Non-Covalent Synthesis of Metal Oxide Nanoparticle–Heparin Hybrid Systems: A New Approach to Bioactive Nanoparticles


 , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
3. Experimental Section
3.1. Synthesis of MO@heparin NPs (Core@Shell: MO@heparin)
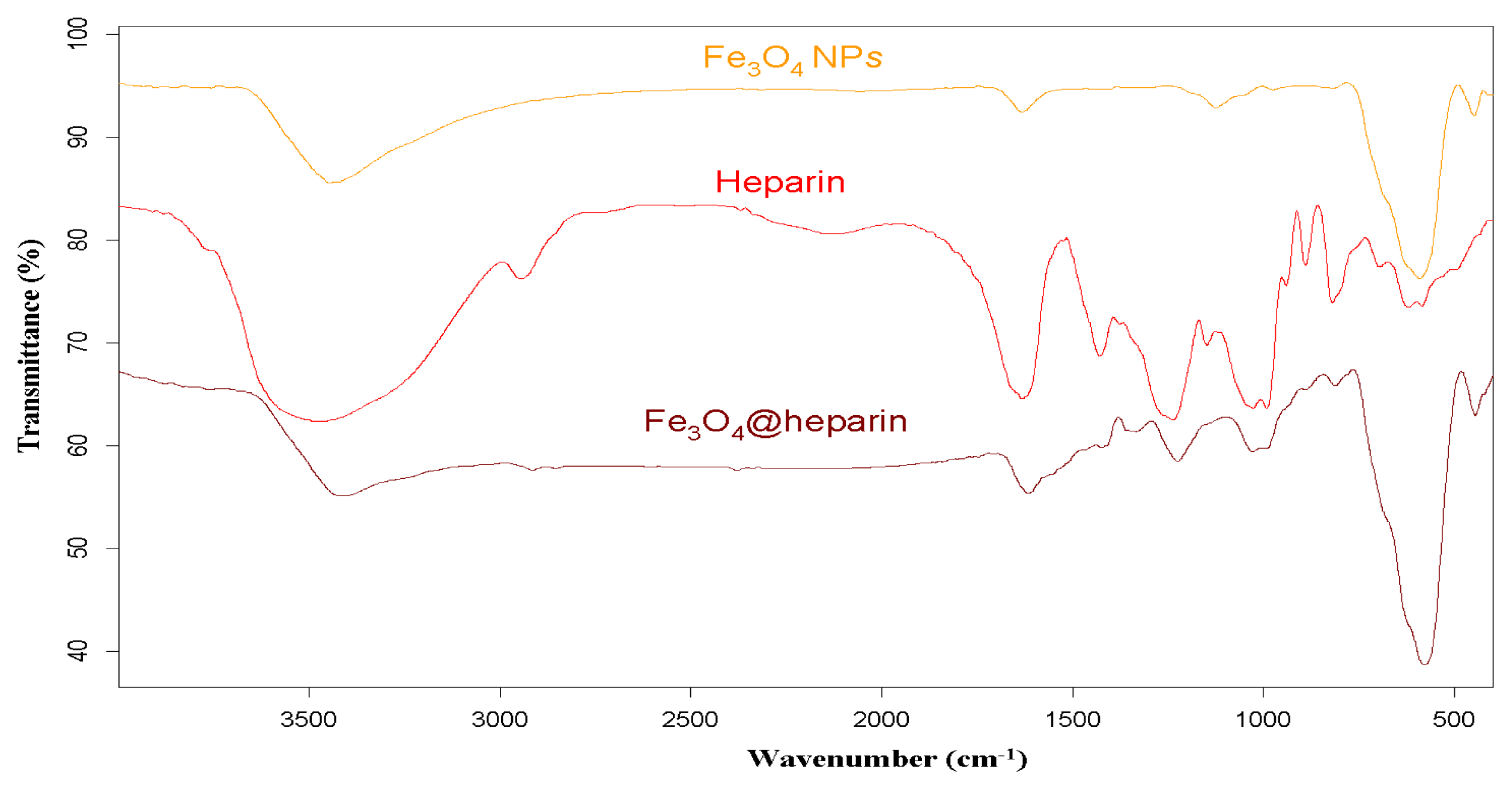
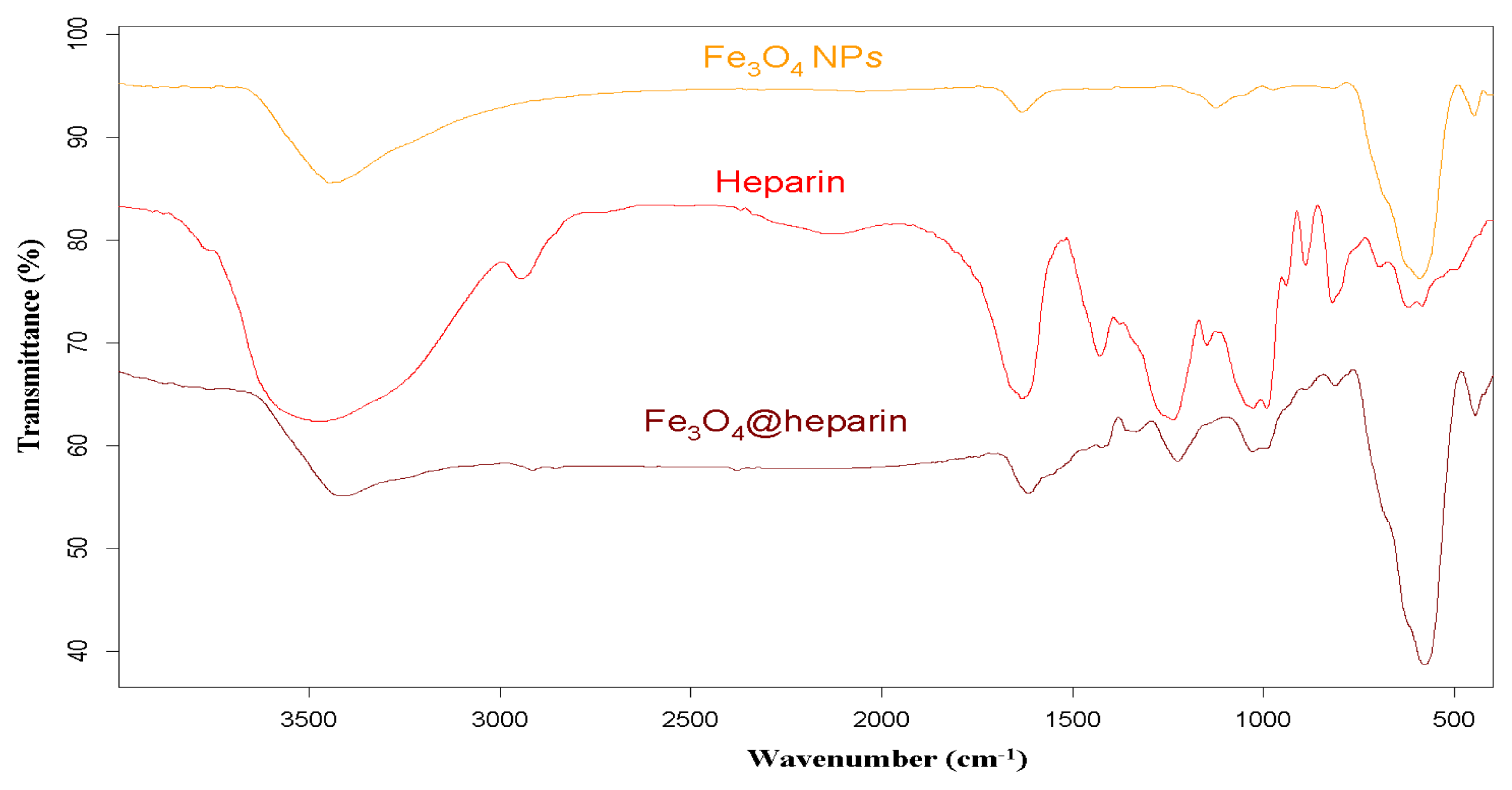
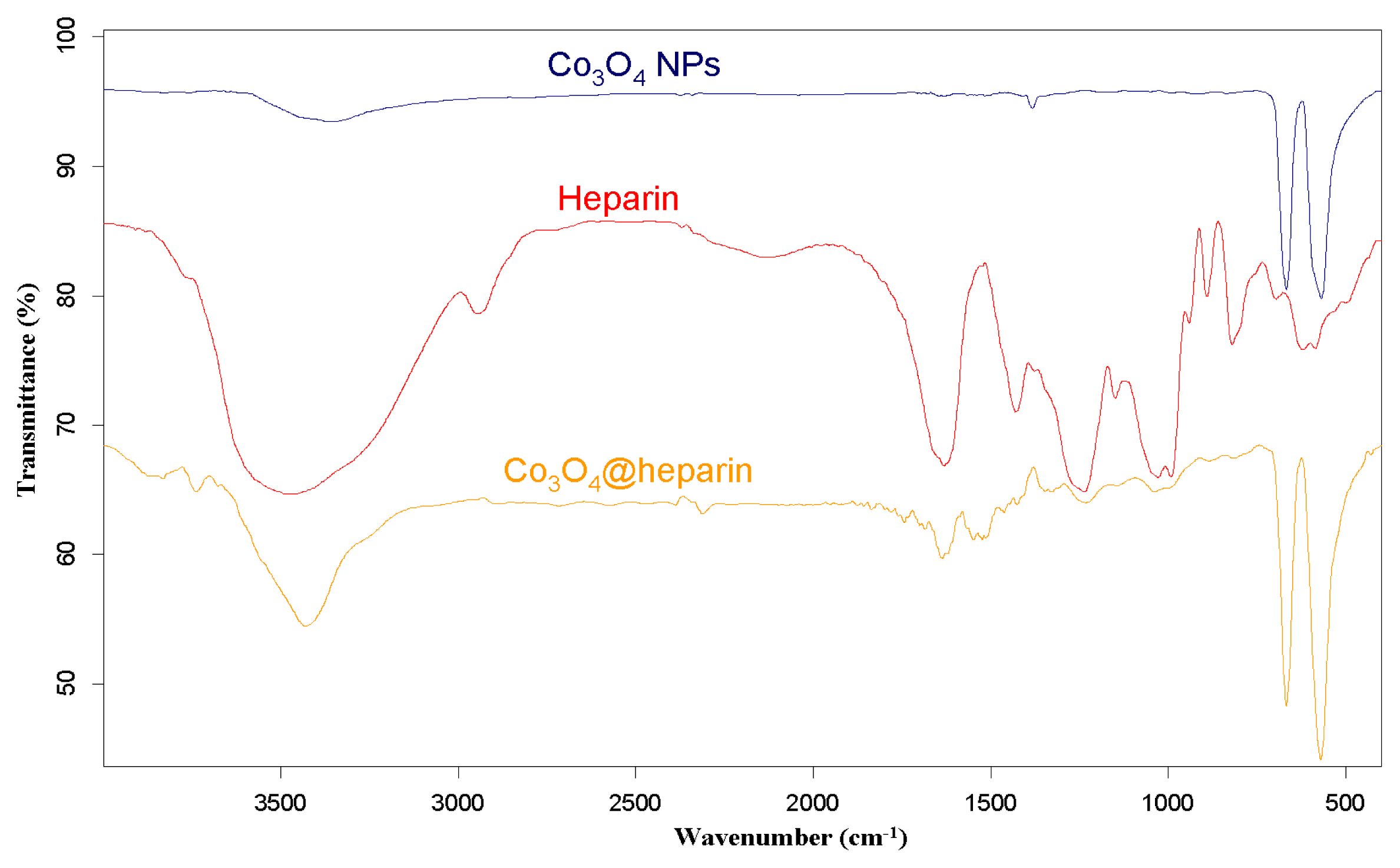
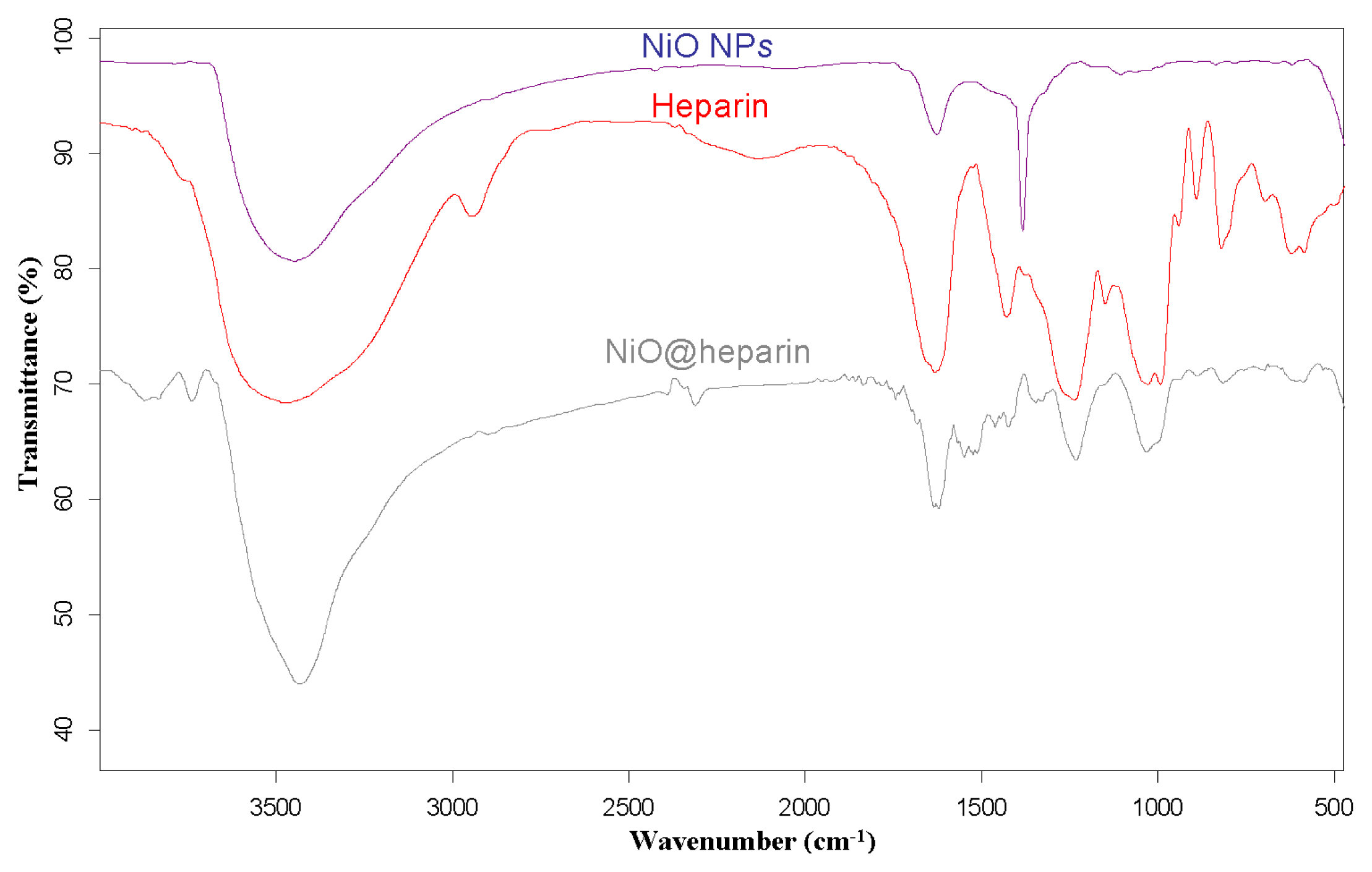
3.2. IR Characterisation of MO@heparin NPs
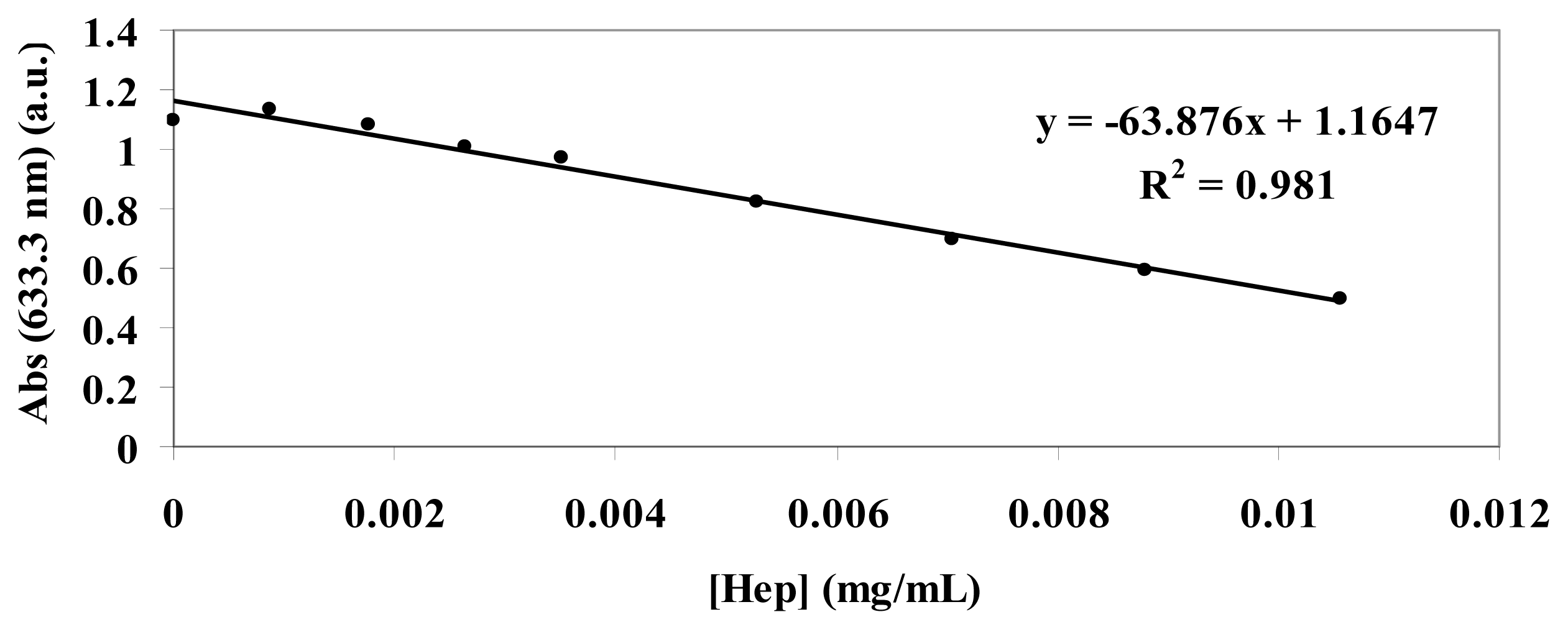
3.3. Standard Curve for the Toluidine Blue (TB) Assay
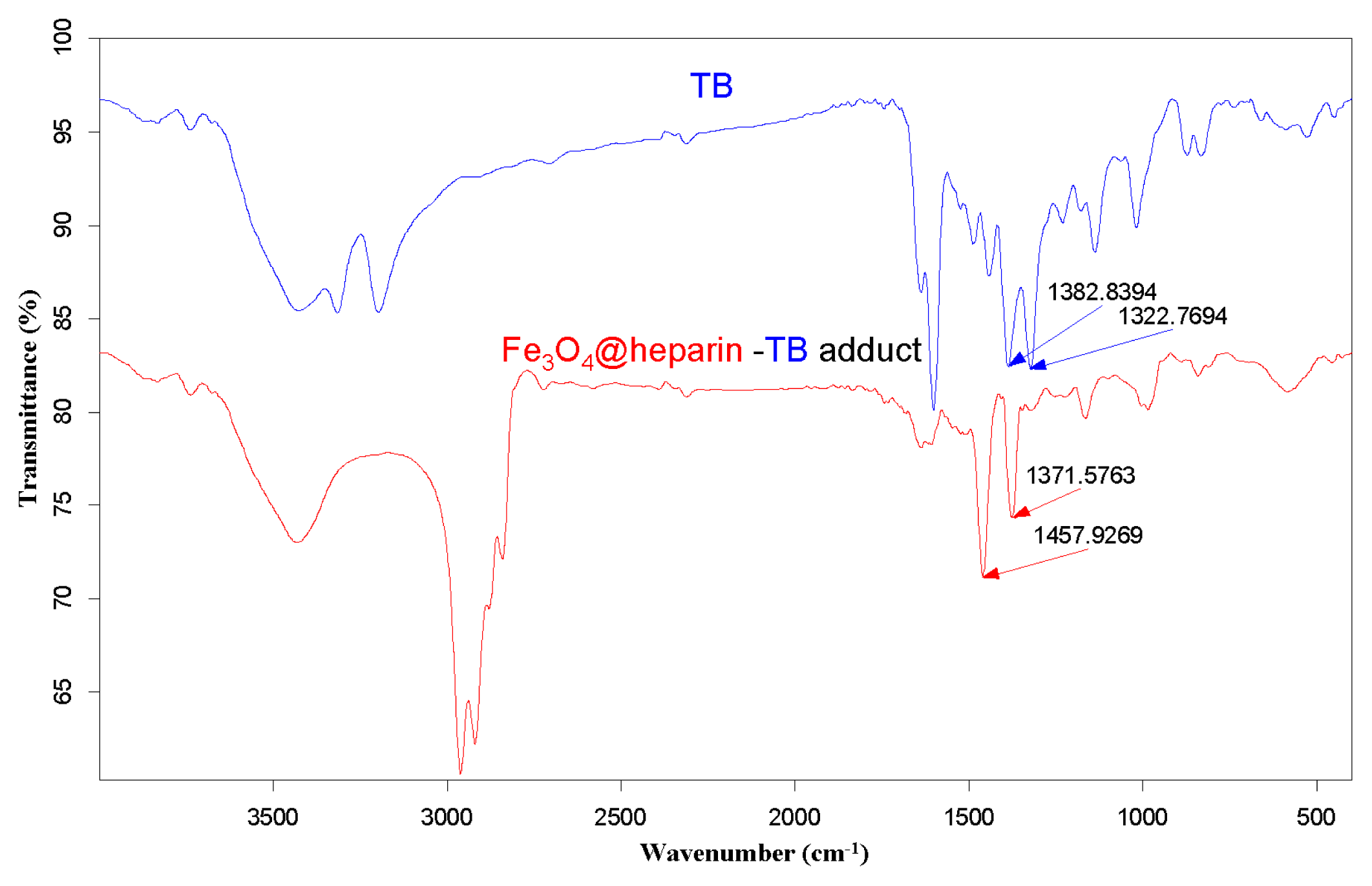
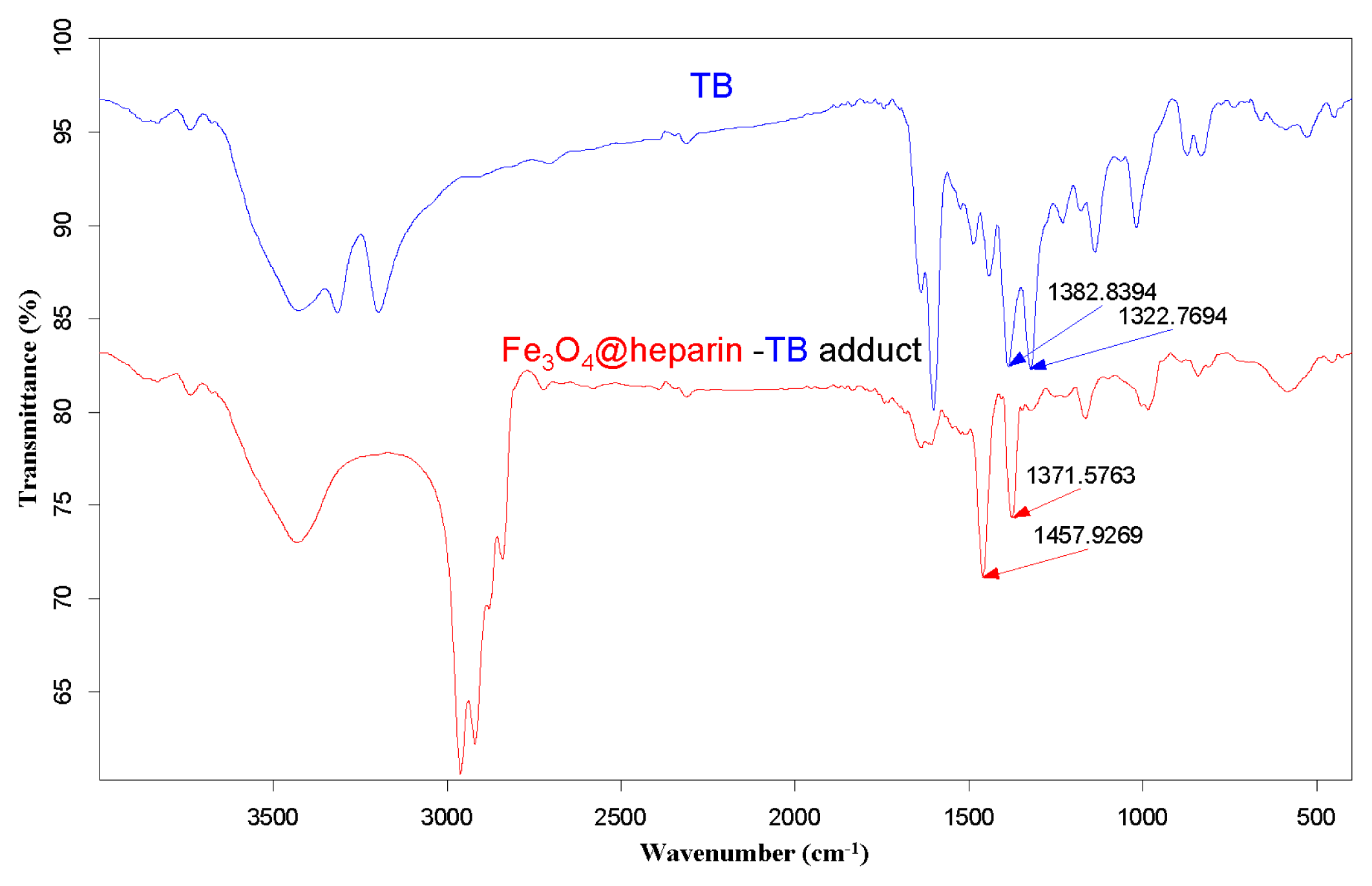
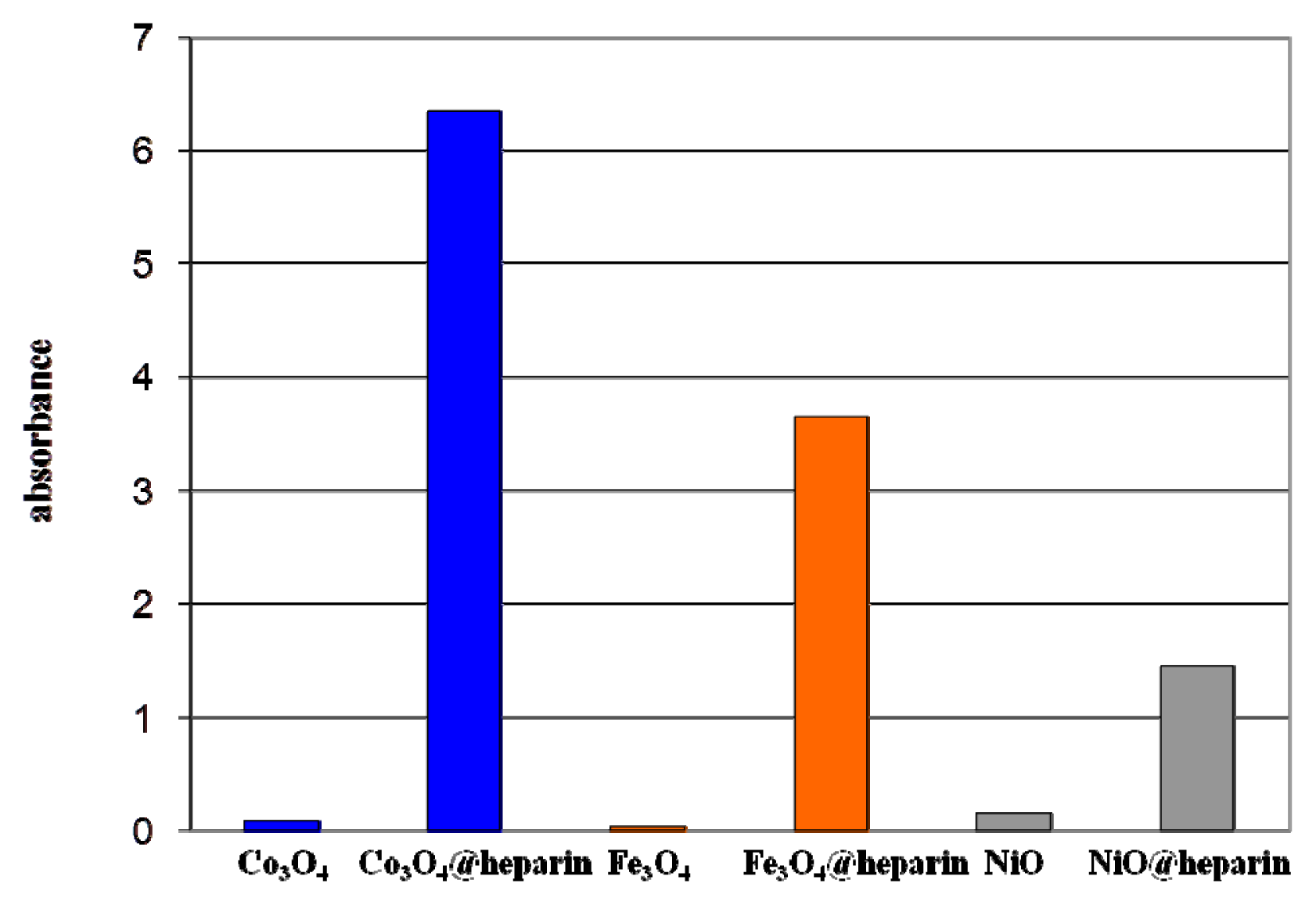
3.4. TB Assay Heparin Content in MO@heparin NPs and IR Characterisation of MO@heparin-TB Adducts
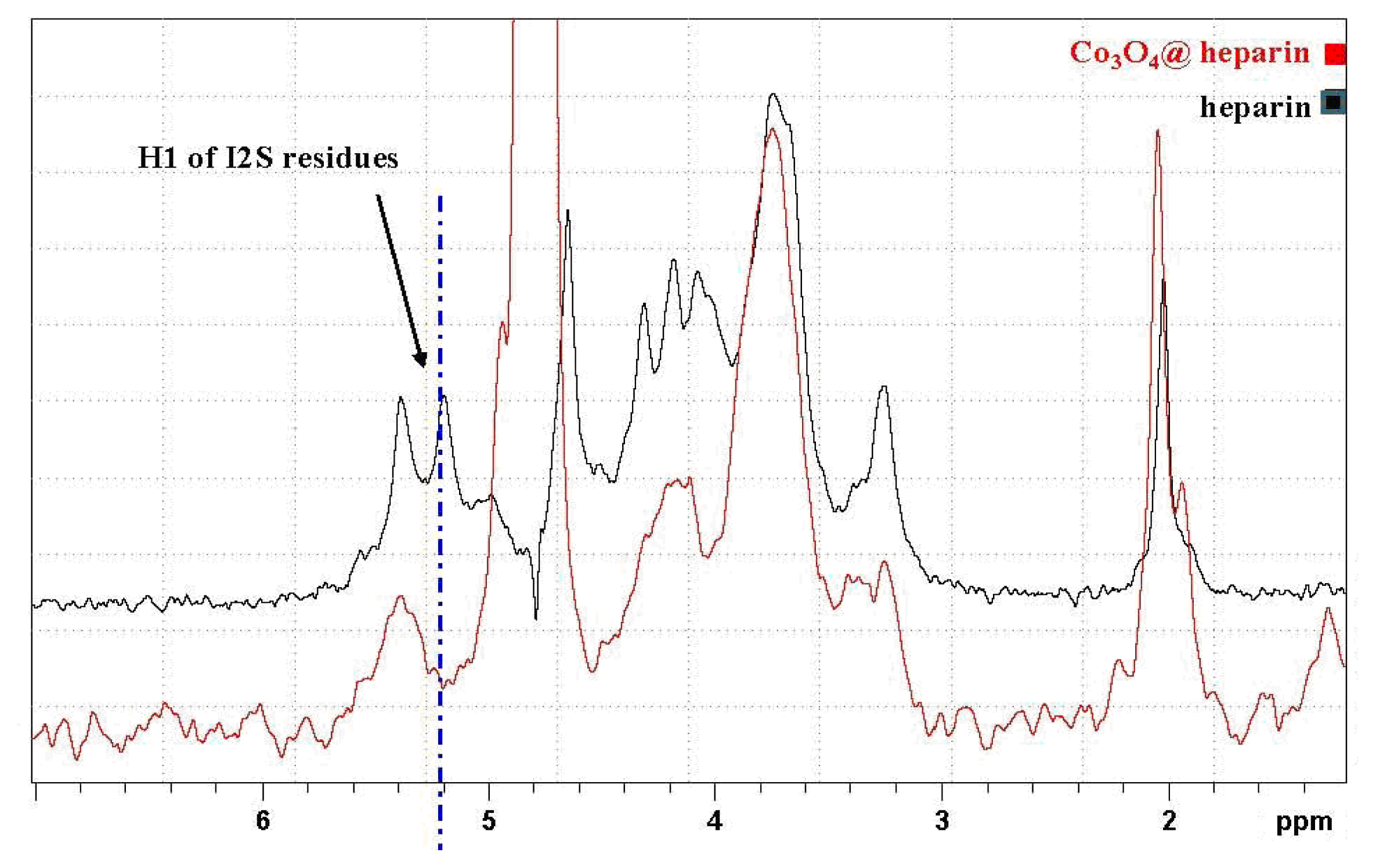
3.5. NMR Characterisation of Co3O4@heparin
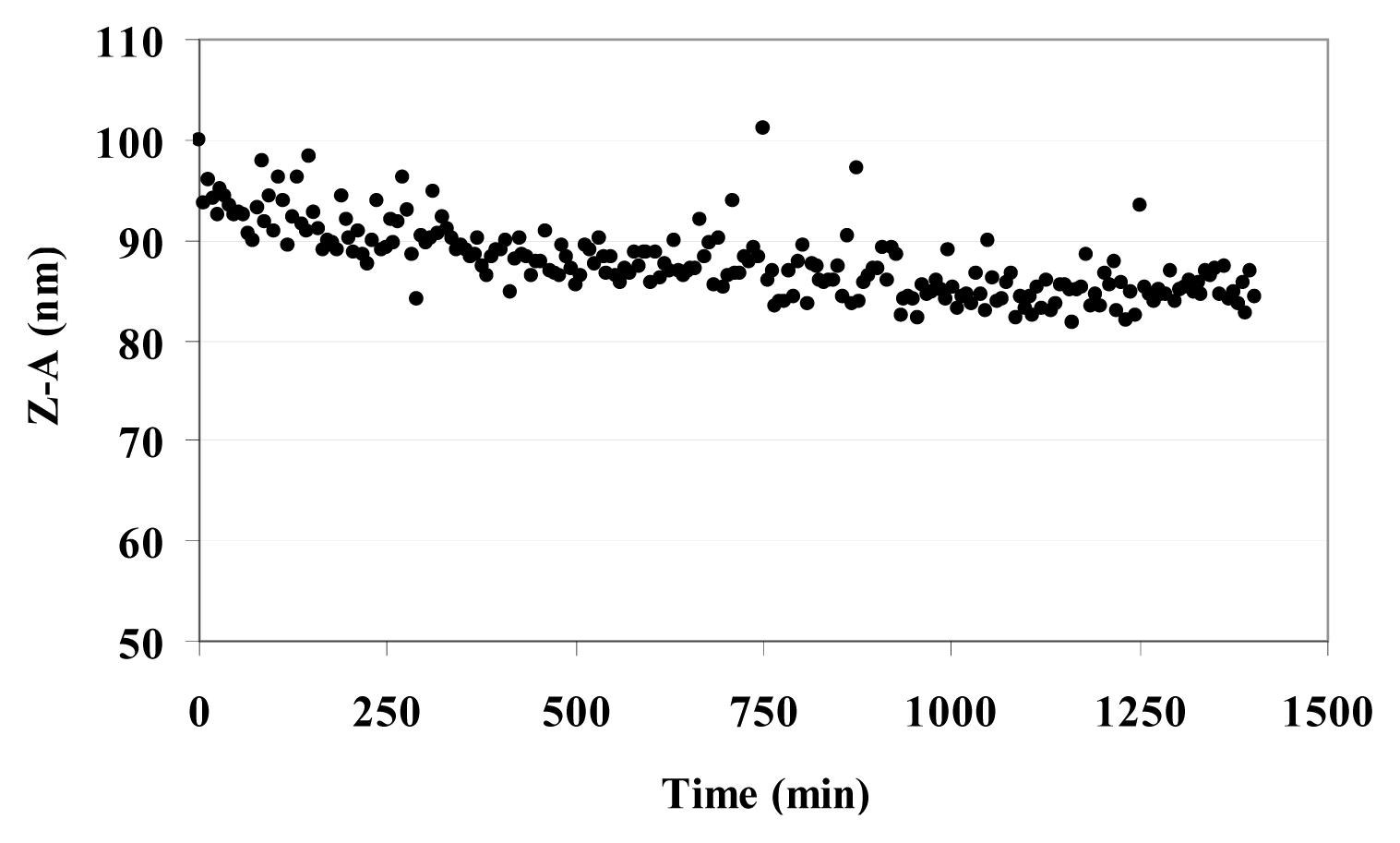
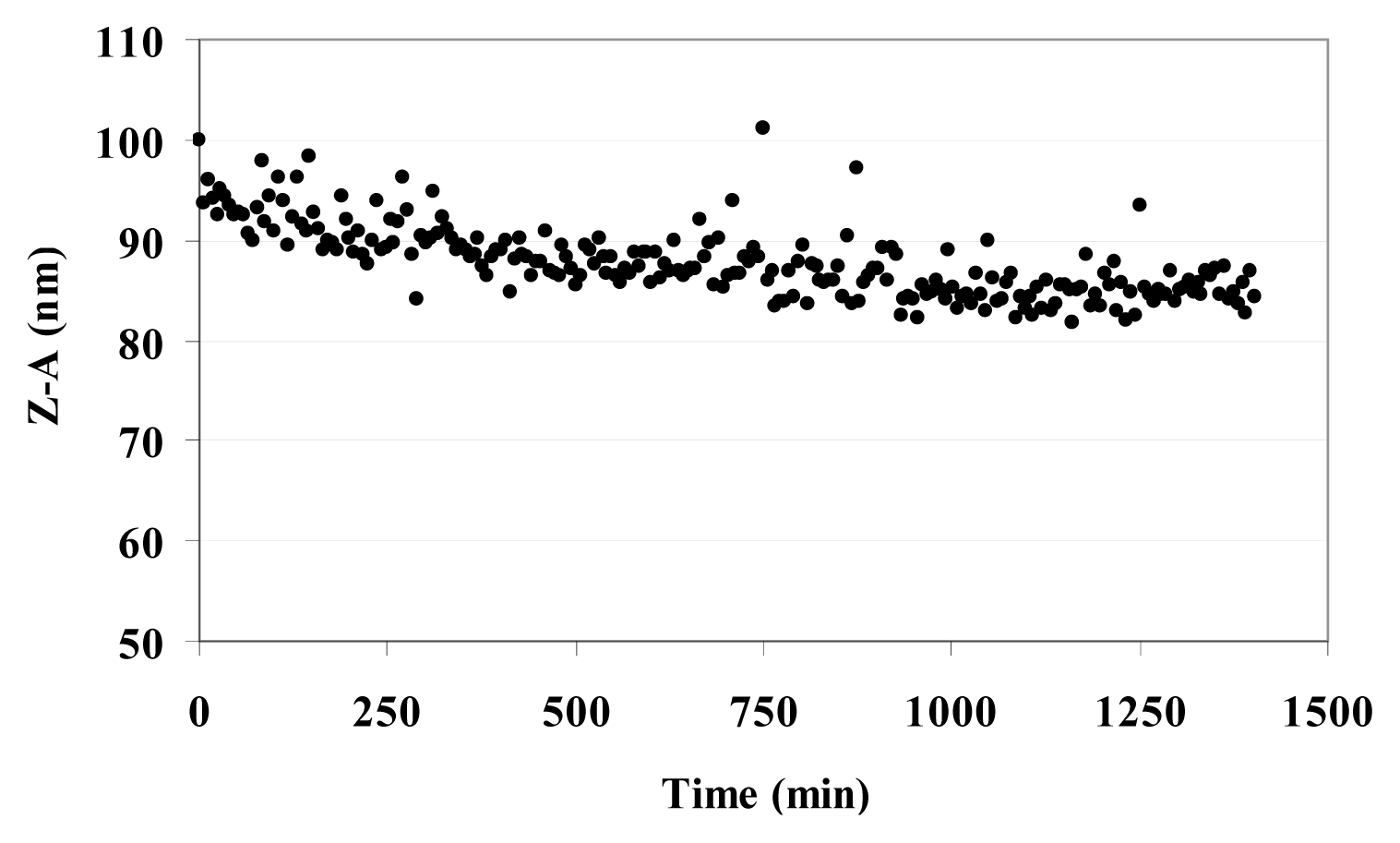
3.6. Dynamic Light Scattering (DLS) and Zeta Potential (ζ) of MO@heparin NPs and of Bare NPs
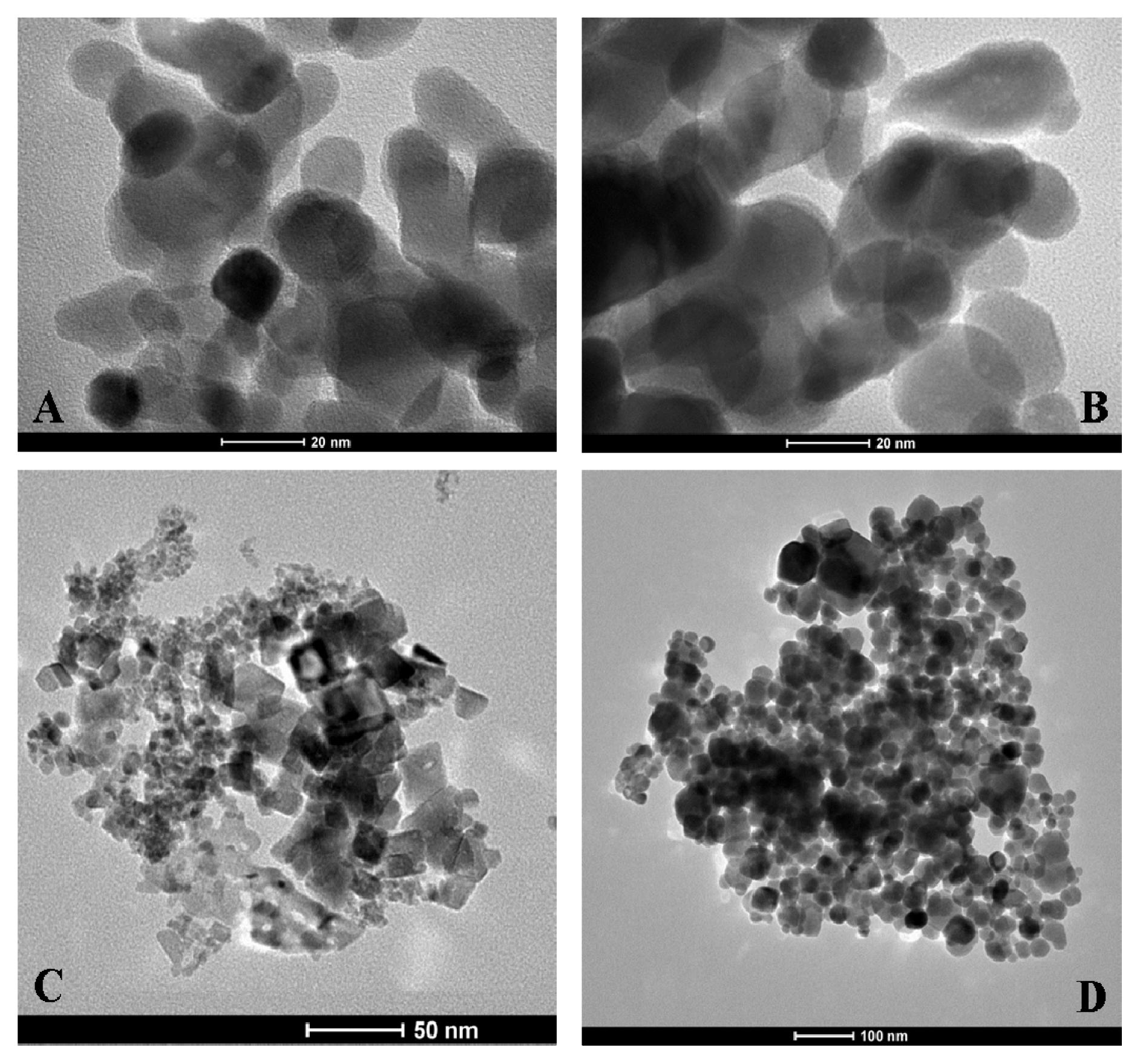
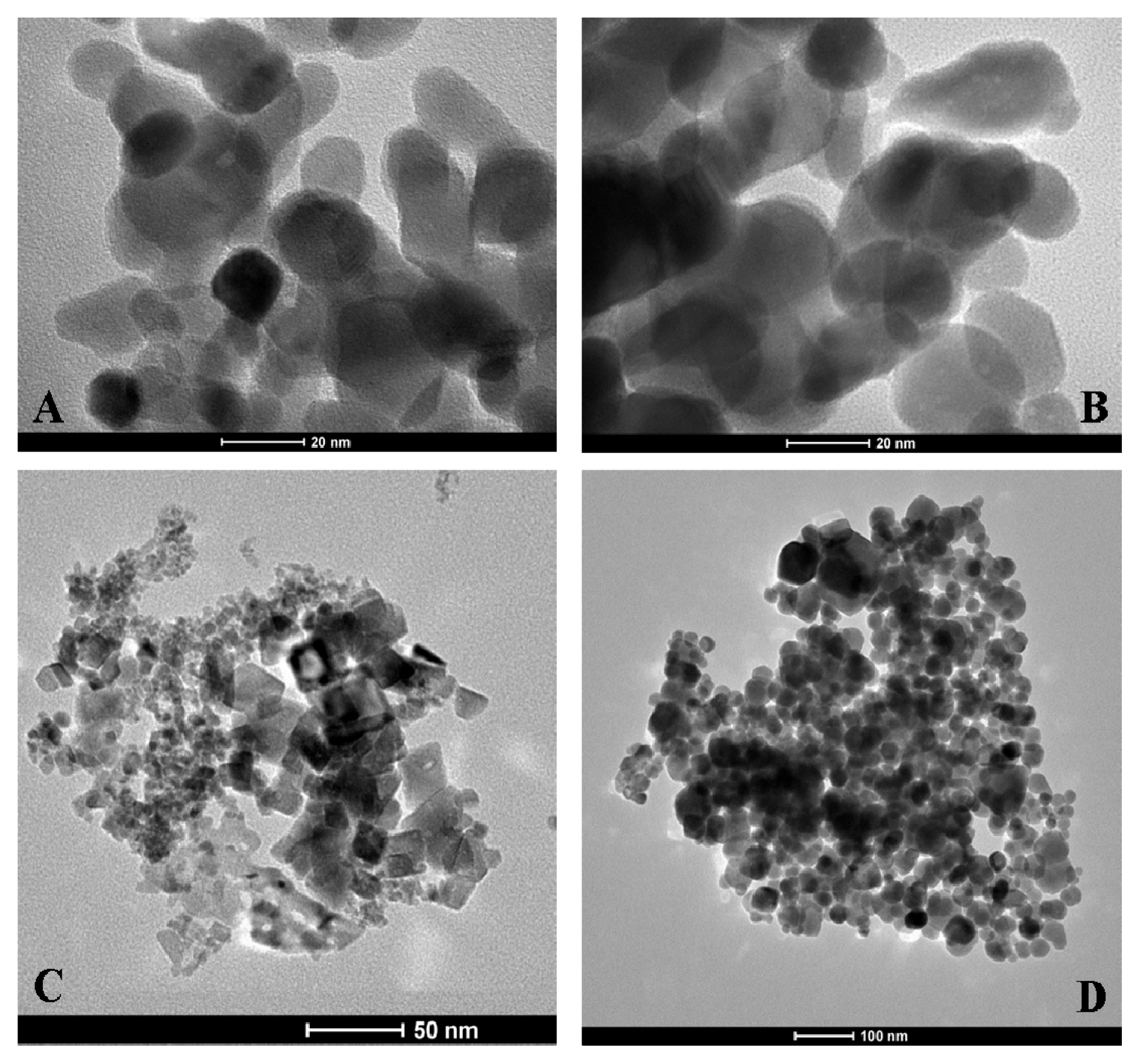
3.7. Transmission Electron Microscope (TEM) Characterisation of MO@heparin NPs and of Bare NPs
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Ghosh, C.R.; Paria, S. Core/shell nanoparticles: Classes, properties, synthesis mechanisms, characterization, and applications. Chem. Rev 2012, 112, 2373–2433. [Google Scholar]
- Janczak, C.M.; Aspinwall, C.A. Composite nanoparticles: The best of two worlds. Anal. Bioanal. Chem 2012, 402, 83–89. [Google Scholar]
- Katz, E.; Willner, I. Integrated Nanoparticles-biomolecule hybrid systems: Synthesis, properties and applications. Angew. Chem. Int. Ed 2004, 43, 6042–6108. [Google Scholar]
- Lu, A.H.; Salabas, E.L.; Schüth, F. Magnetic nanoparticles: Synthesis, protection, functionalization and application. Angew. Chem. Int. Ed 2007, 46, 1222–1244. [Google Scholar]
