
Meta-Xylene-Based Diamines with Protected Benzyl Sites: Potential NCN Pincer Ligands with Tunable Steric Profiles


Abstract
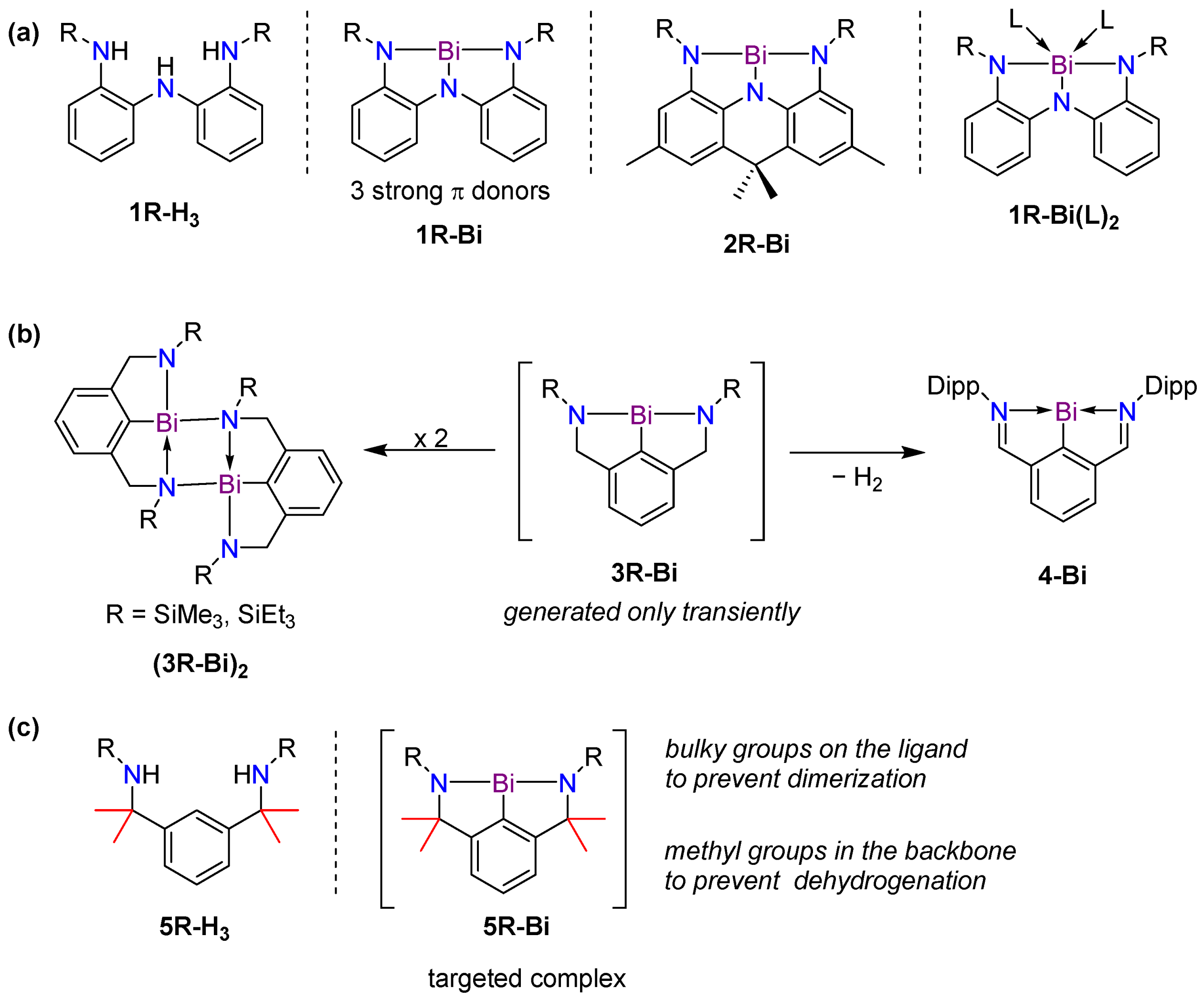
1. Introduction
2. Results and Discussion
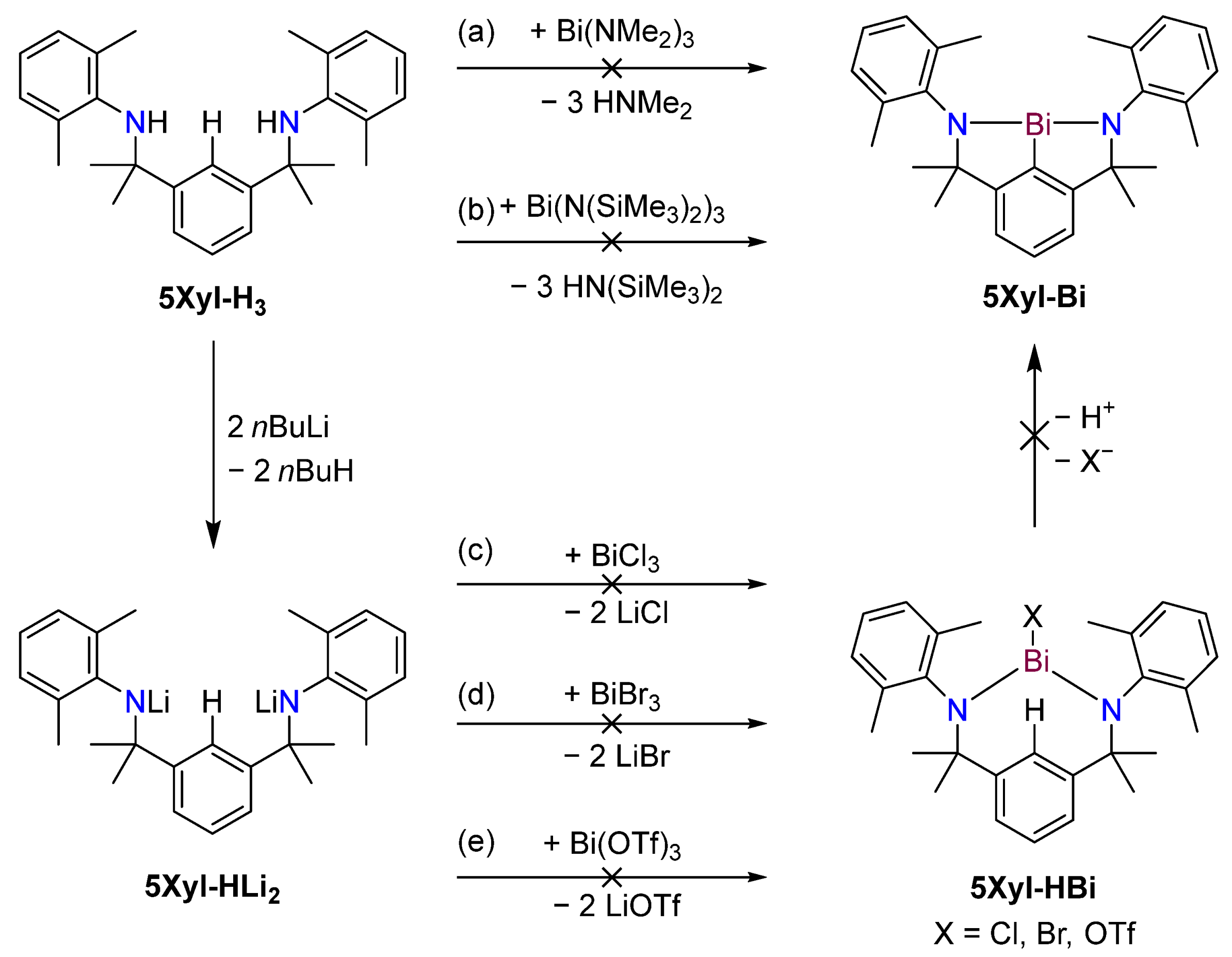
2.1. Synthesis
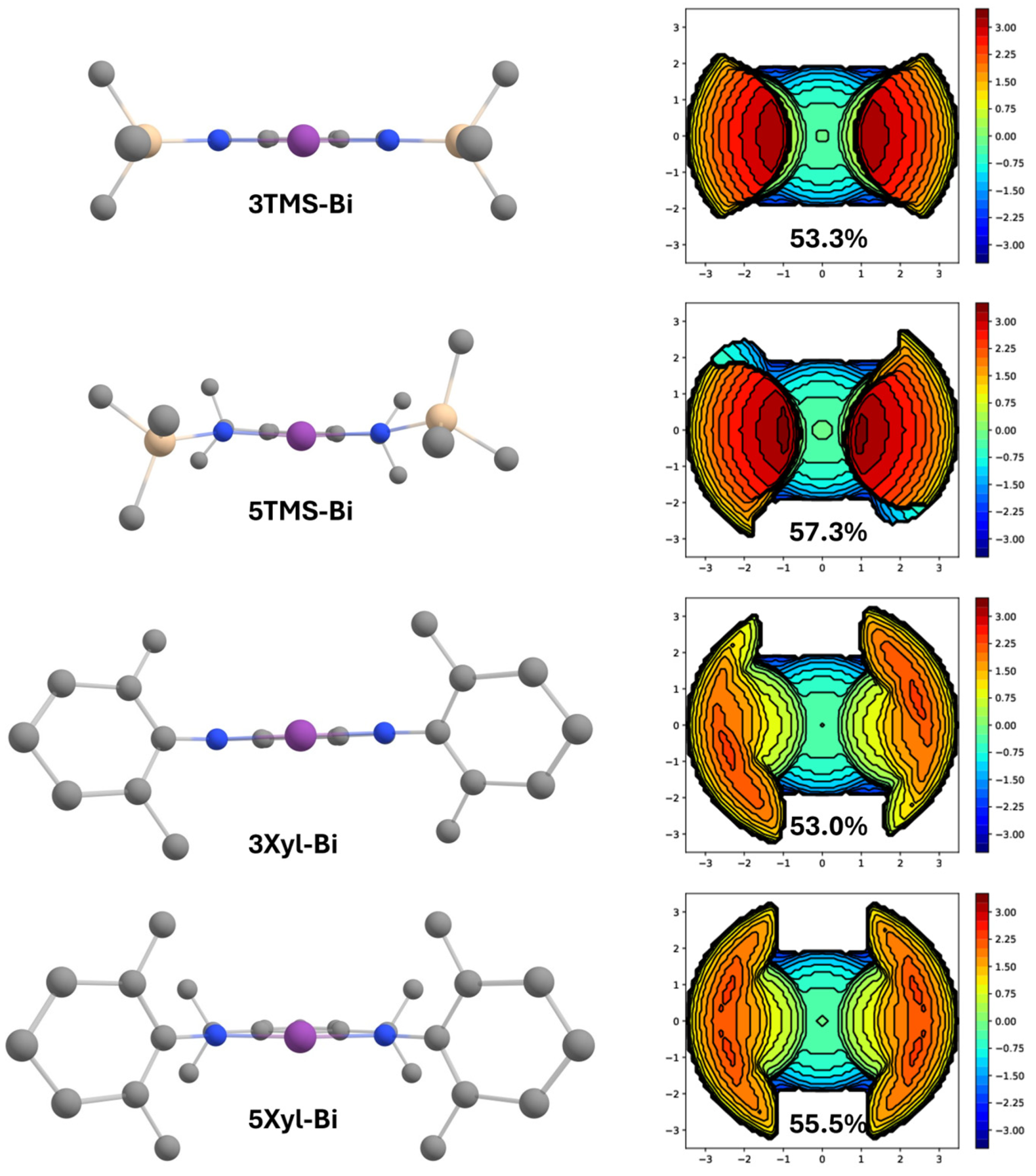
2.2. Steric Impact of the Methyl-Substituted Ligand Arms
2.3. Attempted Synthesis of 5R-Bi
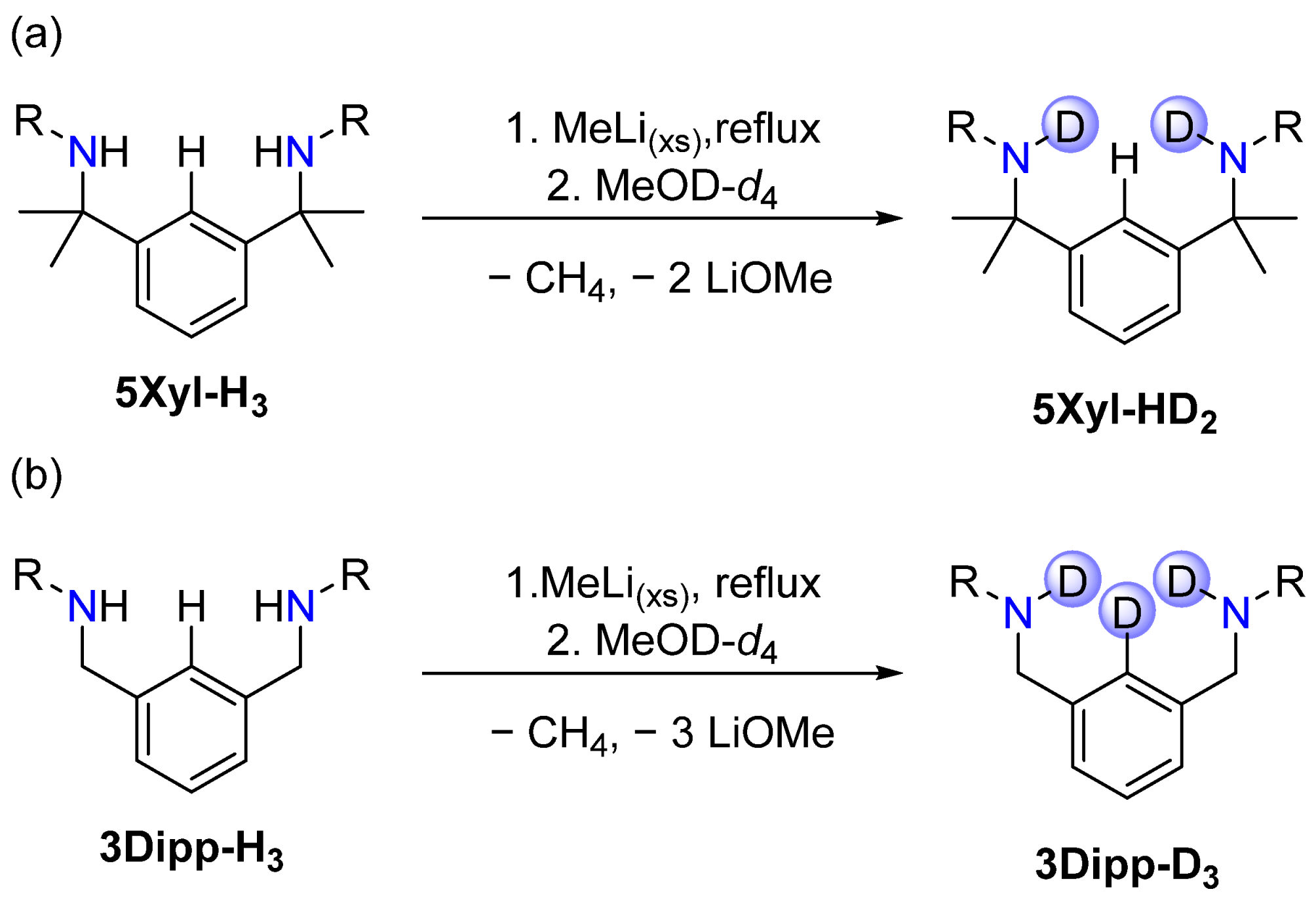
2.4. Impact of the Methyl Side Arms on Deprotonation
3. Materials and Methods
3.1. General Synthetic Procedures
3.2. X-Ray Crystallography
3.3. Theoretical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Peris, E.; Crabtree, R.H. Key factors in pincer ligand design. Chem. Soc. Rev. 2018, 47, 1959–1968. [Google Scholar] [CrossRef] [PubMed]
- Koten, G.v. Tuning the reactivity of metals held in a rigid ligand environment. Pure Appl. Chem. 1989, 61, 1681–1694. [Google Scholar] [CrossRef]
- Chase, P.A.; Gossage, R.A.; van Koten, G. Modern Organometallic Multidentate Ligand Design Strategies: The Birth of the Privileged “Pincer” Ligand Platform. In The Privileged Pincer-Metal Platform: Coordination Chemistry & Applications; van Koten, G., Gossage, R.A., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–15. [Google Scholar]
- Jambor, R.; Dostál, L. The Chemistry of Pincer Complexes of 13–15 Main Group Elements. In Organometallic Pincer Chemistry; van Koten, G., Milstein, D., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 175–202. [Google Scholar]
- Gunanathan, C.; Milstein, D. Bond Activation and Catalysis by Ruthenium Pincer Complexes. Chem. Rev. 2014, 114, 12024–12087. [Google Scholar] [CrossRef]
