
Facile Synthetic Access Towards Sulfur- and Selenium-Functionalized Boron-Based Multiresonance TADF Emitters


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
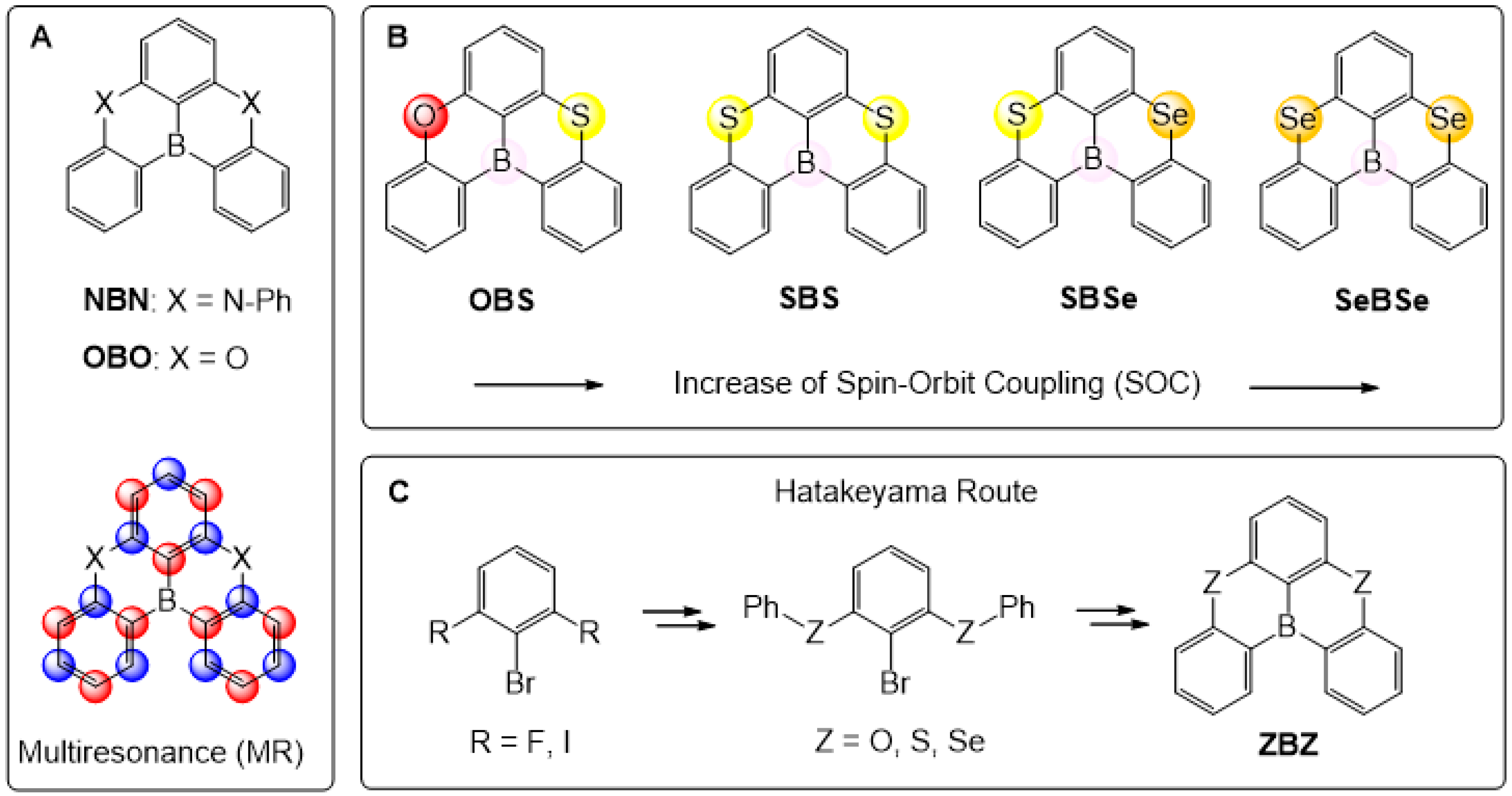
1. Introduction
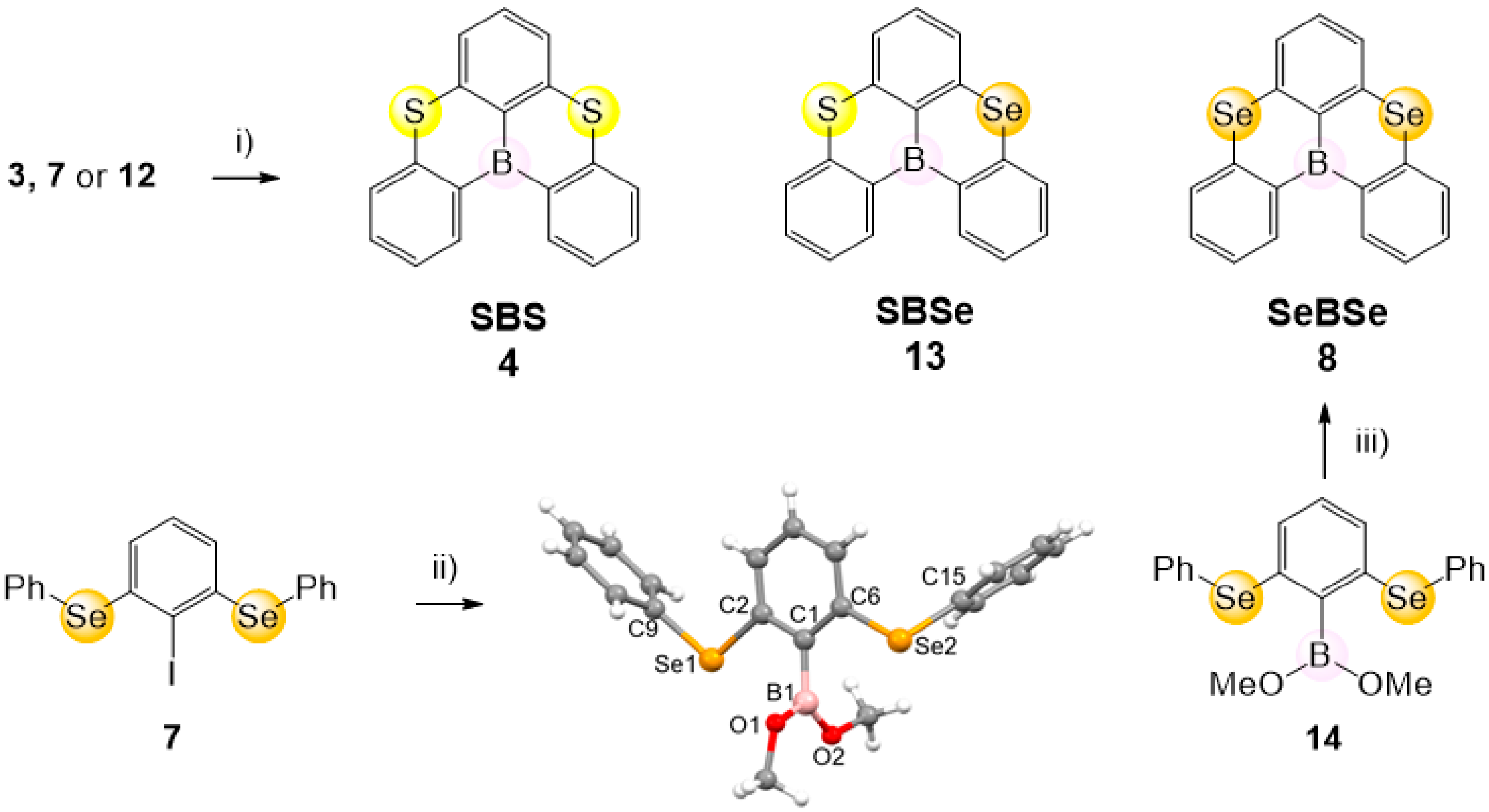
2. Results and Discussion
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tang, C.W.; VanSlyke, S.A. Organic electroluminescent diodes. Appl. Phys. Lett. 1987, 51, 913–915. [Google Scholar] [CrossRef]
- Zou, S.-J.; Shen, Y.; Xie, F.-M.; Chen, J.-D.; Li, Y.-Q.; Tang, J.-X. Recent advances in organic light-emitting diodes: Toward smart lighting and displays. Mater. Chem. Front. 2020, 4, 788–820. [Google Scholar] [CrossRef]
- Song, J.; Lee, H.; Jeong, E.G.; Choi, K.C.; Yoo, S. Organic light-emitting diodes: Pushing toward the limits and beyond. Adv. Mater. 2020, 32, 1907539. [Google Scholar] [CrossRef] [PubMed]
- Pode, R. Organic light emitting diode devices: An energy efficient solid state lighting for applications. Renew. Sustain. Energy Rev. 2020, 133, 110043. [Google Scholar] [CrossRef]
- Chen, H.-W.; Lee, J.-H.; Lin, B.-Y.; Chen, S.; Wu, S.-T. Liquid crystal display and organic light-emitting diode display: Present status and future perspectives. Light Sci. Appl. 2018, 7, 17168. [Google Scholar] [CrossRef]
- Geffroy, B.; Le Roy, P.; Prat, C. Organic light-emitting diode (OLED) technology: Materials, devices and display technologies. Polym. Int. 2006, 55, 572–582. [Google Scholar] [CrossRef]
- Yersin, H.; Monkowius, U. Thermally activated delayed fluorescence and beyond. Photophysics and material design strategies. Adv. Photonics Res. 2024, 2400111. [Google Scholar] [CrossRef]
- Shi, Y.-Z.; Wu, H.; Wang, K.; Yu, J.; Oua, X.-M.; Zhang, X.-H. Recent progress in thermally activated delayed fluorescence emitters for nondoped organic light-emitting diodes. Chem. Sci. 2022, 13, 3625–3651. [Google Scholar] [CrossRef]