- De, M.; Ghosh, P.S.; Rotello, V.M. Applications of nanoparticles in biology. Adv. Mater 2008, 20, 1–17. [Google Scholar]
- Moyano, D.F.; Rotello, V.M. Nano meets biology: Structure and function at the nanoparticle interface. Langmuir 2011, 27, 10376–10385. [Google Scholar]
- Thanh, N.T.K.; Green, L.A.W. Functionalisation of nanoparticles for biomedical applications. Nano Today 2010, 5, 213–230. [Google Scholar]
- Lisi, F.; Falcaro, P.; Buso, D.; Hill, A.J.; Barr, J.A.; Crameri, G.; Nguyen, T.-L.; Wang, L.-F.; Mulvaney, P. Rapid detection of hendra virus using magnetic particles and quantum dots. Adv. Health. Mater 2012, 1, 631–634. [Google Scholar]
- Chen, X.; Gambhir, S.S.; Cheon, J. Theranostic nanomedicine. Acc. Chem. Res 2011, 44, 841–1134. [Google Scholar]
- Cattaneo, A.G.; Gornati, R.; Sabbioni, E.; Chiriva-Internati, M.; Cobos, E.; Jenkins, M.R.; Bernardini, G. Nanotechnology and human health: Risks and benefits. J. Appl. Toxicol 2010, 30, 730–744. [Google Scholar]
- Garg, H.G.; Linhardt, R.J.; Hales, C.A. Chemistry and Biology of Heparin and Heparan Sulfate; Elsevier: New York, NY, USA, 2005. [Google Scholar]
- Hostettler, N.; Naggi, A.; Torri, G.; Isahai-Michaeli, R.; Casu, B.; Vlodavsky, I.; Borsig, L. P-selectin- and heparanase-dependent antimetastatic activity of non-anticoagulant heparins. FASEB J 2007, 21, 3562–3572. [Google Scholar] [Green Version]
- Bendas, G.; Lubor, B. Cancer cell adhesion and metastasis: Selectins, integrins, and the inhibitory potential of heparins. Int. J. Cell. Biol. 2012. [Google Scholar] [CrossRef]
- Casu, B.; Vlodavsky, I.; Sanderson, R.D. Non-anticoagulant heparins and inhibition of cancer. Pathophysiol. Haemost. Thromb 2007, 36, 195–203. [Google Scholar]
- Gupta, A.K.; Naregalkar, R.R.; Vaidya, V.D.; Gupta, M. Recent advances on surface engineering of magnetic iron oxide nanoparticles and their biomedical applications. Nanomedicine 2007, 2, 23–39. [Google Scholar]
- Mornet, S.; Vasseur, S.; Grasset, F.; Duguet, E. Magnetic nanoparticle design for medical diagnosis and therapy. J. Mater. Chem 2004, 14, 2161–2175. [Google Scholar]
