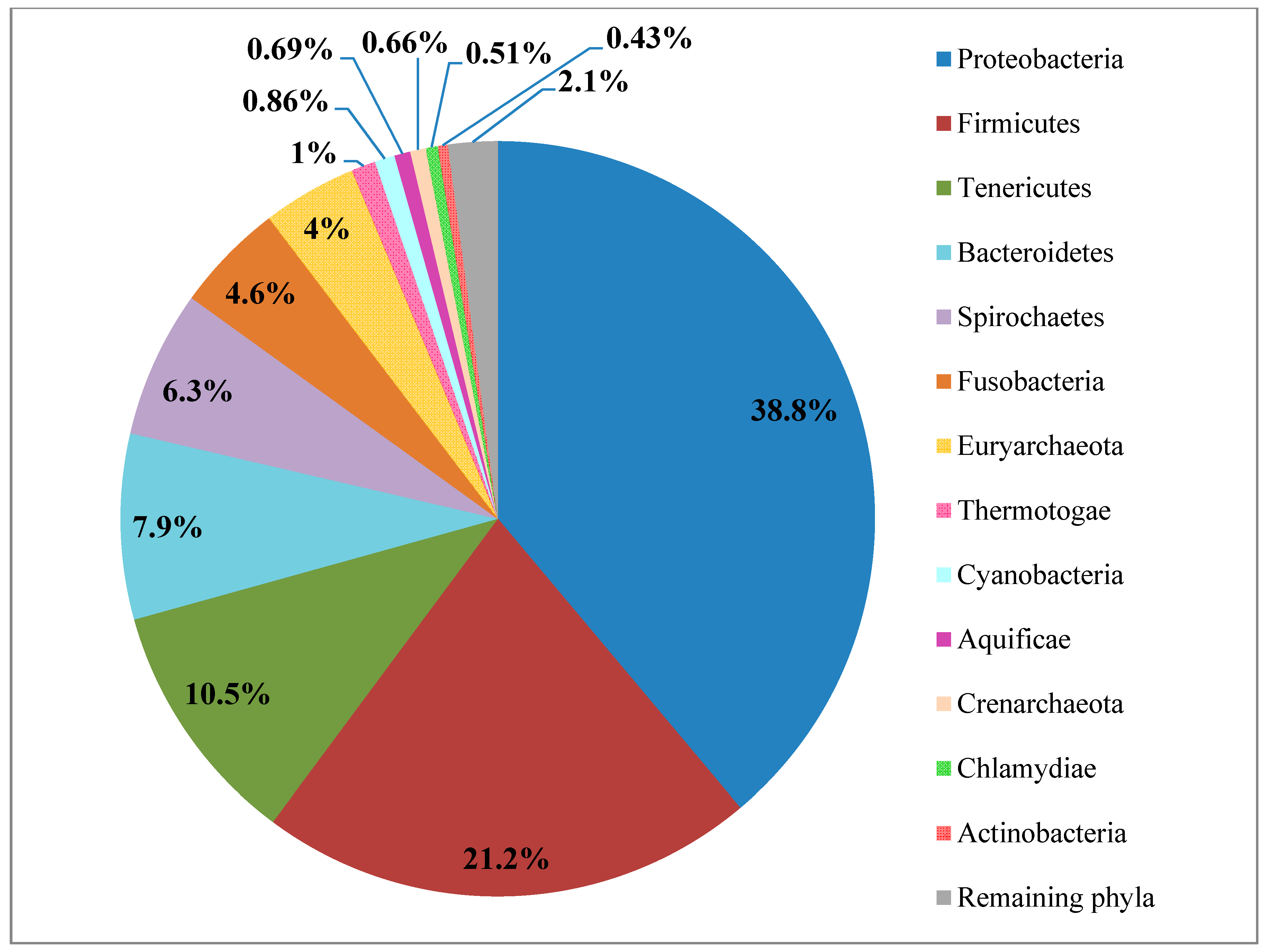
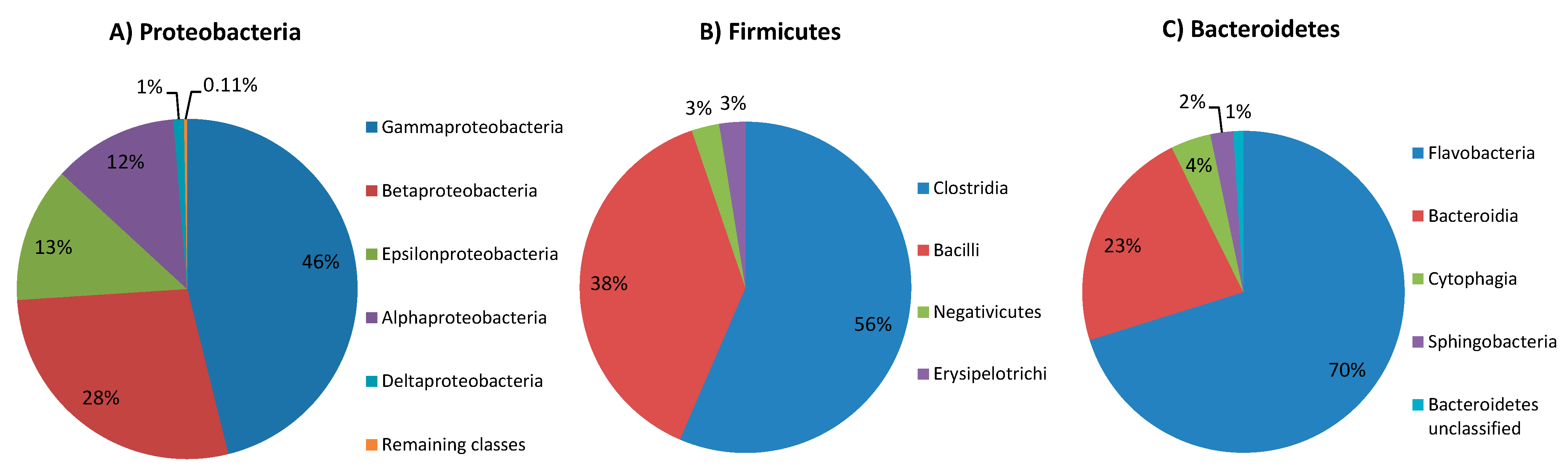
2.1. Proteobacteria

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | The Most Abundant Order (% of Class) | The Most Abundant Family (% of Order) | The Most Abundant Genus/Species | Functional Role/Habitat | Ref. |
|---|---|---|---|---|---|
| Gammaproteobacteria | Enterobacteriales (76.34) | Enterobacteriaceae (100) | “ Candidatus Carsonella”/ “Candidatus Carsonella rudii” | nutrients supply/obligate endosymbiont of psyllids | [32,34] |
| Pasteurellales (4.84) | Pasteurellaceae (100) | Haemophilus/ Haemophilus influenzae | pathogenic/human and animals | [42] | |
| Thiotrichales (3.45) | Francisellaceae (87.09) | Francisella/ Francisella tularenisis | [43] | ||
| Betaproteobacteria | Burkholderiales (94.65) | Oxalobacteraceae (98.63) | “ Candidatus Zinderia”/ “Candidatus Zinderia” (unclassified) | nutrients supply/obligate endosymbiont of spittlebug | [33] |
| Neisseriales (4.7) | Neisseriaceae (100) | Neisseria/ Neisseria meningitides | pathogenic/human origin | [44] | |
| Nitrosomonadales (0.27) | Nitrosomonadaceae (100) | Nitrosomonas/ Nitrosomonas europea | ammonia oxidation/sewage plants disposal; water; soil | [41,45] | |
| Epsilonoproteobacteria | Campylobacterales (81.6) | Campylobacteraceae (64.21) | Campylobacter/ Campylobacter lari | pathogenic/gastrointestinal of human and animals | [46] |
| Nautiliales (16.6) | Nautilaceae (100) | Caminibacter/ Caminibacter mediatlanticus | nitrate and sulphur reduction/deep-sea hydrothermal systems | [37] | |
| Epsilonoproteo-bacteria (unclassified) (1.72) | Nitratiruptor (62.5) | Nitratiruptor/ Nitratiruptor (unclassified) | [38] | ||
| Alphaproteobacteria | Rickettsiales (76.67) | Rickettsiaceae (60.5) | Rickettsia/ Rickettsia bellii | pathogenic/ human and animals | [47] |
| Rhizobiales (19.65) | Bartonellaceae (45.05) | Bartonella/ Bartonella henselae | pathogenic/ human and animals | [48] | |
| Brucellaceae (38.46) | Brucella/ Brucella abortus | [49] | |||
| Deltaproteobacteria | Desulfovibrionales (41.02) | Desulfovibrionaceae (87.5) | Desulfovibrio/ Desulfovibrio magneticus | sulphate-reduction/marine sediments; gastrointestinal of human and animals | [50,51] |
| Desulfobacterales (17.94) | Desulfobulbaceae (42.85) | Desulfotalea/Desulfotalea psychrophila | [52] | ||
| Bdellovibrionales (17.94) | Bacteriovoraceae (85.7) | Bacteriovorax/ Bacteriovorax marinus | predatory/marine environment | [53] |
2.2. Firmicutes
| Class | The Most Abundant Order (% of Class) | The Most Abundant Family (% of Order) | The Most Abundant Genus/Species | Functional Role/Habitat | Ref. |
|---|---|---|---|---|---|
| Clostridia | Clostridiales (72.62) | Clostridiaceae (49.71) | Clostridium/ Clostridium butyricum | polysaccharides degradation and fermentation; pathogenic/gastrointestinal of human and animals; feaces; soil; water | [54,63] |
| Clostridiales Family XI Incertae sedis (47.23) | Anaerococcus/ Anaerococcus vaginalis | polysaccharides degradation and fermentation/clinical specimens of human origin | [64] | ||
| Thermoanaerobacterales (22.71) | Thermoanaero- bacterales Familly III Incertae sedis (53.84) | Caldicellulosiruptor/Caldicellulosiruptor kronotskyensis | polysaccharides degradation and fermentation/hot springs; deep-sea hydrothermal systems | [65] | |
| Thermoanaero- bacteraceae (45.42) | Thermoanaerobacter/Thermoanaerobacter ethanolicus | [66] | |||
| Bacilli | Lactobacillales (51.96) | Streptococcaceae (40.89) | Streptococcus/ Streptococcus bovis | pathogenic/clinical specimens of human and animals origin | [67,68] |
| Lactobacillaceae (34.51) | Lactobacillus/ Lactobacillus iners | polysaccharides fermentation/gastrointestinal of human and animals; water; soil | [61,62] | ||
| Bacillales (48.03) | Bacillaceae (36.82) | Bacillus/Bacillus thuringiensis | polysaccharides degradation and fermentation/ gastrointestinal of human and animals; water; soil | [69] | |