- Teng, J.-M.; Wang, Y.-W.; Chen, C.-F. Recent progress of narrowband TADF emitters and their applications in OLEDs. J. Mater. Chem. C 2020, 8, 11340–11353. [Google Scholar] [CrossRef]
- Wang, T.; Cheng, X.; Yang, C. Thermally activated delayed fluorescence polymers and their application in organic light-emitting diodes. Prog. Polym. Sci. 2024, 158, 101892. [Google Scholar] [CrossRef]
- Wong, M.Y.; Zysman-Colman, E. Purely Organic thermally activated delayed fluorescence materials for organic light-emitting diodes. Adv. Mater. 2017, 29, 1605444. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Chen, Y.; Li, N.; Huang, Z.; Yang, C. Versatile boron-based thermally activated delayed fluorescence materials for organic light-emitting diodes. Aggregate 2022, 3, e182. [Google Scholar] [CrossRef]
- Kim, H.J.; Yasuda, T. Narrowband emissive thermally activated delayed fluorescence materials. Adv. Opt. Mater. 2022, 10, 2201714. [Google Scholar] [CrossRef]
- Du, M.; Zhou, J.; Luo, X.; Duan, L.; Zhang, D. A perspective on boron-based multiple resonance narrowband emitters and devices. Moore More 2024, 1, 5. [Google Scholar] [CrossRef]
- Naveen, K.R.; Konidena, R.K.; Keerthika, P. Neoteric advances in oxygen bridged triaryl boron-based delayed fluorescent materials for organic light emitting diodes. Chem. Rec. 2023, 23, e202300208. [Google Scholar] [CrossRef]
- Ahn, D.H.; Kim, S.W.; Lee, H.; Ko, I.J.; Karthik, D.; Lee, J.Y.; Kwon, J.H. Highly efficient blue thermally activated delayed fluorescence emitters based on symmetrical and rigid oxygen-bridged boron acceptors. Nat. Photonics 2019, 13, 540–546. [Google Scholar] [CrossRef]