- Don, H.; Sun, X.; Sun, S. Monodisperse magnetic nanoparticles for theranostic applications. Acc. Chem. Res 2011, 44, 875–882. [Google Scholar]
- Mahmoudi, M.; Simchi, A.; Milani, A.S.; Stroeve, P. Cell toxicity of superparamagnetic iron oxide nanoparticles. J. Colloid Interf. Sci 2009, 336, 510–518. [Google Scholar]
- Li, Z.; Kawashita, M.; Araki, N.; Mitsumori, M.; Hiraoka, M.; Doi, M. Magnetite nanoparticles with high heating efficiencies for application in the hyperthermia of cancer. Mater. Sci. Eng. C 2010, 30, 990–996. [Google Scholar]
- Munnier, E.; Cohen-Jonathan, S.; Herve, K.; Linassier, C.; Souce, M.; Dubois, P.; Chourpa, I. Doxorubicin delivered to MCF-7 cancer cells by superparamagnetic iron oxide nanoparticles: Effects on subcellular distribution and cytotoxicity. J. Nanopart. Res 2011, 13, 959–971. [Google Scholar]
- Kievit, F.M.; Zhang, M. Surface engineering of iron oxide nanoparticles was studied for targeted cancer therapy. Acc. Chem. Res 2011, 44, 853–862. [Google Scholar]
- Klostergaard, J.; Seeney, C.E. Magnetic nanovectors for drug delivery. Nanomed. Nanotech. Biol. Med 2012, 8, S37–S50. [Google Scholar]
- Papis, E.; Gornati, R.; Prati, M.; Ponti, J.; Sabbioni, E.; Bernardini, G. Gene expression in nanotoxicology research: Analysis by differential display in BALB3T3 fibroblasts exposed to cobalt particles and ions. Toxicol. Lett 2007, 170, 185–192. [Google Scholar]
- Papis, E.; Rossi, F.; Raspanti, M.; Dalle-Donne, I.; Colombo, G.; Milzani, A.; Bernardini, G.; Gornati, R. Engineered cobalt oxide nanoparticles readily enter cells. Toxicol. Lett 2009, 189, 253–259. [Google Scholar]
- Sabbioni, E.; Fortaner, S.; Farina, M.; del Torchio, R.; Petrarca, C.; Bernardini, G.; Mariani-Costantini, R.; Perconti, S.; di Giampaolo, L.; Gornati, R.; et al. Interaction with culture medium components, cellular uptake and intracellular distribution of cobalt nanoparticles, microparticles and ions in Balb/3T3 mouse fibroblasts. Nanotoxicology 2012. [Google Scholar] [CrossRef]
