Assessing the Accuracy of Quantitative Molecular Microbial Profiling

 and
and
Abstract
:1. Introduction
2. Results and Discussion
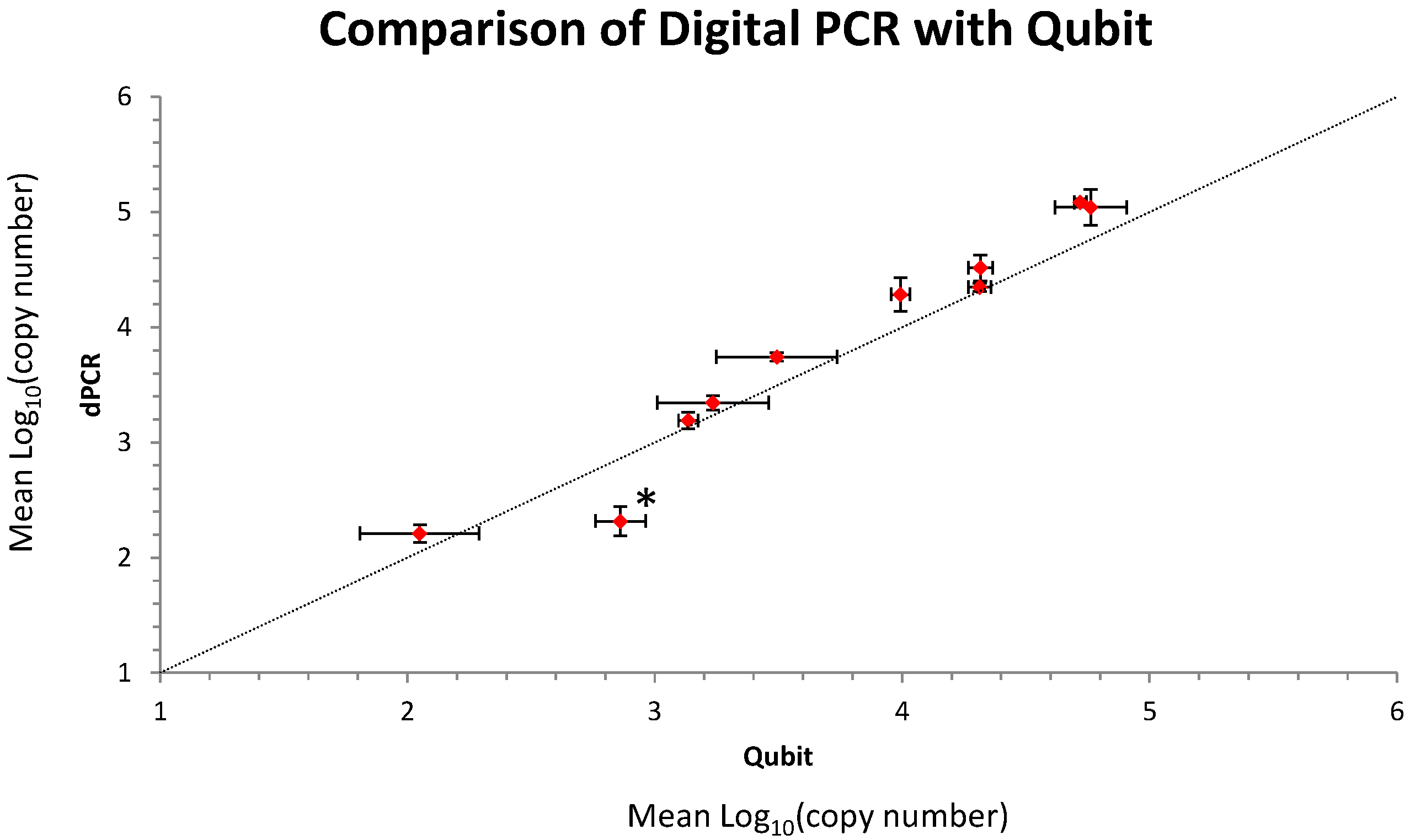
2.1. Initial Analysis and Characterization of the Metagenomic Control Material (MCM)
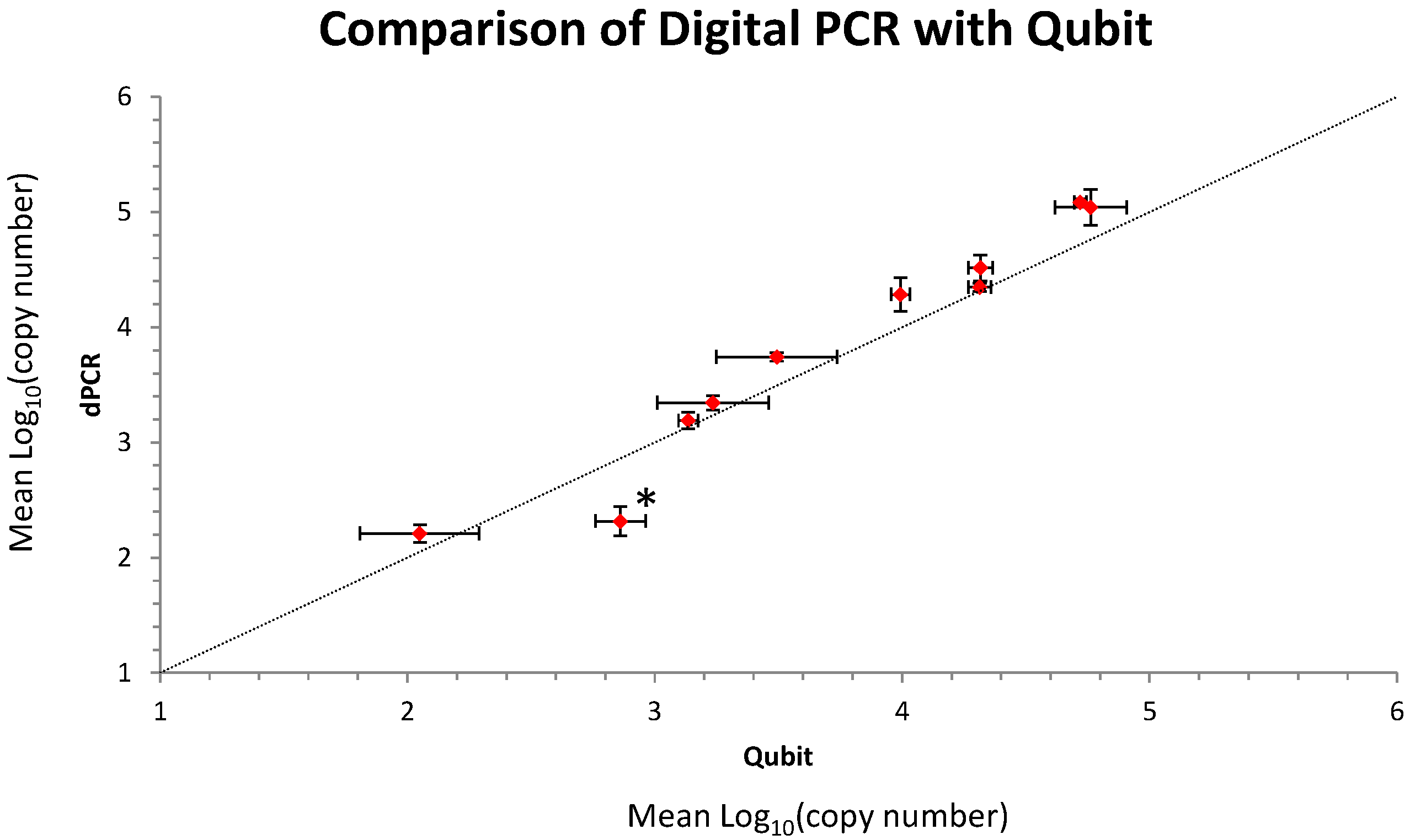