- Valdés, H.; García-Eleno, M.A.; Canseco-Gonzalez, D.; Morales-Morales, D. Recent Advances in Catalysis with Transition-Metal Pincer Compounds. ChemCatChem 2018, 10, 3136–3172. [Google Scholar] [CrossRef]
- Werkmeister, S.; Neumann, J.; Junge, K.; Beller, M. Pincer-Type Complexes for Catalytic (De)Hydrogenation and Transfer (De)Hydrogenation Reactions: Recent Progress. Chem. Eur. J. 2015, 21, 12226–12250. [Google Scholar] [CrossRef]
- Choi, J.; MacArthur, A.H.R.; Brookhart, M.; Goldman, A.S. Dehydrogenation and Related Reactions Catalyzed by Iridium Pincer Complexes. Chem. Rev. 2011, 111, 1761–1779. [Google Scholar] [CrossRef]
- O’Reilly, M.E.; Veige, A.S. Trianionic pincer and pincer-type metal complexes and catalysts. Chem. Soc. Rev. 2014, 43, 6325–6369. [Google Scholar] [CrossRef] [PubMed]
- Hannah, T.J.; Chitnis, S.S. Ligand-enforced geometric constraints and associated reactivity in p-block compounds. Chem. Soc. Rev. 2024, 53, 764–792. [Google Scholar] [CrossRef]
- Šimon, P.; de Proft, F.; Jambor, R.; Růžička, A.; Dostál, L. Monomeric Organoantimony(I) and Organobismuth(I) Compounds Stabilized by an NCN Chelating Ligand: Syntheses and Structures. Angew. Chem. Int. Ed. 2010, 49, 5468–5471. [Google Scholar] [CrossRef]
- Dostál, L. Quest for stable or masked pnictinidenes: Emerging and exciting class of group 15 compounds. Coord. Chem. Rev. 2017, 353, 142–158. [Google Scholar] [CrossRef]
- King, A.J.; Abbenseth, J.; Goicoechea, J.M. Reactivity of a Strictly T-Shaped Phosphine Ligated by an Acridane Derived NNN Pincer Ligand. Chemistry 2023, 29, e202300818. [Google Scholar] [CrossRef] [PubMed]
- Abbenseth, J.; Goicoechea, J.M. Recent developments in the chemistry of non-trigonal pnictogen pincer compounds: From bonding to catalysis. Chem. Sci. 2020, 11, 9728–9740. [Google Scholar] [CrossRef]