| Staphylococcaceae (34.78) | Staphylococcus/ Staphylococcus hominis | polysaccharides fermentation; pathogenic/water; soil; clinical specimens of human and animals origin | [70,71] | ||
| Negativivicutes | Selenomonadales (100) | Veillonellaceae (96.49) | Dialister/Dialister microaerophilus | fermentation/water; soil; gastrointestinal of human and animals; clinical specimens of human origin | [72] |
| Erisipelotrichi | Erisipelotrichales (100) | Erisipelotrichaceae (100) | Coprobacillus/Coprobacillus sp. (unclassified) | fermentation/gastrointestinal of human and animals origin | [73] |
2.3. Bacteroidetes
| Class | The Most Abundant Order (% of Class) | The Most Abundant Family (% of Order) | The Most Abundant Genera/Species | Functional Role/Habitat | Ref. |
|---|---|---|---|---|---|
| Flavobacteriia | Flavobacteriales (100) | Flavobacteriales (unclassified) (54) | “ Candidatus Sulcia”/ “Candidatus Sulcia muelleri” | nutrients supply/obligate endosymbiont of sharpshooters | [33] |
| Flavobacteriaceae (36) | Cellulophaga/ Cellulophaga lytica | polysaccharides degradation/diatoms; algae; seawater | [77] | ||
| Blattabacteriaceae (10) | Blattabacterium/ Blattabacterium sp. (unclassified) | nutrients supply/obligate endosymbiont of cockroaches and termites | [80] | ||
| Bacteroidia | Bacteroidales (100) | Bacteroidaceae (52) | Bacteroides/ Bacteroides Xylanisolvens | polysaccharides degradation and fermentation/human and animals gastrointestinal | [78,81] |
| Prevotellaceae (24) | Prevotella/ Prevotella amnii | polysaccharides degradation and fermentation/human and animals gastrointestinal | [51,82] | ||
| Porphyromonadaceae (14) | Paludibacter/ Paludibacter propionicigenes | polysaccharides fermentation/plant residue | [83] | ||
| Cytophagia | Cytophagales (100) | Cytophagaceae (54) | Cytophaga/Cytophaga hutchinsonii | polysaccharides degradation/soil | [79] |
| Flammeovirgaceae (26) | Marivirga/ Marivirga tractuosa | polysaccharides degradation/water; mud; sand | [84] | ||
| Cyclobacteriaceae (24) | Algoriphagus/ Algoriphagus unclassified | polysaccharides degradation/marine solar saltern | [85] | ||
| Sphingobacteriia | Sphingobacteriales (100) | Sphingobacteriaceae (92) | Pedobacter/ Pedobacter saltans | sulphates degradation/soil; water; fish | [86] |
| Sphingobacterium/ Sphingobacterium spiritivorum | synthesis of antimicrobials/specimens of human origin | [87] | |||
| Mucilaginibacter/ Mucilaginibacter paludis | polysaccharides degradation/sphagnum peat bog | [88] | |||
| Chitinophagacea (7) | Chitinophaga/ Chitinophaga pinensis | polysaccharides degradation/soil | [89] | ||
| Bacteroidetes unclasified | Bacteroidetes (unclassified) (100) | Bacteroidetes (unclassified) (100) | “ Candidatus Amoebophilus”/ “Candidatus Amoebophilus” (unclassified) | nutrients supply/obligate endosymbiont of amoeba | [76] |
2.4. Remaining Phyla
3. Materials and Methods
3.1. P. pellucida Collection and Maintenance
3.2. Indirect Extraction of Metagenomic DNA
3.3. Metagenomic DNA Sequencing, Assembling and Taxonomic Profiling of the Corresponding Microbial Community
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jefreys, J.G. British Conchology: or, an Account of the Mollusca Which Now Inhabit the British Isles and the Surrounding Seas; J. Van Voorst: London, UK, 1865; pp. 242–245. [Google Scholar]
- Da Silva, C.M.; Landau, B.M.; Domènech, R.; Martinell, J. Pliocene Atlanto-Mediterranean biogeography of Patella pellucida (Gastropoda, Patellidae): Palaeoceanographic implications. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2006, 233, 225–234. [Google Scholar] [CrossRef]
- McGrath, D. Recruitment and Growth of the Blue-Rayed Limpet, Helcion Pellucidum (L.), in South East Ireland. J. Molluscan Stud. 1992, 58, 425–431. [Google Scholar]