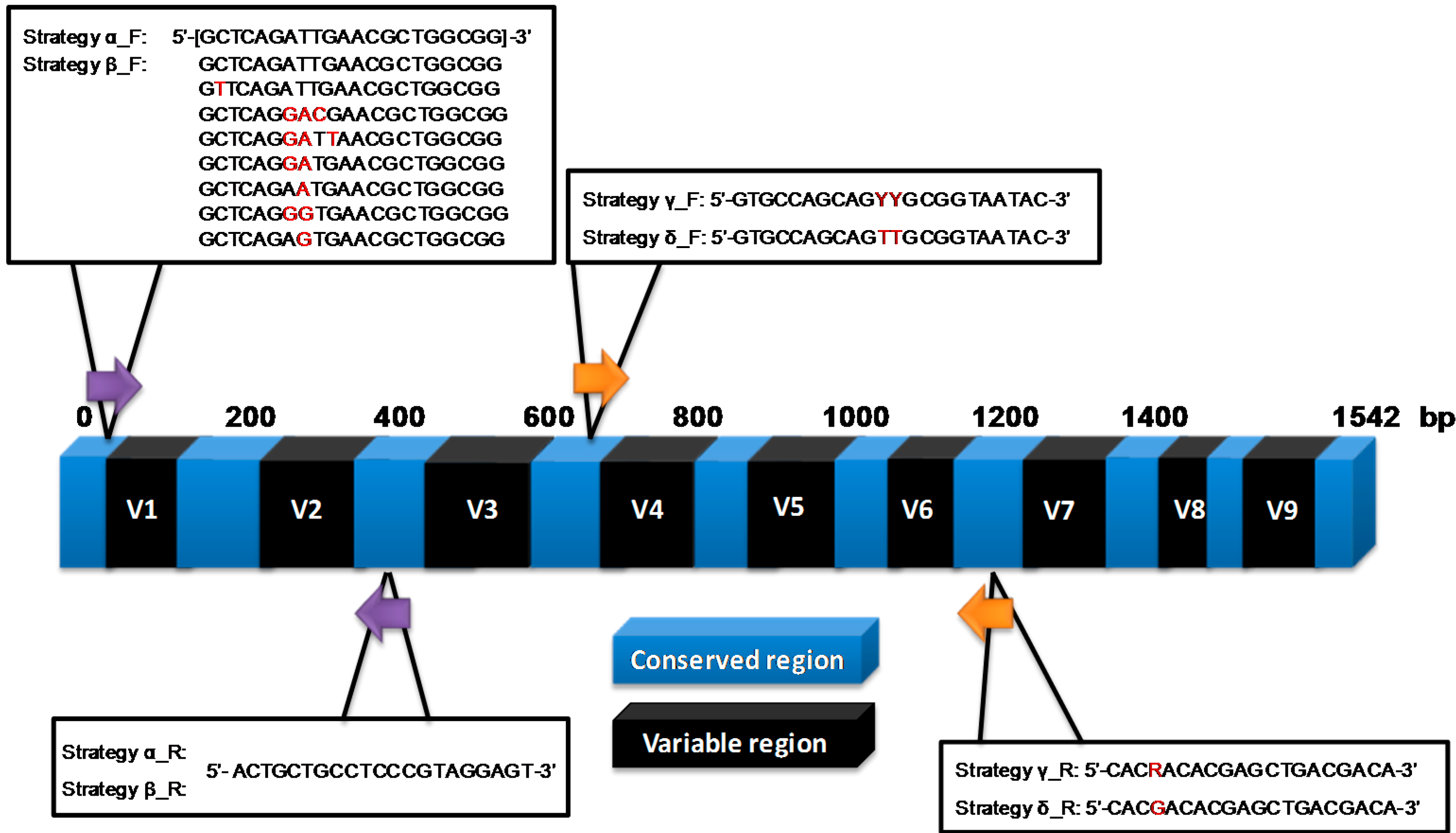
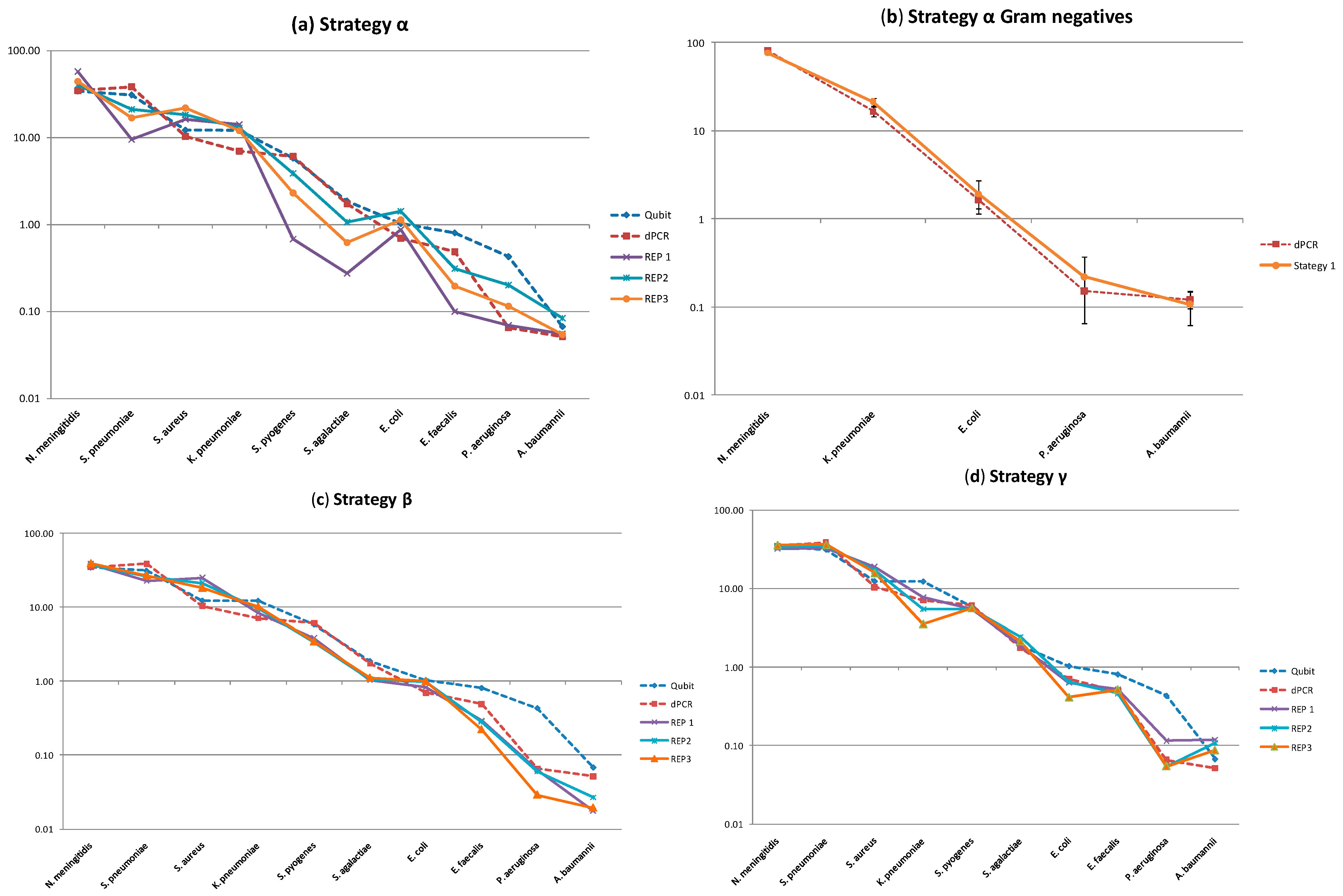
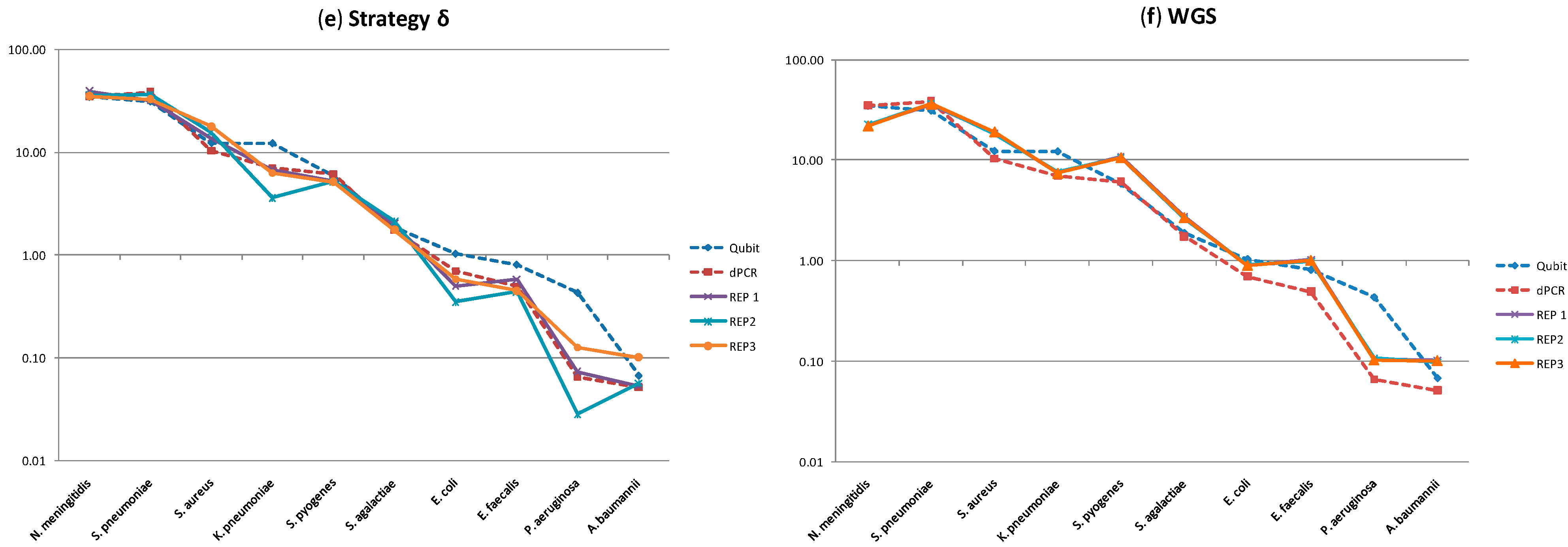
2.2. Molecular Profiling Using Different Sequencing Strategies
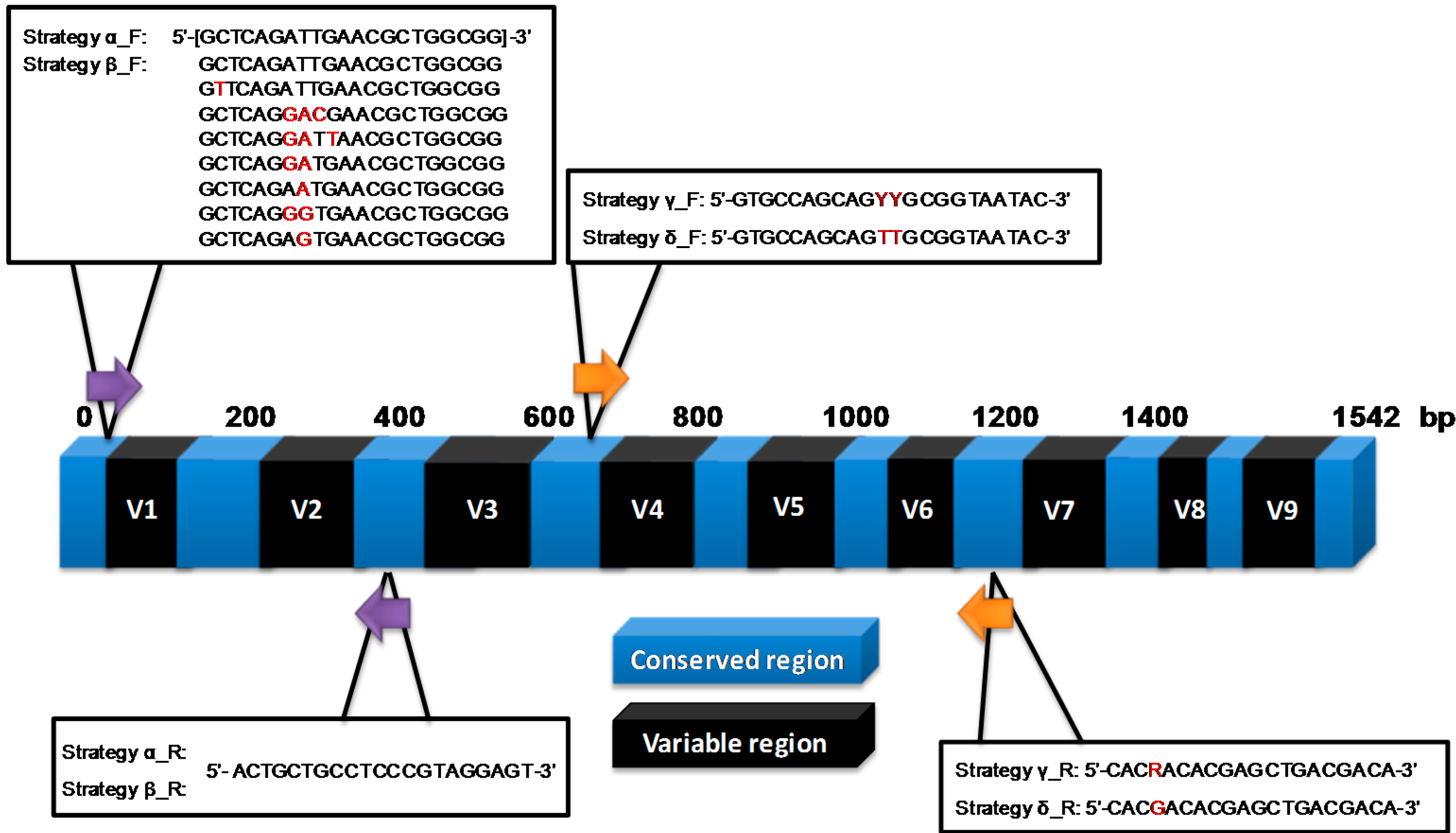

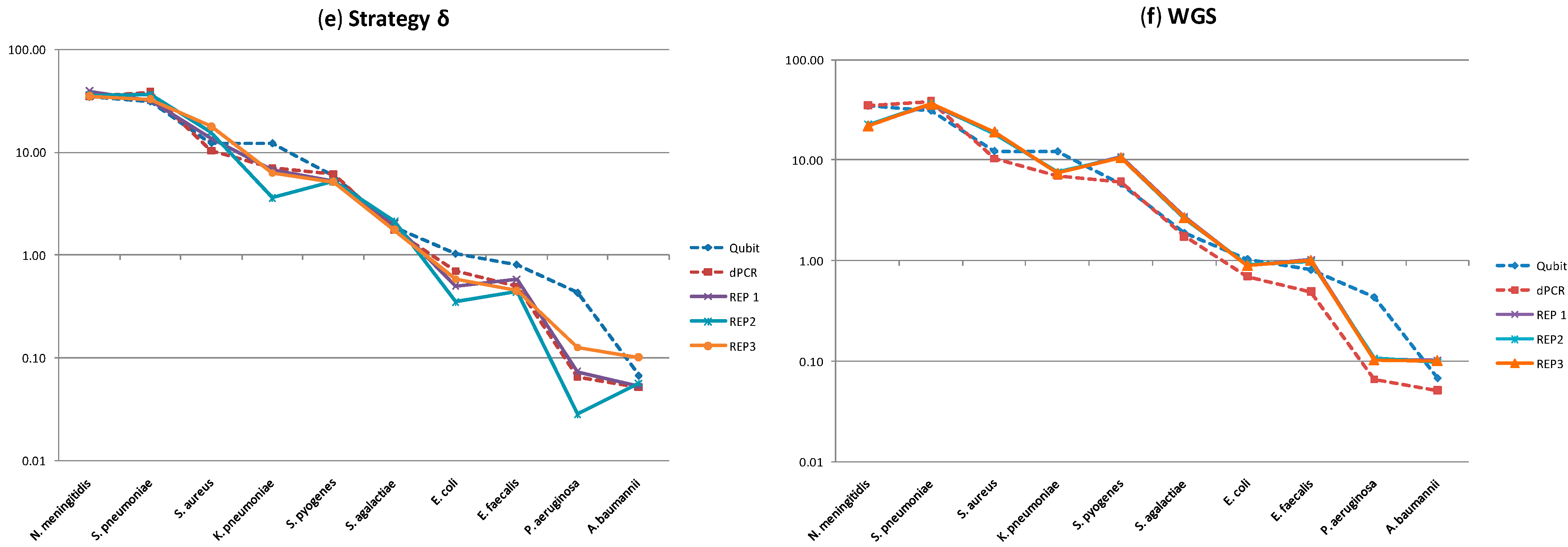

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gram | Species | Strategies (% CV) | ||||