- Pan, Y.; Du, X.; Zhao, F.; Xu, B. Magnetic nanoparticles for the manipulation of proteins and cells. Chem. Soc. Rev 2012, 41, 2912–2942. [Google Scholar]
- Apátiga, L.M.; Castaňo, V.M. Magnetic behavior of cobalt oxide films prepared by pulsed liquid injection chemical vapor deposition from a metal-organic precursor. Thin Solid Films 2006, 496, 576–579. [Google Scholar]
- Ctistis, G.; Papaioanno, E.; Patok, P.; Gute, J.; Fumagalli, P.; Giersig, M. Optical and magnetic properties of hexagonal arrays of subwavelength holes in optically thin cobalt films. Nano Lett 2009, 9, 1–6. [Google Scholar]
- Salavati-Niasari, M.; Khansari, A.; Davar, F. Synthesis and characterization of cobalt oxide nanoparticles by thermal treatment process. Inorg. Chim. Acta 2009, 362, 4937–4942. [Google Scholar]
- Wolff, A.; Frese, K.; Wißbrock, M.; Eckstädt, K.; Ennen, I.; Hetaba, W.; Löffler, S.; Regtmeier, A.; Thomas, P.; Sewald, N.; et al. Influence of the synthetic polypeptide c25-mms6 on cobalt ferrite nanoparticle formation. J. Nanopart. Res 2012, 14, 1161–1171. [Google Scholar]
- Zhao, J.; Deng, M.; Zeng, J.; Huang, Z.; Yin, G.; Liao, X.; Gub, J.; Huang, J. Preparation of Fe3O4 and CoFe2O4 nanoparticles with cellular compatibility via the histidine assistance. Colloid Surf. A 2012, 401, 54–60. [Google Scholar]
- Moghaddam, A.B.; Ganjali, M.R.; Dinarvand, R.; Razavi, T.; Saboury, A.A.; Moosavi-Movahedi, A.A.; Norouzi, P. Direct electrochemistry of cytochrome C on electrodeposited nickel oxide nanoparticles. J. Electroanal. Chem 2008, 614, 83–92. [Google Scholar]
- Lee, K.S.; Lee, I.S. Decoration of superparamagnetic iron oxide nanoparticles with Ni2+: Agent to bind and separate histidine-tagged proteins. Chem. Commun 2008, 709–711. [Google Scholar]
- Luo, L.; Li, F.; Zhu, L.; Ding, Y.; Zhang, Z.; Deng, D.; Lu, B. Nonenzymatic glucose sensor based on nickel(II)oxide/ordered mesoporous carbon modified glassy carbon electrode. Colloid Surf. B 2013, 102, 307–311. [Google Scholar]