- Maiti, A.; Yadav, R.; Greb, L. Chapter Eight—Structural constraint effects on p-block elements: Recent advances. In Advances in Inorganic Chemistry; Meyer, K., van Eldik, R., Eds.; Academic Press: Cambridge, MA, USA, 2023; Volume 82, pp. 261–299. [Google Scholar]
- Kindervater, M.B.; Marczenko, K.M.; Werner-Zwanziger, U.; Chitnis, S.S. A Redox-Confused Bismuth(I/III) Triamide with a T-Shaped Planar Ground State. Angew Chem Int. Ed. 2019, 58, 7850–7855. [Google Scholar] [CrossRef] [PubMed]
- Coburger, P.; Buzanich, A.G.; Emmerling, F.; Abbenseth, J. Combining geometric constraint and redox non-innocence within an ambiphilic PBiP pincer ligand. Chem. Sci. 2024, 15, 6036–6043. [Google Scholar] [CrossRef]
- Chval, Z. Ir(I)–Bi(III) Donor–Acceptor Adducts Stabilized by Dispersion Interactions between the Metal Pincer Ligands and Their Possible Self-Assembly Forming Molecular 1D Semiconductors. Inorg. Chem. 2024, 63, 12417–12425. [Google Scholar] [CrossRef]
- Hannah, T.J.; McCarvell, W.M.; Kirsch, T.; Bedard, J.; Hynes, T.; Mayho, J.; Bamford, K.L.; Vos, C.W.; Kozak, C.M.; George, T.; et al. Planar bismuth triamides: A tunable platform for main group Lewis acidity and polymerization catalysis. Chem. Sci. 2023, 14, 4549–4563. [Google Scholar] [CrossRef]
- Hynes, T.; Masuda, J.D.; Chitnis, S.S. Mesomeric Tuning at Planar Bi centres: Unexpected Dimerization and Benzyl C−H Activation in [CN2]Bi Complexes. ChemPlusChem 2022, 87, e202200244. [Google Scholar] [CrossRef] [PubMed]
- Goodman, J.T.; Schrock, R.R. Kinetic Investigation of the Polymerization of 1-Hexene by the {[t-BuNON] ZrMe}[B (C6F5) 4] Initiator. Organometallics 2001, 20, 5205–5211. [Google Scholar] [CrossRef]