- Taylor, P.; Percival, E. The polysaccharides of green, red and brown seaweeds: Their basic structure, biosynthesis and function. Br. Phycol. J. 1979, 14, 103–117. [Google Scholar]
- Jami, E.; Mizrahi, I. Composition and similarity of bovine rumen microbiota across individual animals. PLoS One 2012, 7, e33306. [Google Scholar] [PubMed]
- Hong, P.Y.; Wheeler, E.; Cann, I.K.O.; Mackie, R.I. Phylogenetic analysis of the fecal microbial community in herbivorous land and marine iguanas of the Galápagos Islands using 16S rRNA-based pyrosequencing. ISME J. 2011, 5, 1461–1470. [Google Scholar] [CrossRef]
- Eigeland, K. Bacterial community structure in the hindgut of wild and captive dugongs (dugong dugon). Aquat. Mamm. 2012, 38, 402–411. [Google Scholar] [CrossRef]
- Haygood, M.G.; Schmidt, E.W.; Davidson, S.K.; Faulkner, D.J. Microbial symbionts of marine invertebrates: Opportunities for microbial biotechnology. J. Mol. Microbiol. Biotechnol. 1999, 1, 33–43. [Google Scholar]
- Gomare, S.; Kim, H.A.; Ha, J.H.; Lee, M.W.; Park, J.M. Isolation of the polysaccharidase-producing bacteria from the gut of sea snail, Batillus cornutus. Korean J. Chem. Eng. 2011, 28, 1252–1259. [Google Scholar] [CrossRef]
- Bakunina, I.I.; Nedashkovskaia, O.I.; Alekseeva, S.A.; Ivanova, E.P.; Romanenko, L.A; Gorshkova, N.M.; Isakov, V.V.; Zviagintseva, T.N.; Mikhaĭlov, V.V. Degradation of fucoidan by the marine proteobacterium Pseudoalteromonas citrea. Mikrobiologiia 2002, 71, 49–55. [Google Scholar]
- Gurgui, C.; Piel, J. Metagenomic Approaches to Identify and Isolate Bioactive Natural Products from Microbiota of Marine Sponges. Methods Mol. Biol. Methods Protoc. 2010, 668, 247–264. [Google Scholar]
- Zhang, C.; Kim, S.K. Research and application of marine microbial enzymes: Status and prospects. Mar. Drugs 2010, 8, 1920–1934. [Google Scholar]
- Demirbas, M.F. Biorefineries for biofuel upgrading: A critical review. Appl. Energy 2009, 86, 151–161. [Google Scholar] [CrossRef]
- Buonocore, F. Marine Biotechnology: Developments and Perspectives. J. Aquac. Res. Dev. 2012, 4. [Google Scholar] [CrossRef]
- Williams, P.G. Panning for chemical gold: Marine bacteria as a source of new therapeutics. Trends Biotechnol. 2009, 27, 45–52. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Dupont, C.L. Microbial Metagenomics: Beyond the Genome. Annu. Rev. Mar. Sci. 2011, 3, 347–371. [Google Scholar] [CrossRef]
- Handelsman, J. Metagenomics: Application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 2004, 68, 669–685. [Google Scholar] [CrossRef]
- Kleiner, M.; Wentrup, C.; Lott, C.; Teeling, H.; Wetzel, S.; Young, J.; Chang, Y.J.; Shah, M; VerBerkmoes, N.C.; Zarzycki, J.; et al. Metaproteomics of a gutless marine worm and its symbiotic microbial community reveal unusual pathways for carbon and energy use. Proc. Natl. Acad. Sci. USA 2012, 109, 1173–1182. [Google Scholar]
- Svanevik, C.S.; Lunestad, B.T. Characterisation of the microbiota of Atlantic mackerel (Scomber scombrus). Int. J. Food Microbiol. 2011, 151, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Gilbert, J.; Meyer, F. Metagenomics—A guide from sampling to data analysis. Microb. Inform. Exp. 2012, 2. [Google Scholar] [CrossRef]
- Kunin, V.; Copeland, A.; Lapidus, A.; Mavromatis, K.; Hugenholtz, P. A bioinformatician’s guide to metagenomics. Microbiol. Mol. Biol. Rev. 2008, 72, 557–578. [Google Scholar] [CrossRef]
- Tringe, S.G.; Rubin, E.M. Metagenomics: DNA sequencing of environmental samples. Nat. Rev. Genet. 2005, 6, 805–814. [Google Scholar] [CrossRef]
- Harnpicharnchai, P.; Thongaram, T.; Sriprang, R.; Champreda, V.; Tanapongpipat, S.; Eurwilaichitr, L. An efficient purification and fractionation of genomic DNA from soil by modified troughing method. Lett. Appl. Microbiol. 2007, 45, 387–391. [Google Scholar] [CrossRef]