- Hatakeyama, T.; Shiren, K.; Nakajima, K.; Nomura, S.; Nakatsuka, S.; Kinoshita, K.; Ni, J.; Ono, Y.; Ikuta, T. Ultrapure blue thermally activated delayed fluorescence molecules: Efficient HOMO–LUMO separation by the multiple resonance effect. Adv. Mater. 2016, 28, 2777–2781. [Google Scholar] [CrossRef]
- Hirai, H.; Nakajima, K.; Nakatsuka, S.; Shiren, K.; Ni, J.; Nomura, S.; Ikuta, T.; Hatakeyama, T. One-step borylation of 1,3-diaryloxybenzenes towards efficient materials for organic light-emitting diodes. Angew. Chem. Int. Ed. 2015, 54, 13581–13585. [Google Scholar] [CrossRef]
- Mamada, M.; Hayakawa, M.; Ochi, J.; Hatakeyama, T. Organoboron-based multiple-resonance emitters: Synthesis, structure–property correlations, and prospects. Chem. Soc. Rev 2024, 53, 1624–1692. [Google Scholar] [CrossRef]
- Hu, Y.X.; Miao, J.; Hua, T.; Huang, Z.; Qi, Y.; Zou, Y.; Qiu, Y.; Xia, H.; Liu, H.; Cao, X.; et al. Efficient selenium-integrated TADF OLEDs with reduced roll-off. Nat. Photonics 2022, 16, 803–810. [Google Scholar] [CrossRef]
- Nagata, M.; Min, H.; Watanabe, E.; Fukumoto, H.; Mizuhata, Y.; Tokitoh, N.; Agou, T.; Yasuda, T. Fused-nonacyclic multi-resonance delayed fluorescence emitter based on ladder-thiaborin exhibiting narrowband sky-blue emission with accelerated reverse intersystem crossing. Angew. Chem. Int. Ed. 2021, 60, 20280–20285. [Google Scholar] [CrossRef] [PubMed]
- Park, I.S.; Min, H.; Yasuda, T. Ultrafast triplet–singlet exciton interconversion in narrowband blue organoboron emitters doped with heavy chalcogens. Angew. Chem. Int. Ed. 2022, 61, e202205684. [Google Scholar] [CrossRef] [PubMed]