|---|---|---|---|---|---|---|
| α | β | γ | δ | WGS | ||
| Negative | N. meningitidis | 19 | 2 | 5 | 6 | 1 |
| K. pnuemoniae | 8 | 10 | 37 | 31 | 2 | |
| E. coli | 24 | 10 | 23 | 25 | 1 | |
| P. aeruginosa | 52 | 37 | 47 | 64 | 2 | |
| A. baumannii | 26 | 23 | 15 | 38 | 2 | |
| Positive | S. pnuemoniae | 37 | 10 | 5 | 7 | 1 |
| S. aureus | 15 | 16 | 9 | 13 | 3 | |
| S. pyogenes | 70 | 7 | 2 | 1 | 1 | |
| S. agalactiae | 61 | 3 | 13 | 10 | 2 | |
| E. faecalis | 52 | 14 | 7 | 16 | 2 | |
| Average | 36 | 13 | 16 | 21 | 2 | |
2.3. Evaluation of the Effect of Database Composition
3. Experimental Section
3.1. Preparation of Metagenomic Control Material
3.2. Microfluidic Digital PCR
3.3. Amplicon Sequencing
3.3.1. PCR
3.3.2. Amplicon Sequencing
3.3.3. Data Analysis
3.4. Whole Genome Sequencing
Data Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yooseph, S.; Sutton, G.; Rusch, D.B.; Halpern, A.L.; Williamson, S.J.; Remington, K.; Eisen, J.A.; Heidelberg, K.B.; Manning, G.; Li, W.; et al. The Sorcerer II Global Ocean Sampling expedition: Expanding the universe of protein families. PLoS Biol. 2007, 5, e16. [Google Scholar] [CrossRef]
- Human Microbiome Project. Available online: http://hmpdacc.org/ (accessed on 10 February 2014).