- Mehrkhodavandi, P.; Schrock, R.R. Cationic hafnium alkyl complexes that are stable toward β-hydride elimination below 10 C and active as initiators for the living polymerization of 1-hexene. J. Am. Chem. Soc. 2001, 123, 10746–10747. [Google Scholar] [CrossRef]
- Harney, M.B.; Keaton, R.J.; Sita, L.R. End-group-confined chain walking within a group 4 living polyolefin and well-defined cationic zirconium alkyl complexes for modeling this behavior. J. Am. Chem. Soc. 2004, 126, 4536–4537. [Google Scholar] [CrossRef]
- Shultz, L.H.; Brookhart, M. Measurement of the barrier to β-hydride elimination in a β-agostic palladium−ethyl complex: A model for the energetics of chain-walking in (α-diimine) PdR+ olefin polymerization catalysts. Organometallics 2001, 20, 3975–3982. [Google Scholar] [CrossRef]
- Tellmann, K.P.; Humphries, M.J.; Rzepa, H.S.; Gibson, V.C. Experimental and computational study of β-H transfer between cobalt (I) alkyl complexes and 1-alkenes. Organometallics 2004, 23, 5503–5513. [Google Scholar] [CrossRef]
- Leung, D.H.; Ziller, J.W.; Guan, Z. Axial donating ligands: A new strategy for late transition metal olefin polymerization catalysis. J. Am. Chem. Soc. 2008, 130, 7538–7539. [Google Scholar] [CrossRef] [PubMed]
- Noda, S.; Nakamura, A.; Kochi, T.; Chung, L.W.; Morokuma, K.; Nozaki, K. Mechanistic studies on the formation of linear polyethylene chain catalyzed by palladium phosphine–sulfonate complexes: Experiment and theoretical studies. J. Am. Chem. Soc. 2009, 131, 14088–14100. [Google Scholar] [CrossRef]