- Cardoso, A.M.; Cavalcante, J.J.; Cantão, M.E.; Thompson, C.E.; Flatschart, R.B.; Glogauer, A.; Scapin, S.M.; Sade, Y.B.; Beltrão, P.J.; Gerber, A.L.; et al. Metagenomic analysis of the microbiota from the crop of an invasive snail reveals a rich reservoir of novel genes. PLoS One 2012, 7, e48505. [Google Scholar]
- Segata, N.; Boernigen, D.; Tickle, T.L.; Morgan, X.C.; Garrett, W.S.; Huttenhower, C. Computational meta’omics for microbial community studies. Mol. Syst. Biol. 2013, 9. [Google Scholar] [CrossRef]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef]
- Devine, S.P.; Pelletreau, K.N.; Rumpho, M.E. 16S rDNA-based metagenomic analysis of bacterial diversity associated with two populations of the kleptoplastic sea slug Elysia chlorotica and its algal prey Vaucheria litorea. Biol. Bull. 2012, 223, 138–154. [Google Scholar]
- Davis, J.; Fricke, W.F.; Haman, M.T.; Esquenazi, E.; Dorrestein, P.C.; Hill, R.T. Characterization of the bacterial community of the chemically defended Hawaiian sacoglossan Elysia rufescens. Appl. Environ. Microbiol. 2013, 79, 7073–7081. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.W.Y.; Jumars, P.A.; Armbrust, E.V. Genetic diversity of attached bacteria in the hindgut of the deposit-feeding shrimp Neotrypaea (formerly Callianassa) californiensis (decapoda: Thalassinidae). Microb. Ecol. 2002, 43, 455–466. [Google Scholar] [CrossRef]
- Taylor, M.W.; Radax, R.; Stege, D.R.; Wagner, M. Sponge-associated microorganisms: Evolution, ecology, and biotechnological potential. Microbiol. Mol. Biol. Rev. 2007, 71, 295–347. [Google Scholar] [CrossRef]
- Isnansetyo, A.; Kamei, Y. Bioactive substances produced by marine isolates of Pseudomonas. J. Ind. Microbiol. Biotechnol. 2009, 36, 1239–1248. [Google Scholar] [PubMed]
- Tamames, J.; Gil, R.; Latorre, A.; Peretó, J.; Silva, F.J.; Moya, A. The frontier between cell and organelle: Genome analysis of “Candidatus Carsonella ruddii”. BMC Evol. Biol. 2007, 7. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, J.P.; Moran, N.A. Functional convergence in reduced genomes of bacterial symbionts spanning 200 My of evolution. Genome Biol. Evol. 2010, 2, 708–718. [Google Scholar]
- McCutcheon, J.P.; Moran, N.A. Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 2012, 10, 13–26. [Google Scholar]
- Lim, G.E.; Haygood, M.G. “Candidatus Endobugula glebosa”, a specific bacterial symbiont of the marine bryozoan Bugula simplex. Appl. Environ. Microbiol. 2004, 70, 4921–4929. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.K.; Allen, S.W.; Lim, G.E.; Anderson, C.M.; Haygood, M.G. Evidence for the biosynthesis of bryostatins by the bacterial symbiont “Candidatus Endobugula sertula” of the bryozoan Bugula neritina. Appl. Environ. Microbiol. 2001, 67, 4531–4537. [Google Scholar] [CrossRef]
- Voordeckers, J.W.; Starovoyto, V.V.; Vetriani, C. Caminibacter mediatlanticus sp. nov., a thermophilic, chemolithoautotrophic, nitrate-ammonifying bacterium isolated from a deep-sea hydrothermal vent on the Mid-Atlantic Ridge. Int. J. Syst. Evol. Microbiol. 2005, 55, 773–779. [Google Scholar]
- Nakagawa, S.; Takai, K.; Inagaki, F.; Horikoshi, K.; Sako, Y. Nitratiruptor tergarcus gen. nov., sp. nov. and Nitratifractor salsuginis gen. nov., sp. nov., nitrate-reducing chemolithoautotrophs of the epsilon-Proteobacteria isolated from a deep-sea hydrothermal system in the Mid-Okinawa Trough. Int. J. Syst. Evol. Microbiol. 2005, 55, 925–933. [Google Scholar]
- Alazard, D.; Dukan, S.; Urios, A.; Verhé, F.; Bouabida, N.; Morel, F.; Thomas, P.; Garcia, J.L.; Ollivier, B. Desulfovibrio hydrothermalis sp. nov., a novel sulfate-reducing bacterium isolated from hydrothermal vents. Int. J. Syst. Evol. Microbiol. 2003, 53, 173–178. [Google Scholar]
- Cabrera, G.; Pérez, R.; Gómez, J.M.; Ábalos, A.; Cantero, D. Toxic effects of dissolved heavy metals on Desulfovibrio vulgaris and Desulfovibrio sp. strains. J. Hazard. Mater. 2006, 135, 40–46. [Google Scholar]