- Tassa, C.; Shaw, S.Y.; Weissleder, R. Dextran-coated iron oxide nanoparticles: A versatile platform for targeted molecular imaging, molecular diagnostics, and therapy. Acc. Chem. Res 2011, 44, 842–852. [Google Scholar]
- Chertok, B.; Moffat, B.A.; David, A.E.; Yu, F.; Bergemann, C.; Ross, B.D.; Yang, V.C. Iron oxide nanoparticles as a drug delivery vehicle for MRI monitored magnetic targeting of brain tumors. Biomaterials 2008, 29, 487–496. [Google Scholar]
- Kemp, M.M.; Linhardt, R.J. Heparin-based nanoparticles. Nanomed. Nanobiotechnol 2010, 2, 77–87. [Google Scholar]
- Lee, J.; Jung, M.J.; Hwang, Y.H.; Lee, Y.J.; Lee, S.S.; Lee, D.Y.; Shin, H. MRI of transplanted surface-labeled pancreatic islets with heparinized superparamagnetic iron oxide nanoparticles. Biomaterials 2011, 32, 9391–9400. [Google Scholar]
- Hong, R.; Fischer, N.O.; Verma, A.; Goodman, C.M.; Emrick, T.; Rotello, V.M. Control of protein structure and function through surface recognition by tailored nanoparticle scaffolds. J. Am. Chem. Soc. 2004, 126, 739–743. [Google Scholar]
- Irrgang, J.; Ksienczyk, J.; Lapiene, V.; Niemeyer, C.M. Analysis of non-covalent bioconjugation of colloidal nanoparticles by means of atomic force microscopy and data clustering. ChemPhysChem 2009, 10, 1483–1491. [Google Scholar]
- Medintz, I.L.; Uyeda, H.T.; Goldmann, E.R.; Mattoussi, H. Quantum dot bioconjugates for imaging labelling and sensing. Nat. Mater 2005, 4, 435–446. [Google Scholar]
- Clapp, A.R.; Medintz, I.L.; Mattoussi, H. Förster resonance energy transfer investigations using quantum-dot fluorophores. ChemPhysChem 2006, 7, 47–57. [Google Scholar]
- Khurshid, H.; Kim, S.H.; Bonder, M.J.; Colak, L.; Ali, B.; Shah, S.I.; Kiick, K.L.; Hadjipanayis, G.C. Development of heparin-coated magnetic nanoparticles for targeted drug delivery applications. J. Appl. Phys 2009, 105. [Google Scholar] [CrossRef]