- Dahlenburg, L.; Treffert, H.; Heinemann, F.W. 1,3-Bis(α-aminoisopropyl)benzene, meta-C6H4(CMe2NH2)2: An N,N-bridging and N,C,N-cyclometalating ligand. Inorg. Chim. Acta 2008, 361, 1311–1318. [Google Scholar] [CrossRef]
- Daniele, S.; Hitchcock, P.B.; Lappert, M.F.; Nile, T.A.; Zdanski, C.M. Synthesis and structures of dinuclear low-coordinate lithium and zirconium(iv) complexes derived from the diamido ligands 1,3-(CH2NC6H3R12)2C6H4 (R1 = Me or Pri). J. Chem. Soc. Dalton Trans. 2002, 21, 3980–3984. [Google Scholar] [CrossRef]
- Mantina, M.; Chamberlin, A.C.; Valero, R.; Cramer, C.J.; Truhlar, D.G. Consistent van der Waals Radii for the Whole Main Group. J. Phys. Chem. A 2009, 113, 5806–5812. [Google Scholar] [CrossRef]
- Pethes, I.; Bakó, I.; Pusztai, L. Chloride ions as integral parts of hydrogen bonded networks in aqueous salt solutions: The appearance of solvent separated anion pairs. Phys. Chem. Chem. Phys. 2020, 22, 11038–11044. [Google Scholar] [CrossRef]
- Pandey, D.K.; Khaskin, E.; Khusnutdinova, J.R. PNP-Pincer Ligands Armed With Methyls: New Tools To Control Sterics and Non-Innocence. ChemCatChem 2023, 15, e202300644. [Google Scholar] [CrossRef]
- Deolka, S.; Fayzullin, R.R.; Khaskin, E. Bulky PNP Ligands Blocking Metal-Ligand Cooperation Allow for Isolation of Ru(0), and Lead to Catalytically Active Ru Complexes in Acceptorless Alcohol Dehydrogenation. Chem. Eur. J. 2022, 28, e202103778. [Google Scholar] [CrossRef]
- Rivada-Wheelaghan, O.; Chakraborty, S.; Shimon, L.J.W.; Ben-David, Y.; Milstein, D. Z-Selective (Cross-)Dimerization of Terminal Alkynes Catalyzed by an Iron Complex. Angew. Chem. Int. Ed. 2016, 55, 6942–6945. [Google Scholar] [CrossRef]