- Schmidt, I. Chemoorganoheterotrophic growth of Nitrosomonas europaea and Nitrosomonas eutropha. Curr. Microbiol. 2009, 59, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Musher, D.M. Medical Microbiology, 4th ed.; Baron, S., Ed.; The University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Barns, S.M.; Grow, C.C.; Okinaka, R.T.; Keim, P.; Kuske, C.R. Detection of diverse new Francisella-like bacteria in environmental samples. Appl. Environ. Microbiol. 2005, 71, 5494–5500. [Google Scholar] [CrossRef]
- Gasparini, R.; Amicizia, D.; Lai, P.L.; Panatto, D. Neisseria meningitidis, pathogenetic mechanisms to overcome the human immune defences. J. Prev. Med. Hyg. 2012, 53, 50–55. [Google Scholar]
- Bollmann, A.; Sedlacek, C.J.; Norton, J.; Laanbroek, H.J.; Suwa, Y.; Stein, L.Y.; Klotz, M.G.; Arp, D.; Sayavedra-Soto, L.; Lu, M.; et al. Complete genome sequence of Nitrosomonas sp. Is79, an ammonia oxidizing bacterium adapted to low ammonium concentrations. Stand. Genomic Sci. 2013, 7, 469–482. [Google Scholar]
- Foster, G.; Holmes, B.; Steigerwalt, A.G.; Lawson, P.A.; Thorne, P.; Byrer, D.E.; Ross, H.M.; Xerry, J.; Thompson, P.M.; Collins, M.D. Campylobacter insulaenigrae sp. nov., isolated from marine mammals. Int. J. Syst. Evol. Microbiol. 2004, 54, 2369–2373. [Google Scholar] [CrossRef]
- Perlman, S.J.; Hunter, M.S.; Zchori-Fein, E. The emerging diversity of Rickettsia. Proc. Biol. Sci. 2006, 273, 2097–2106. [Google Scholar] [CrossRef]
- Maggi, R.G.; Raverty, S.A.; Lester, S.J.; Huff, D.G.; Haulena, M.; Ford, S.L.; Nielsen, O.; Robinson, J.H.; Breitschwerdt, E.B. Bartonella henselae in captive and hunter-harvested beluga (Delphinapterus leucas). J. Wildl. Dis. 2008, 44, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E.; Cloeckaert, A.; Moriyón, I. Brucella evolution and taxonomy. Vet. Microbiol. 2002, 90, 209–222. [Google Scholar]
- Sakaguchi, T.; Arakaki, A.; Matsunaga, T. Desulfovibrio magneticus sp. nov., a novel sulfate-reducing bacterium that produces intracellular single-domain-sized magnetite particles. Int. J. Syst. Evol. Microbiol. 2002, 52, 215–221. [Google Scholar]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Rabus, R.; Ruepp, A.; Frickey, T.; Rattei, T.; Fartmann, B.; Stark, M.; Bauer, M.; Zibat, A.; Lombardot, T.; Becker, I. The genome of Desulfotalea psychrophila, a sulfate-reducing bacterium from permanently cold Arctic sediments. Environ. Microbiol. 2004, 6, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Crossman, L.C.; Chen, H.; Cerdeño-Tárraga, A.M.; Brooks, K.; Quail, M.A.; Pineiro, S.A.; Hobley, L.; Sockett, R.E.; Bentley, S.D.; Parkhill, J.; et al. A small predatory core genome in the divergent marine Bacteriovorax marinus SJ and the terrestrial Bdellovibrio bacteriovorus. ISME J. 2012, 1, 148–160. [Google Scholar]
- Salf, N.A.L.; Brazier, J.S. The distribution of Clostridium difficile in the environment of South Wales. J. Med. Microbiol. 1996, 45, 133–137. [Google Scholar] [PubMed]
- Songsiriritthigul, C.; Lapboonrueng, S.; Pechsrichuang, P.; Pesatcha, P.; Yamabhai, M. Expression and characterization of Bacillus licheniformis chitinase (ChiA), suitable for bioconversion of chitin waste. Bioresour. Technol. 2010, 101, 4096–4103. [Google Scholar] [CrossRef] [PubMed]
- Trincone, A. Potential biocatalysts originating from sea environments. J. Mol. Catal. B Enzym. 2010, 66, 241–256. [Google Scholar] [CrossRef]
- Trincone, A. Marine biocatalysts: Enzymatic features and applications. Mar. Drugs 2011, 9, 478–499. [Google Scholar] [CrossRef]
- Turner, P.; Mamo, G.; Karlsson, E.N. Potential and utilization of thermophiles and thermostable enzymes in biorefining. Microb. Cell Fact. 2007, 23, 1–23. [Google Scholar]
- Jang, S.; Shirai, Y.; Uchida, M.; Wakisaka, M. Production of mono sugar from acid hydrolysis of seaweed. J. Biotechnol. 2012, 11, 1953–1963. [Google Scholar]