- International Soil Metagenome Sequencing Consortium. Available online: http://www.terragenome.org/ (accessed on 22 January 2014).
- Earth Microbiome Project. Available online: http://www.earthmicrobiome.org/ (accessed on 10 February 2014).
- Woese, C.R. Bacterial evolution. Microbiol. Rev. 1987, 51, 221–271. [Google Scholar] [PubMed]
- Baker, G.C.; Smith, J.J.; Cowan, D.A. Review and re-analysis of domain-specific 16S primers. J. Microbiol. Methods 2003, 55, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Oberauner, L.; Zachow, C.; Lackner, S.; Högenauer, C.; Smolle, K.-H.; Berg, G. The ignored diversity: Complex bacterial communities in intensive care units revealed by 16S pyrosequencing. Sci. Rep. 2013, 3, 1413–1425. [Google Scholar] [CrossRef] [PubMed]
- Luna, R.A.; Fasciano, L.R.; Jones, S.C.; Boyanton, B.L.; Ton, T.T.; Versalovic, J. DNA pyrosequencing-based bacterial pathogen identification in a pediatric hospital setting. J. Clin. Microbiol. 2007, 45, 2985–2992. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Thermes, C. Library preparation methods for next-generetaion sequencing: Tone down the bias. Exp. Cell Res. 2014, 322, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, T.; Hayashizaki, Y.; Daub, C.O. SAMStat: Monitoring biases in next generation sequencing data. Bioinformatics 2011, 27, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Willner, D.; Daly, J.; Whiley, D.; Grimwood, K.; Wainwright, C.E.; Hugenholtz, P. Comparison of DNA extraction methods for microbial community profiling with an application to pediatric bronchoalveolar lavage samples. PLoS One 2012, 7, e34605. [Google Scholar] [CrossRef] [PubMed]
- (JCGM) J.C.f.G.I.M. Evaluation of measurement data—Guide to the expression of uncertainty in measurement (GUM). 2008. Available online at http://www.bipm.org/utils/common/documents/jcgm/JCGM_100_2008_E.pdf (accessed on 19 November 2014).
- Yilmaz, P.; Kottmann, R.; Field, D.; Knight, R.; Cole, J.R.; Amaral-Zettler, L.; Gilbert, J.A.; Karsch-Mizrachi, I.; Johnston, A.; Cochrane, G.; et al. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat. Biotech. 2011, 29, 415–420. [Google Scholar] [CrossRef]
- Shakya, M.; Quince, C.; Campbell, J.H.; Yang, Z.K.; Schadt, C.W.; Podar, M. Comparative metagenomic and rRNA microbial diversity characterization using archaeal and bacterial synthetic communities. Environ. Microbiol. 2013, 15, 1882–1899. [Google Scholar] [CrossRef] [PubMed]
- Huggett, J.; Laver, T.; Tamisak, S.; Nixon, G.; O’Sullivan, D.; Elaswarapu, R.; Studholme, D.; Foy, C. Considerations for the development and application of control materials to improve metagenomic microbial community profiling. Accredit. Qual. Assur. 2013, 18, 77–83. [Google Scholar] [CrossRef]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef]
- Corbisier, P.; Bhat, S.; Partis, L.; Xie, V.R.D.; Emslie, K. Absolute quantification of genetically modified MON810 maize (Zea mays L.) by digital polymerase chain reaction. Anal. Bioanal. Chem. 2010, 396, 2143–2150. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. PNAS 1999, 96, 9236–9241. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.; McLaughlin, J.L.H.; Emslie, K.R. Effect of sustained elevated temperature prior to amplification on template copy number estimation using digital polymerase chain reaction. Analyst 2011, 136, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Jumpstart Consortium Human Microbiome Project Data Generation Working Group. Evaluation of 16S rDNA-based community profiling for human microbiome research. PLoS One 2012, 7, e39315. [Google Scholar]