- Pandey, D.K.; Khaskin, E.; Pal, S.; Fayzullin, R.R.; Khusnutdinova, J.R. Efficient Fe-Catalyzed Terminal Alkyne Semihydrogenation by H2: Selectivity Control via a Bulky PNP Pincer Ligand. ACS Catal. 2023, 13, 375–381. [Google Scholar] [CrossRef]
- Gorgas, N.; Brünig, J.; Stöger, B.; Vanicek, S.; Tilset, M.; Veiros, L.F.; Kirchner, K. Efficient Z-Selective Semihydrogenation of Internal Alkynes Catalyzed by Cationic Iron(II) Hydride Complexes. J. Am. Chem. Soc. 2019, 141, 17452–17458. [Google Scholar] [CrossRef]
- Falivene, L.; Cao, Z.; Petta, A.; Serra, L.; Poater, A.; Oliva, R.; Scarano, V.; Cavallo, L. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 2019, 11, 872–879. [Google Scholar] [CrossRef]
- Poater, A.; Ragone, F.; Giudice, S.; Costabile, C.; Dorta, R.; Nolan, S.P.; Cavallo, L. Thermodynamics of N-Heterocyclic Carbene Dimerization: The Balance of Sterics and Electronics. Organometallics 2008, 27, 2679–2681. [Google Scholar] [CrossRef]
- Poater, A.; Ragone, F.; Mariz, R.; Dorta, R.; Cavallo, L. Comparing the Enantioselective Power of Steric and Electrostatic Effects in Transition-Metal-Catalyzed Asymmetric Synthesis. Chem. Eur. J. 2010, 16, 14348–14353. [Google Scholar] [CrossRef]
- Hannah, T.J.; Kirsch, T.Z.; Chitnis, S.S. Why Are Some Pnictogen(III) Pincer Complexes Planar and Others Pyramidal? Chem. Eur. J. 2024, 30, e202402851. [Google Scholar] [CrossRef]
- Koller, J.; Sarkar, S.; Abboud, K.A.; Veige, A.S. Synthesis and Characterization of (2,6-iPrNCN)HfCl2− and (3,5-MeNCN)2Hf2− (where NCN = 2,6-bis[phenylazanidyl]methylphenyl): New Trianionic Pincer Ligands. Organometallics 2007, 26, 5438–5441. [Google Scholar] [CrossRef]