- Van der Wal, H.; Sperber, B.L.; Houweling-Tan, B.; Bakker, R.R.; Brandenburg, W.; López-Contreras, A.M. Production of acetone, butanol, and ethanol from biomass of the green seaweed Ulva lactuca. Bioresour. Technol. 2013, 128, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Kathiresan, K.; Thiruneelakandan, G. Prospects of lactic acid bacteria of marine origin. Indian J. Biotechnol. 2008, 7, 170–177. [Google Scholar]
- Hwang, H.J.; Lee, S.Y.; Kim, S.M.; Lee, S.B. Fermentation of seaweed sugars by Lactobacillus species and the potential of seaweed as a biomass feedstock. Biotechnol. Bioprocess Eng. 2011, 16, 1231–1239. [Google Scholar]
- Williams, A.G.; Withers, S.; Sutherland, A.D. The potential of bacteria isolated from ruminal contents of seaweed-eating North Ronaldsay sheep to hydrolyse seaweed components and produce methane by anaerobic digestion in vitro. Microb. Biotechnol. 2013, 6, 45–52. [Google Scholar] [CrossRef]
- Hugon, P.; Mishra, A.K.; Robert, C.; Raoult, D.; Fournier, P.E. Non-contiguous finished genome sequence and description of Anaerococcus vaginalis. Stand. Genomic Sci. 2012, 6, 356–365. [Google Scholar] [CrossRef]
- Miroshnichenko, M.L.; Kublanov, I.V.; Kostrikina, N.A.; Tourova, T.P.; Kolganova, T.V.; Birkeland, N.K.; Bonch-Osmolovskaya, E.A. Caldicellulosiruptor kronotskyensis sp. nov. and Caldicellulosiruptor hydrothermalis sp. nov., two extremely thermophilic, cellulolytic, anaerobic bacteria from Kamchatka thermal springs. Int. J. Syst. Evol. Microbiol. 2008, 58, 1492–1496. [Google Scholar]
- Wiegel, J.; Ljungdahl, L.G. Thermoanaerobacter ethanolicus gen. nov., spec. nov., a new, extreme thermophilic, anaerobic bacterium. Arch. Microbiol. 1981, 128, 343–348. [Google Scholar]
- Herrera, P.; Kwon, Y.M.; Ricke, S.C. Ecology and pathogenicity of gastrointestinal Streptococcus bovis. Anaerobe 2009, 15, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Murray, H.W.; Roberts, R.B. Streptococcus bovis bacteremia and underlying gastrointestinal disease. Arch. Intern. Med. 1978, 138, 1097–1099. [Google Scholar] [CrossRef]
- Ivanova, E.P.; Vysotskii, M.V.; Svetashev, V.I.; Nedashkovskaya, O.I.; Gorshkova, N.M.; Mikhailov, V.V.; Yumoto, N.; Shigeri, Y.; Taguchi, T.; Yoshikawa, S. Characterization of Bacillus strains of marine origin. Int. Microbiol. 1999, 2, 267–271. [Google Scholar]
- Gunn, B.A.; Colwell, R.R. Numerical taxonomy of Staphylococci isolated from the marine environment. Int. J. Syst. Bacteriol. 1983, 33, 751–759. [Google Scholar]
- Vuong, C.; Otto, M. Staphylococcus epidermidis infections. Microbes Infect. 2002, 4, 481–489. [Google Scholar] [CrossRef]
- Jumas-Bilak, E.; Jean-Pierre, H.; Carlier, J.P.; Teyssier, C.; Bernard, K.; Gay, B.; Campos, J.; Morio, F.; Marchandin, H. Dialister micraerophilus sp. nov. and Dialister propionicifaciens sp. nov., isolated from human clinical samples. Int. J. Syst. Evol. Microbiol. 2005, 55, 2471–2478. [Google Scholar]
- Kageyama, A.; Benno, Y. Coprobacillus catenaformis gen. nov., sp. nov., a new genus and species isolated from human feces. Microbiol. Immunol. 2000, 44, 23–28. [Google Scholar]
- Barbeyron, T.; L’Haridon, S.; Corre, E.; Kloareg, B.; Potin, P. Zobellia galactanovorans gen. nov., sp. nov., a marine species of Flavobacteriaceae isolated from a red alga, and classification of [Cytophaga] uliginosa (ZoBell and Upham 1944) Reichenbach 1989 as Zobellia uliginosa gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 2001, 51, 985–997. [Google Scholar]
- Fernández-Gómez, B.; Richter, M.; Schüler, M.; Pinhassi, J.; Acinas, S.G.; González, J.M.; Pedrós-Alió, C. Ecology of marine Bacteroidetes: A comparative genomics approach. ISME J. 2013, 7, 1026–1037. [Google Scholar] [CrossRef]