- Smith, P.K.; Mallia, A.K.; Hermanson, G.T. Colorimetric method for the assay of heparin content in immobilized heparin preparations. Anal. Biochem 1980, 109, 466–473. [Google Scholar]
- Legrand, L.; Sagon, G.; Lecomte, S.; Chausse, A.; Messina, R. A raman and infrared study of a new carbonate green rust obtained by electrochemical way. Corros. Sci 2001, 43, 1739–1749. [Google Scholar]
- Ishikawa, T.; Ueno, T.; Yasukawa, A.; Kandori, K.; Nakayama, T.; Tsubota, T. Influence of metal ions on the structure of poorly crystallized iron oxide rusts. Corros. Sci 2003, 45, 1037–1049. [Google Scholar]
- Deraz, N.M. Production and characterization of pure and doped copper ferrite nanoparticles. J. Anal. Appl. Pyrolysis 2008, 82, 212–222. [Google Scholar]
- Simpson, A.J.; Kingery, W.L.; Shaw, D.R.; Spraul, M.; Humpfer, E.; Dvortsak, P. The application of 1H HR-MAS NMR spectroscopy for the study of structures and associations of organic components at the solid-aqueous interface of a whole soil. Environ. Sci. Technol 2001, 35, 3321–3325. [Google Scholar]
- Neville, G.A.; Mori, F.; Holme, K.R.; Perlin, A.S. Monitoring the purity of pharmaceutical heparin preparations by high-field 1H-nuclear magnetic resonance spectroscopy. J. Pharm. Sci 1989, 78, 101–104. [Google Scholar]
- Smernik, R.J.; Oades, J.M. Effects of added paramagnetic ions on the 13C CP/MAS NMR spectrum of a de-ashed soil. Geoderma 1999, 89, 219–248. [Google Scholar]
- Rudd, T.R.; Guimond, S.E.; Skidmore, M.A.; Duchesne, L.; Guerrini, M.; Torri, G.; Cosentino, C.; Brown, A.; Clarke, D.T.; Turnbull, J.E.; et al. Influence of substitution pattern and cation binding on conformation and activity in heparin derivatives. Glycobiology 2007, 17, 983–993. [Google Scholar]
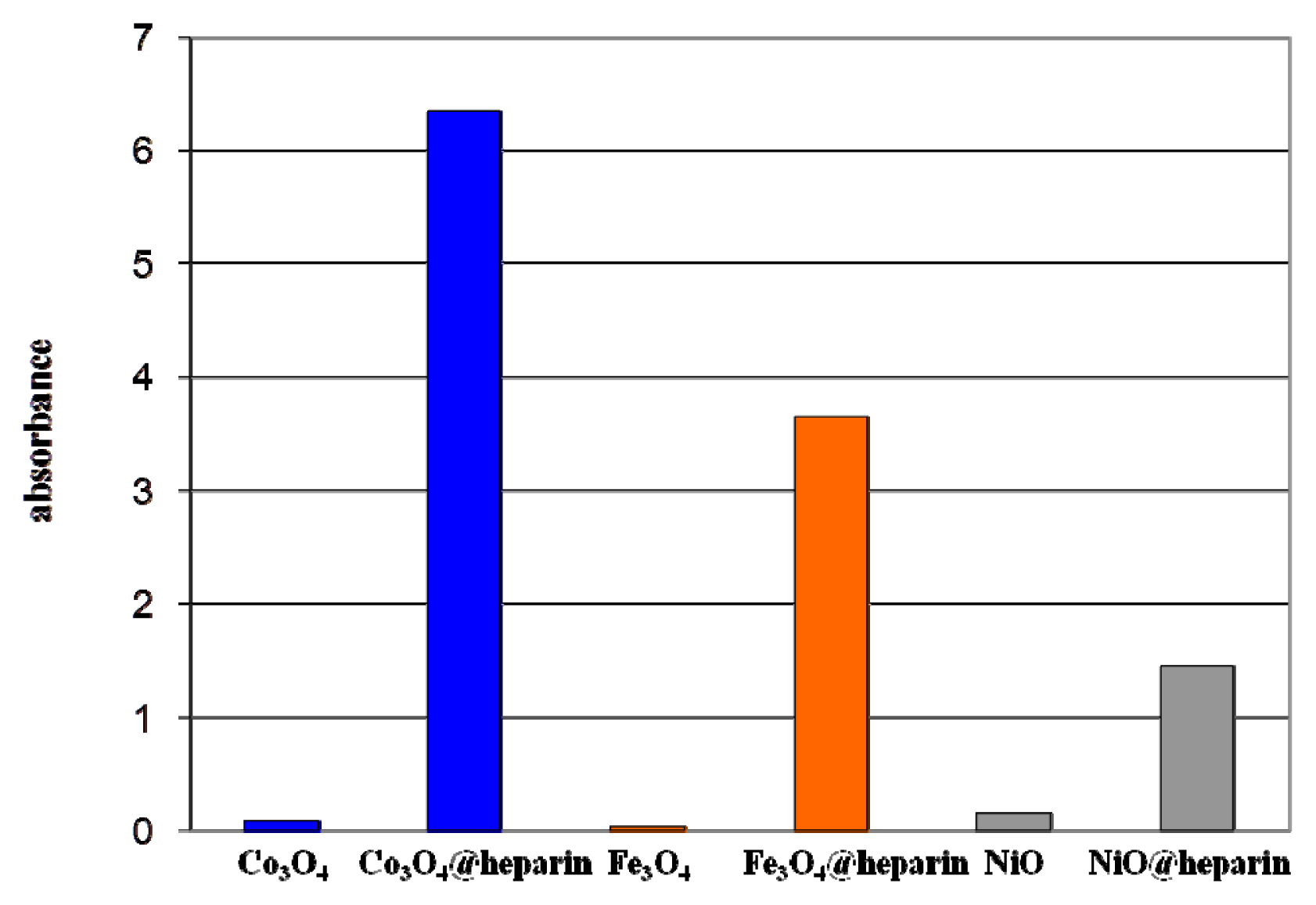
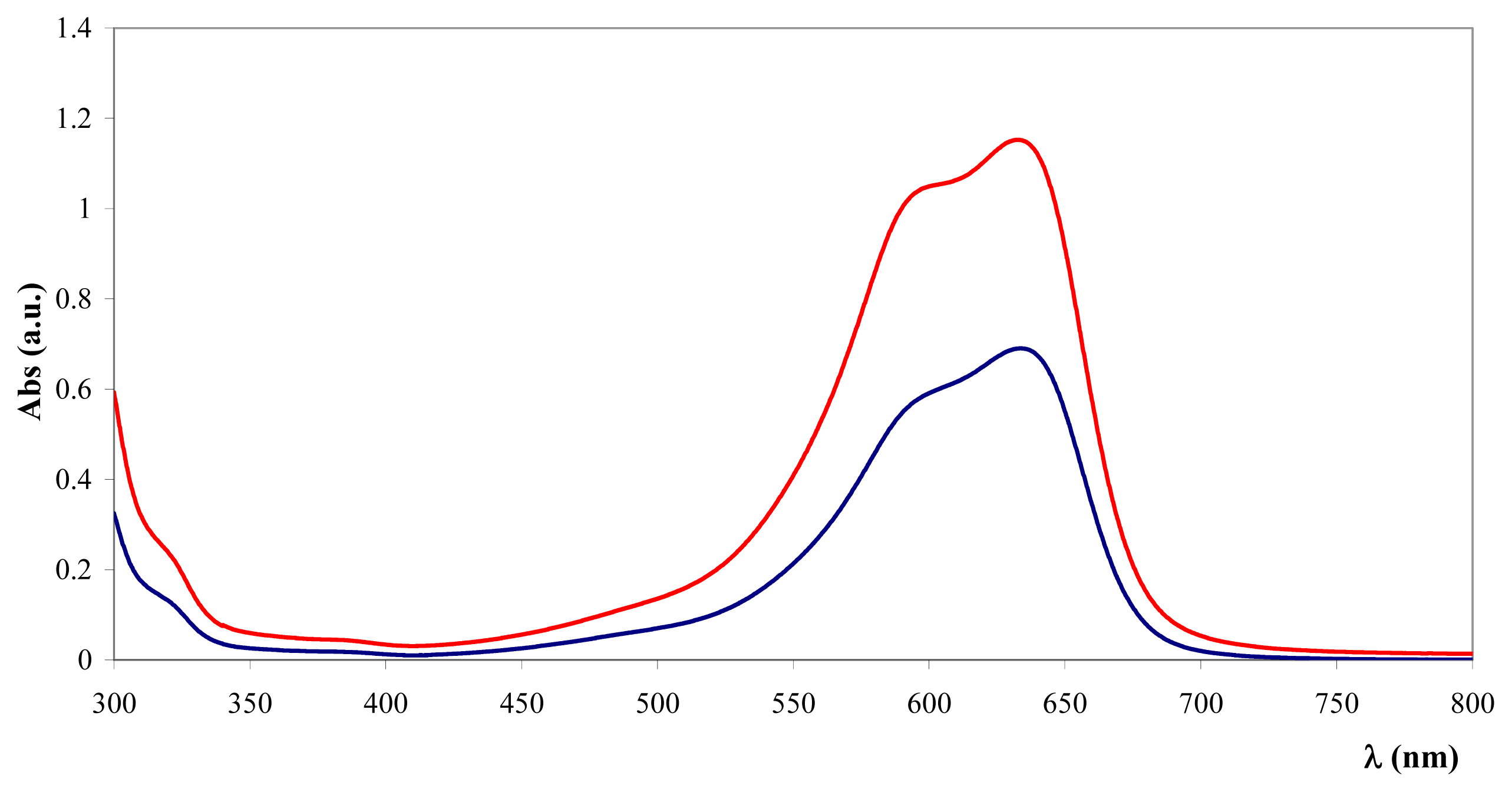
 ) and after (
) and after (
 ) the addition of Fe3O4@heparin NP. The comparison highlights the depletion of TB in the supernatant upon the addition of Fe3O4@heparin NP.