- Szakács, Z.; Kraszni, M.; Noszál, B. Determination of microscopic acid–base parameters from NMR–pH titrations. Anal. Bioanal. Chem. 2004, 378, 1428–1448. [Google Scholar] [CrossRef]
- Abraham, M.H.; Abraham, R.J.; Byrne, J.; Griffiths, L. NMR Method for the Determination of Solute Hydrogen Bond Acidity. J. Org. Chem. 2006, 71, 3389–3394. [Google Scholar] [CrossRef]
- Benoit, R.L.; Fréchette, M.; Lefebvre, D. 2,6-Di-tert-butylpyridine: An unusually weak base in dimethylsulfoxide. Can. J. Chem. 1988, 66, 1159–1162. [Google Scholar] [CrossRef]
- Su, C.; Hopson, R.; Williard, P.G. Mixed Aggregates of an Alkyl Lithium Reagent and a Chiral Lithium Amide Derived from N-Ethyl-O-triisopropylsilyl Valinol. J. Am. Chem. Soc. 2013, 135, 14367–14379. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Honma, T.; Sugai, S. Synthesis and Herbicidal Activities of 1,2-Benzisoxazole-3-acetamide Derivatives. Agric. Biol. Chem. 1985, 49, 3563–3567. [Google Scholar] [CrossRef]
- Bruker, A.S.; Bruker, A. Inc., Madison, Wisconsin, USA, 2004; (b) GM Sheldrick. Acta Cryst. A 1990, 46, 467–473. [Google Scholar]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Ho, C.Y.; Chan, C.W.; He, L. Catalytic Asymmetric Hydroalkenylation of Vinylarenes: Electronic Effects of Substrates and Chiral N-Heterocyclic Carbene Ligands. Angew. Chem. Int. Ed. 2015, 54, 4512–4516. [Google Scholar] [CrossRef]
- McGarrity, J.F.; Ogle, C.A. High-Field Proton Nmr Study of the Aggregation and Complexation of N-Butyllithium in Tetrahydrofuran. J. Am. Chem. Soc. 1985, 107, 1805–1810. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Lengths and Angles | [Å] and [°] |
|---|---|
| C1-C2 | 1.400 (1) |
| C2-C3 | 1.397 (1) |
| C3-C10 | 1.525 (1) |
| C10-C12 | 1.525 (1) |
| C10-C11 | 1.531 (1) |