- Harris, K.A.; Hartley, J.C. Development of broad-range 16S rDNA PCR for use in the routine diagnostic clinical microbiology service. J. Med. Microbiol. 2003, 52, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qian, P.Y. Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS One 2009, 4, e7401. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, G.; Muniyappa, K. Hoogsteen base-pairing revisited: Resolving a role in normal biological processes and human diseases. Biochem. Biophys. Res. Commun. 2006, 343, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.T.; Giovannoni, S.J. Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl. Environ. Microbiol. 1996, 62, 625–630. [Google Scholar] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Kunin, V.; Engelbrektson, A.; Ochman, H.; Hugenholtz, P. Wrinkles in the rare biosphere: Pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ. Microbiol. 2010, 12, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Vergin, K.L.; Beszteri, B.; Monier, A.; Cameron Thrash, J.; Temperton, B.; Treusch, A.H.; Kilpert, F.; Worden, A.Z.; Giovannoni, S.J. High-resolution SAR11 ecotype dynamics at the Bermuda Atlantic Time-series Study site by phylogenetic placement of pyrosequences. ISME J. 2013, 7, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Huggett, J.F.; Foy, C.A.; Benes, V.; Emslie, K.; Garson, J.A.; Haynes, R.; Hellemans, J.; Kubista, M.; Mueller, R.D.; Nolan, T.; et al. The digital MIQE guidelines: Minimum information for publication of quantitative digital PCR experiments. Clin. Chem. 2013, 59, 892–902. [Google Scholar] [CrossRef]
- The Human Microbiome Project Consortium. A framework for human microbiome research. Nature 2012, 486, 215–221. [Google Scholar]
- Morgulis, A.; Coulouris, G.; Raytselis, Y.; Madden, T.L.; Agarwala, R.; Schäffer, A.A. Database indexing for production MegaBLAST searches. Bioinformatics 2008, 24, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.-J.; Weber, N.; Schuster, S.C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. Fast QC: A quality control tool for high throughput sequence data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 12 March 2014).
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS One 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Aronesty, E. Comparison of sequencing utility programs. Open Bioinform. J. 2013, 7, 1–8. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Gascuel, O. Bionj: An improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 1997, 14, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Galtier, N.; Gouy, M.; Gautier, C. Seaview and Phylo_win: Two graphic tools for sequence alignment and molecular phylogeny. Comput. Appl. Biosci. CABIOS 1996, 12, 543–548. [Google Scholar]
- Lee, D.-Y.; Shannon, K.; Beaudette, L.A. Detection of bacterial pathogens in municipal wastewater using an oligonucleotide microarray and real-time quantitative PCR. J. Microbiol. Methods 2006, 65, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Hartman, L.J.; Selby, E.B.; Whitehouse, C.A.; Coyne, S.R.; Jaissle, J.G.; Twenhafel, N.A.; Burke, R.L.; Kulesh, D.A. Rapid real-time PCR assays for detection of Klebsiella pneumoniae with the rmpA or magA genes associated with the hypermucoviscosity phenotype: Screening of nonhuman primates. J. Mol. Diagn. 2009, 11, 464–471. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
O'Sullivan, D.M.; Laver, T.; Temisak, S.; Redshaw, N.; Harris, K.A.; Foy, C.A.; Studholme, D.J.; Huggett, J.F. Assessing the Accuracy of Quantitative Molecular Microbial Profiling. Int. J. Mol. Sci. 2014, 15, 21476-21491. https://doi.org/10.3390/ijms151121476
O'Sullivan DM, Laver T, Temisak S, Redshaw N, Harris KA, Foy CA, Studholme DJ, Huggett JF. Assessing the Accuracy of Quantitative Molecular Microbial Profiling. International Journal of Molecular Sciences. 2014; 15(11):21476-21491. https://doi.org/10.3390/ijms151121476
Chicago/Turabian StyleO'Sullivan, Denise M., Thomas Laver, Sasithon Temisak, Nicholas Redshaw, Kathryn A. Harris, Carole A. Foy, David J. Studholme, and Jim F. Huggett. 2014. "Assessing the Accuracy of Quantitative Molecular Microbial Profiling" International Journal of Molecular Sciences 15, no. 11: 21476-21491. https://doi.org/10.3390/ijms151121476
APA StyleO'Sullivan, D. M., Laver, T., Temisak, S., Redshaw, N., Harris, K. A., Foy, C. A., Studholme, D. J., & Huggett, J. F. (2014). Assessing the Accuracy of Quantitative Molecular Microbial Profiling. International Journal of Molecular Sciences, 15(11), 21476-21491. https://doi.org/10.3390/ijms151121476