) and after (
) the addition of Fe3O4@heparin NP. The comparison highlights the depletion of TB in the supernatant upon the addition of Fe3O4@heparin NP.
) the addition of Fe3O4@heparin NP. The comparison highlights the depletion of TB in the supernatant upon the addition of Fe3O4@heparin NP.
) and after (
) the addition of Fe3O4@heparin NP. The comparison highlights the depletion of TB in the supernatant upon the addition of Fe3O4@heparin NP.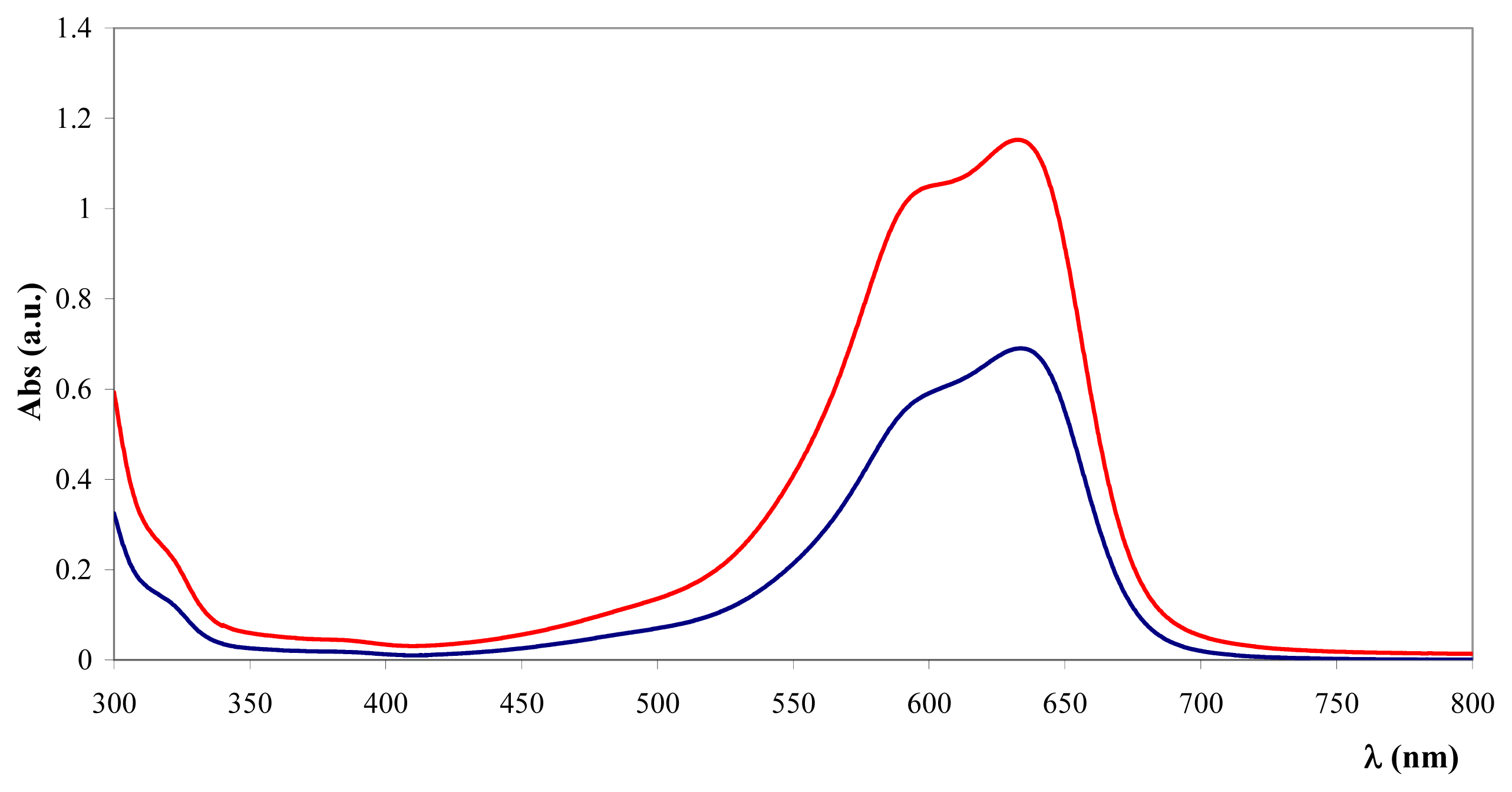

 ) and TB (
).
) and TB (
).
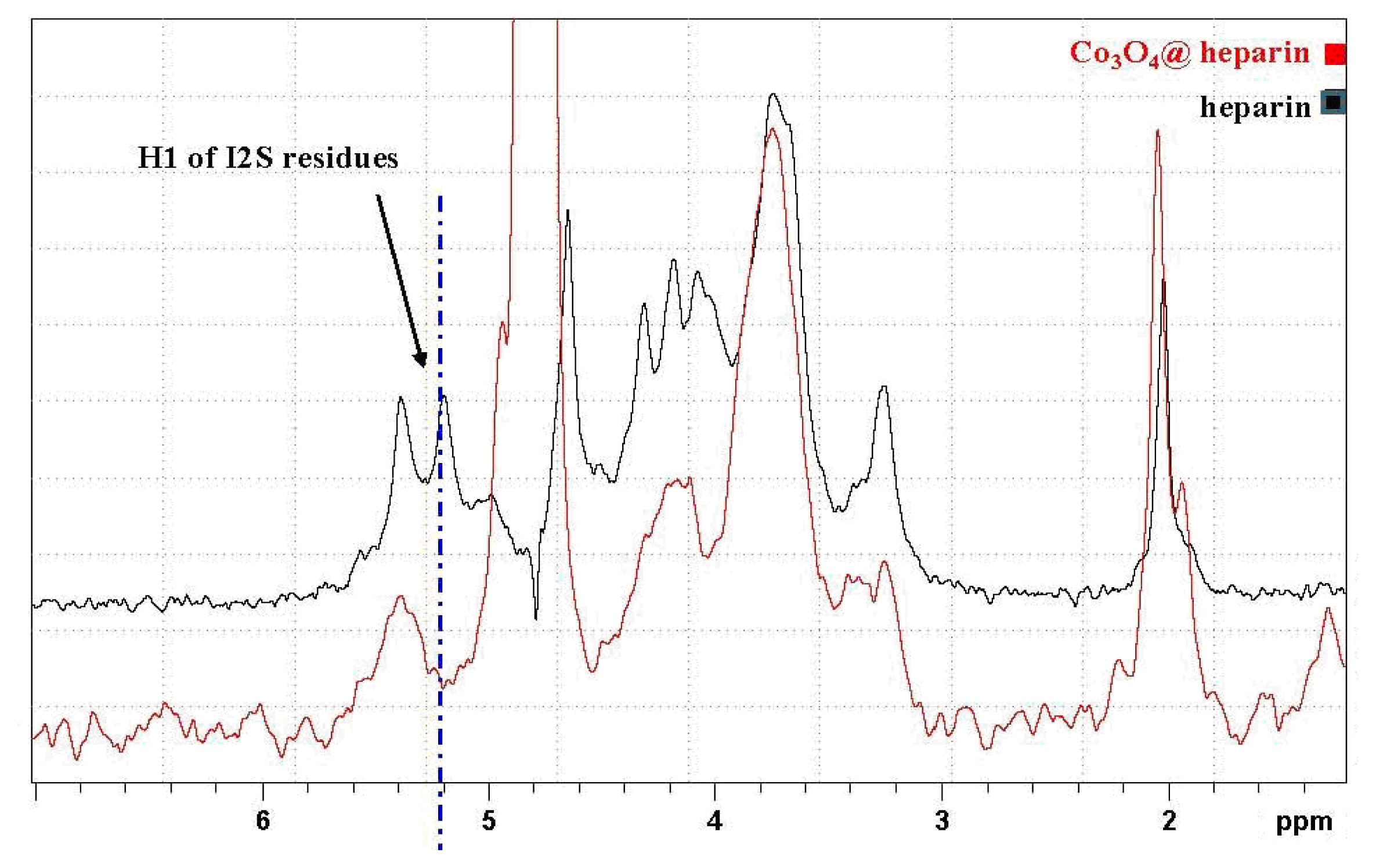
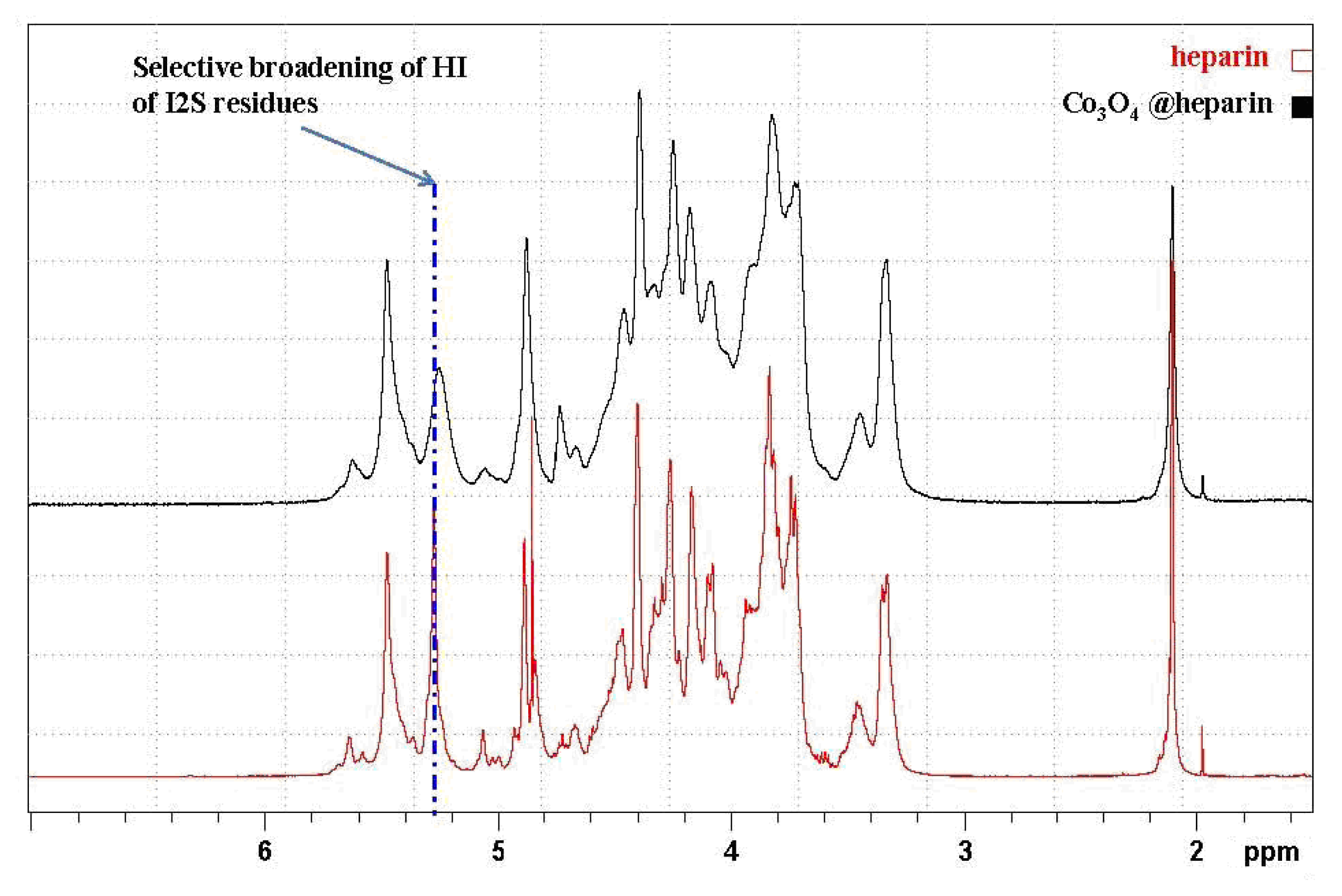
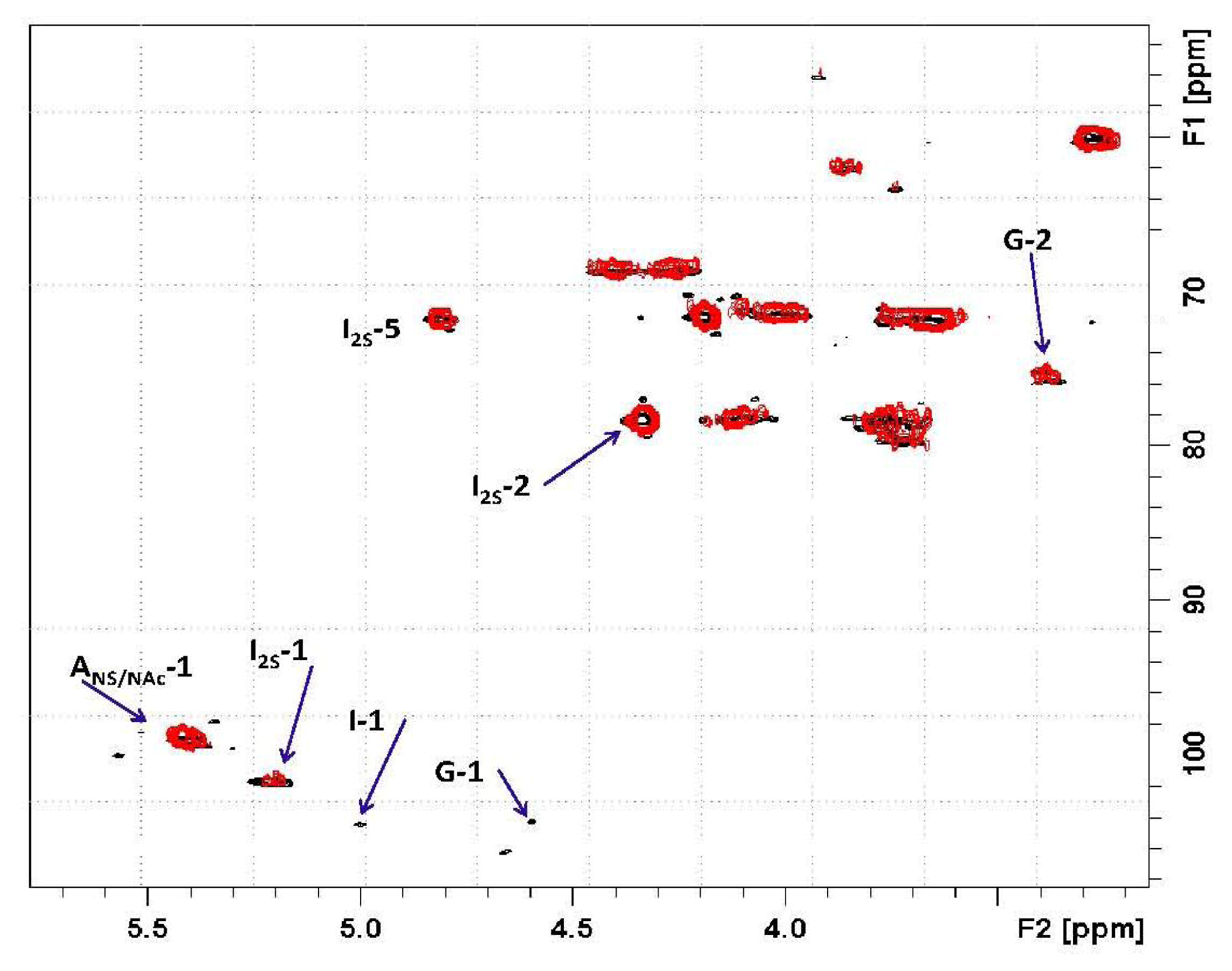 ) overlapped with the mother heparin (black); (Right) Same expansion with signal intensities increased up to the noise level.
) overlapped with the mother heparin (black); (Right) Same expansion with signal intensities increased up to the noise level.
) overlapped with the mother heparin (black); (Right) Same expansion with signal intensities increased up to the noise level.
) overlapped with the mother heparin (black); (Right) Same expansion with signal intensities increased up to the noise level.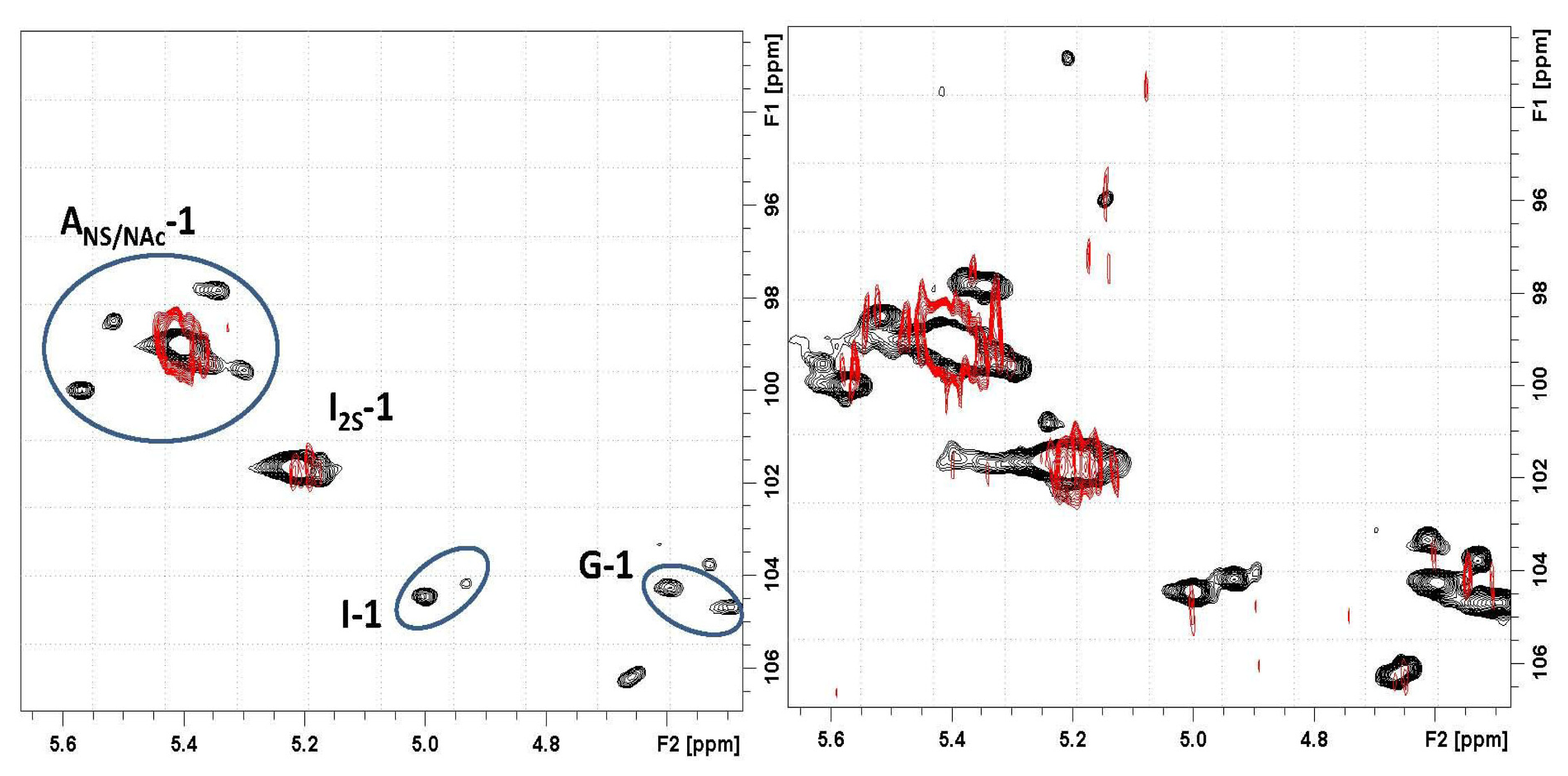

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NP | Starting NP (mg) | MO@heparin weight increase (mg) | TB assay heparin coating (mg) |
|---|---|---|---|
| Co3O4 | 100 | 11 | 10–19 |
| NiO | 100 | 19 | 10–22 |
| Fe3O4 | 100 | 5 | 10–15 |
| Starting Heparin (g/mL) | Starting NP (g/mL) | Heparin/NP w/w | TB assay heparin coating (mg) |
|---|---|---|---|
| 0.033 | 0.003 | 11 | 15 |
| 0.028 | 0.010 | 2.8 | 9 |
| 0.022 | 0.016 | 1.4 | 2 |
| MO@heparin | Z-A (nm) | PDI |
|---|---|---|
| Co3O4@heparin | 103 | 0.17 |
| Fe3O4@heparin | 92 | 0.17 |
| NiO@heparin | 93 | 0.23 |
| NP | ζ (mV) | MO@heparin | ζ (mV) |
|---|---|---|---|
| Co3O4 | −31 | Co3O4@heparin | −70 |
| Fe3O | −14 | Fe3O4@heparin | −61 |
| NiO | +19 | NiO@heparin | −47 |