| C10-N2 | 1.562 (1) |
| N2-C21 | 1.480 (1) |
| C21-C26 | 1.406 (1) |
| C26-C28 | 1.508 (1) |
| C21-C22 | 1.408 (1) |
| C22-C27 | 1.509 (1) |
| C1-C7 | 1.5222 (1) |
| C7-N1 | 1.556 (1) |
| C3-C10-N2 | 108.83 (6) |
| C1-C7-N1 | 111.13 (6) |
| C10-N2-C21 | 118.51 (6) |
| C7-N1-C13 | 119.92 (6) |
| C11-C10-C12 | 108.32 (7) |
| Entry | Reagent | Solvent | Condition |
|---|---|---|---|
| 1 | nBuLi | THF | r.t. |
| 2 | nBuLi | THF | 60 °C |
| 3 | nBuLi | THF | 60 °C, +TMEDA |
| 4 | tBuLi | THF | −30 °C to r.t. |
| 5 | nBuLi | toluene | 30 min at 110 °C |
| 6 | MeLi | toluene | 1 h at 120 °C |
| 7 | secBuLi | THF | −78 °C to r.t. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirsch, T.Z.; Hynes, T.; Masuda, J.D.; Chitnis, S.S. Meta-Xylene-Based Diamines with Protected Benzyl Sites: Potential NCN Pincer Ligands with Tunable Steric Profiles. Molecules 2025, 30, 1331. https://doi.org/10.3390/molecules30061331
Kirsch TZ, Hynes T, Masuda JD, Chitnis SS. Meta-Xylene-Based Diamines with Protected Benzyl Sites: Potential NCN Pincer Ligands with Tunable Steric Profiles. Molecules. 2025; 30(6):1331. https://doi.org/10.3390/molecules30061331
Chicago/Turabian StyleKirsch, Tamina Z., Toren Hynes, Jason D. Masuda, and Saurabh S. Chitnis. 2025. "Meta-Xylene-Based Diamines with Protected Benzyl Sites: Potential NCN Pincer Ligands with Tunable Steric Profiles" Molecules 30, no. 6: 1331. https://doi.org/10.3390/molecules30061331
APA StyleKirsch, T. Z., Hynes, T., Masuda, J. D., & Chitnis, S. S. (2025). Meta-Xylene-Based Diamines with Protected Benzyl Sites: Potential NCN Pincer Ligands with Tunable Steric Profiles. Molecules, 30(6), 1331. https://doi.org/10.3390/molecules30061331