- Thomas, F.; Hehemann, J.H.; Rebuffet, E.; Czjzek, M.; Michel, G. Environmental and gut Bacteroidetes: The food connection. Front. Microbiol. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Pati, A.; Abt, B.; Teshima, H.; Nolan, M.; Lapidus, A.; Lucas, S.; Hammon, N.; Deshpande, S.; Cheng, J.F.; Tapia, R.; et al. Complete genome sequence of Cellulophaga lytica type strain (LIM-21). Stand. Genomic Sci. 2011, 4, 221–232. [Google Scholar]
- Ravcheev, D.A.; Godzik, A.; Osterman, A.L.; Rodionov, D.A. Polysaccharides utilization in human gut bacterium Bacteroides thetaiotaomicron: Comparative genomics reconstruction of metabolic and regulatory networks. BMC Genomics 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Bruce, D.C.; Challacombe, J.F.; Chertkov, O.; Detter, J.C.; Gilna, P.; Han, C.S.; Lucas, S.; Misra, M.; Myers, G.L.; et al. Genome sequence of the cellulolytic gliding bacterium Cytophaga hutchinsonii. Appl. Environ. Microbiol. 2007, 73, 3536–3546. [Google Scholar]
- López-Sánchez, M.J.; Neef, A.; Peretó, J.; Patiño-Navarrete, R.; Pignatelli, M.; Latorre, A.; Moya, A. Evolutionary convergence and nitrogen metabolism in Blattabacterium strain Bge, primary endosymbiont of the cockroach Blattella germanica. PLoS Genet. 2009, 5, e1000721. [Google Scholar] [CrossRef] [PubMed]
- Mirande, C.; Kadlecikova, E.; Matulova, M.; Capek, P.; Bernalier-Donadille, A.; Forano, E.; Béra-Maillet, C. Dietary fibre degradation and fermentation by two xylanolytic bacteria Bacteroides xylanisolvens XB1AT and Roseburia intestinalis XB6B4 from the human intestine. J. Appl. Microbiol. 2010, 109, 451–460. [Google Scholar] [PubMed]
- Lawson, P.A.; Moore, E.; Falsen, E. Prevotella amnii sp. nov., isolated from human amniotic fluid. Int. J. Syst. Evol. Microbiol. 2008, 58, 89–92. [Google Scholar]
- Gronow, S.; Munk, C.; Lapidus, A.; Nolan, M.; Lucas, S.; Hammon, N.; Deshpande, S.; Cheng, J.F.; Tapia, R.; Han, C.; et al. Complete genome sequence of Paludibacter propionicigenes type strain (WB4T). Stand. Genomic Sci. 2011, 4, 36–44. [Google Scholar]
- Pagani, I.; Chertkov, O.; Lapidus, A.; Lucas, S.; del Rio, T.G.; Tice, H.; Copeland, A.; Cheng, J.F.; Nolan, M.; Saunders, E.; et al. Complete genome sequence of Marivirga tractuosa type strain (H-43). Stand. Genomic Sci. 2011, 4, 154–162. [Google Scholar]
- Yoon, J.H.; Kang, S.J.; Oh, T.K. Algoriphagus locisalis sp. nov., isolated from a marine solar saltern. Int. J. Syst. Evol. Microbiol. 2005, 55, 1635–1639. [Google Scholar]
- Liolios, K.; Sikorski, J.; Lu, M.; Nolan, M.; Lapidus, A.; Lucas, S.; Hammon, N.; Deshpande, S.; Cheng, J.F.; Tapia, R. Complete genome sequence of the gliding, heparinolytic Pedobacter saltans type strain (113). Stand. Genomic Sci. 2011, 5, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Yabuuchi, E.; Kaneko, T.; Yano, I.; Moss, C.W.; Miyoshi, N. Sphingobacterium, gen. nov., Sphingobacterium spiritivorum comb. nov., Sphingobacterium multivorum comb. nov., Sphingobacterium mizutae sp. nov., and Flavobacterium indologenes sp. nov.: Glucose-nonfermenting gram-negative rods in CDC groups IIK-2 and IIb. Int. J. Syst. Evol. Microbiol. 1983, 33, 580–598. [Google Scholar]
- Pankratov, T.A.; Tindall, B.J.; Liesack, W.; Dedysh, S.N. Mucilaginibacter paludis gen. nov., sp. nov. and Mucilaginibacter gracilis sp. nov., pectin-, xylan- and laminarin-degrading members of the family Sphingobacteriaceae from acidic Sphagnum peat bog. Int. J. Syst. Evol. Microbiol. 2007; 57, 2349–2354. [Google Scholar]
- Glavina del Rio, T.; Abt, B.; Spring, S.; Lapidus, A.; Nolan, M.; Tice, H.; Copeland, A.; Cheng, J.F.; Chen, F.; Bruce, D. Complete genome sequence of Chitinophaga pinensis type strain (UQM 2034). Stand. Genomic Sci. 2010, 2, 87–95. [Google Scholar] [CrossRef]
- Kendall, M.M.; Liu, Y.; Sieprawska-Lupa, M.; Stetter, K.O.; Whitman, W.B.; Boone, D.R. Methanococcus aeolicus sp. nov., a mesophilic, methanogenic archaeon from shallow and deep marine sediments. Int. J. Syst. Evol. Microbiol. 2006, 56, 1525–1529. [Google Scholar]