| heparin:Fe3O4@heparin (w:w) | Z-A (nm) | PDI | ζ (mV) |
|---|---|---|---|
| 0:1 | 118 | 0.24 | −54 |
| 0.1:1 | 112 | 0.26 | −38 |
| 0,2:1 | 113 | 0.27 | −70 |
| 0.4:1 | 118 | 0.24 | −69 |
| 1:1 | 118 | 0.24 | −73 |
| Reaction Conditions | Starting NP (mg) | Weight increase (mg) | TB assay heparin coating (mg) | Z-A (nm) | PDI | ζ Pot (mV) |
|---|---|---|---|---|---|---|
| Water + Tween 20® | 100 | 5 | 9 | 93 | 0.25 | −46 |
| PBS | 100 | 6 | 9 | - | 0.38 | −47 |
| PBS + Tween 20® | 100 | 8 | 10 | - | 0.32 | −49 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vismara, E.; Valerio, A.; Coletti, A.; Torri, G.; Bertini, S.; Eisele, G.; Gornati, R.; Bernardini, G. Non-Covalent Synthesis of Metal Oxide Nanoparticle–Heparin Hybrid Systems: A New Approach to Bioactive Nanoparticles. Int. J. Mol. Sci. 2013, 14, 13463-13481. https://doi.org/10.3390/ijms140713463
Vismara E, Valerio A, Coletti A, Torri G, Bertini S, Eisele G, Gornati R, Bernardini G. Non-Covalent Synthesis of Metal Oxide Nanoparticle–Heparin Hybrid Systems: A New Approach to Bioactive Nanoparticles. International Journal of Molecular Sciences. 2013; 14(7):13463-13481. https://doi.org/10.3390/ijms140713463
Chicago/Turabian StyleVismara, Elena, Antonio Valerio, Alessia Coletti, Giangiacomo Torri, Sabrina Bertini, Giorgio Eisele, Rosalba Gornati, and Giovanni Bernardini. 2013. "Non-Covalent Synthesis of Metal Oxide Nanoparticle–Heparin Hybrid Systems: A New Approach to Bioactive Nanoparticles" International Journal of Molecular Sciences 14, no. 7: 13463-13481. https://doi.org/10.3390/ijms140713463
APA StyleVismara, E., Valerio, A., Coletti, A., Torri, G., Bertini, S., Eisele, G., Gornati, R., & Bernardini, G. (2013). Non-Covalent Synthesis of Metal Oxide Nanoparticle–Heparin Hybrid Systems: A New Approach to Bioactive Nanoparticles. International Journal of Molecular Sciences, 14(7), 13463-13481. https://doi.org/10.3390/ijms140713463