- Van der Maarel, M.J.; Artz, R.R.; Haanstra, R.; Forney, L.J. Association of marine archaea with the digestive tracts of two marine fish species. Appl. Environ. Microbiol. 1998, 64, 2894–2898. [Google Scholar] [PubMed]
- Preston, C.M.; Wu, K.E.Y.; Molinskit, T.F.; Delong, E.F. A psychrophilic crenarchaeon inhabits a marine sponge: Cenarchaeum symbiosum gen. nov., sp. nov. Proc. Natl. Acad. Sci. USA 1996, 93, 6241–6246. [Google Scholar]
- Caswell, J.L.; Bateman, K.G.; Cai, H.Y.; Castillo-Alcala, F. Mycoplasma bovis in respiratory disease of feedlot cattle. Vet. Clin. N. Am. Food Anim. Pract. 2010, 26, 365–379. [Google Scholar] [CrossRef]
- Gupta, R.S.; Mahmood, S.; Adeolu, M. A phylogenomic and molecular signature based approach for characterization of the phylum Spirochaetes and its major clades: Proposal for a taxonomic revision of the phylum. Front. Microbiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Sitnikova, T.; Michel, E.; Tulupova, Y.; Khanaev, I.; Parfenova, V.; Prozorova, L. Spirochetes in gastropods from Lake Baikal and North American freshwaters: New multi-family, multi-habitat host records. Symbiosis 2012, 56, 103–110. [Google Scholar] [CrossRef]
- Chen, J.G.; Lou, D.; Yang, J.F. Isolation and Identification of Acholeplasma sp. from the Mud Crab, Scylla serrata. Evid. Based Complement. Altern. Med. 2011, 2011. [Google Scholar] [CrossRef]
- Strauss, J.; Kaplan, G.G.; Beck, P.L.; Rioux, K.; Panaccione, R.; Devinney, R.; Lynch, T.; Allen-Vercoe, E. Invasive potential of gut mucosa-derived Fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm. Bowel Dis. 2011, 17, 1971–1978. [Google Scholar] [CrossRef]
- Keenan, S.W.; Engel, A.S.; Elsey, R.M. The alligator gut microbiome and implications for archosaur symbioses. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Van den Burg, B. Extremophiles as a source for novel enzymes. Curr. Opin. Microbiol. 2003, 6, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Bull, A.T.; Stach, J.E.M. Marine Actinobacteria: New opportunities for natural product search and discovery. Trends Microbiol. 2007, 15, 491–499. [Google Scholar] [CrossRef]
- Alex, A.; Vasconcelos, V.; Tamagnini, P.; Santos, A.; Antunes, A. Unusual symbiotic Cyanobacteria association in the genetically diverse intertidal marine sponge Hymeniacidon perlevis (Demospongiae, Halichondrida). PLoS One 2012, 7, e51834. [Google Scholar] [PubMed]
- Ross, L.G.; Ross, B. Anaesthetic and Sedative Techniques for Aquatic Animals, 3rd ed.; Blackwell Publishing: Oxford, UK, 2008. [Google Scholar]
- Aronesty, E. Comparison of sequencing utility programs. Open Bioinform. J. 2013, 7, 1–8. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dudek, M.; Adams, J.; Swain, M.; Hegarty, M.; Huws, S.; Gallagher, J. Metaphylogenomic and Potential Functionality of the Limpet Patella pellucida’s Gastrointestinal Tract Microbiome. Int. J. Mol. Sci. 2014, 15, 18819-18839. https://doi.org/10.3390/ijms151018819
Dudek M, Adams J, Swain M, Hegarty M, Huws S, Gallagher J. Metaphylogenomic and Potential Functionality of the Limpet Patella pellucida’s Gastrointestinal Tract Microbiome. International Journal of Molecular Sciences. 2014; 15(10):18819-18839. https://doi.org/10.3390/ijms151018819
Chicago/Turabian StyleDudek, Magda, Jessica Adams, Martin Swain, Matthew Hegarty, Sharon Huws, and Joe Gallagher. 2014. "Metaphylogenomic and Potential Functionality of the Limpet Patella pellucida’s Gastrointestinal Tract Microbiome" International Journal of Molecular Sciences 15, no. 10: 18819-18839. https://doi.org/10.3390/ijms151018819
APA StyleDudek, M., Adams, J., Swain, M., Hegarty, M., Huws, S., & Gallagher, J. (2014). Metaphylogenomic and Potential Functionality of the Limpet Patella pellucida’s Gastrointestinal Tract Microbiome. International Journal of Molecular Sciences, 15(10), 18819-18839. https://doi.org/10.3390/ijms151018819