- Park, I.S.; Yang, M.; Shibata, H.; Amanokura, N.; Yasuda, T. Achieving ultimate narrowband and ultrapure blue organic light-emitting diodes based on polycyclo-heteraborin multi-resonance delayed-fluorescence emitters. Adv. Mater. 2022, 34, 2107951. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhao, L.; Wang, X.; Yang, Q.; Li, W.; Tian, H.; Shao, S.; Wang, L.; Jing, X.; Wang, F. Novel boron- and sulfur-doped polycyclic aromatic hydrocarbon as multiple resonance emitter for ultrapure blue thermally activated delayed fluorescence polymers. Sci. China Chem. 2021, 64, 547–551. [Google Scholar] [CrossRef]
- Keshri, S.K.; Liu, G.; Yasuda, T. Ultrafast spin- flip exciton conversion and narrowband sky-blue luminescence in a fused polycyclic selenaborin emitter. Front. Chem. 2024, 12, 1375552. [Google Scholar] [CrossRef]
- Pratik, S.M.; Coropceanu, V.; Brédas, J.-L. Purely organic emitters for multiresonant thermally activated delay fluorescence: Design of highly efficient sulfur and selenium derivatives. ACS Mater. Lett. 2022, 4, 440–447. [Google Scholar] [CrossRef]
- Hagai, M.; Inai, N.; Yasuda, T.; Fujimoto, K.J.; Yanai, T. Extended theoretical modeling of reverse intersystem crossing for thermally activated delayed fluorescence materials. Sci. Adv. 2024, 10, eadk3219. [Google Scholar] [CrossRef]
- Chang, Y.; Wu, Y.; Wang, X.; Li, W.; Yang, Q.; Wang, S.; Shao, S.; Wang, L. Boron sulfur-doped polycyclic aromatic hydrocarbon emitters with multiple-resonance-dominated lowest excited states for efficient narrowband deep-blue emission. Chem. Eng. J. 2023, 451, 138545. [Google Scholar] [CrossRef]
- Gao, H.; Li, Z.; Pang, Z.; Qin, Y.; Liu, G.; Gao, T.; Dong, X.; Shen, S.; Xie, X.; Wang, P.; et al. Rational molecular design dtrategy for high-efficiency ultrapure blue TADF emitters: Symmetrical and rigid sulfur-bridged boron based acceptors. ACS Appl. Mater. Interfaces 2023, 15, 5529–5537. [Google Scholar] [CrossRef]
- Chang, Y.; Wu, Y.; Zhang, K.; Wang, S.; Wang, X.; Shao, S.; Wang, L. 1,8-diphenyl-carbazole-based boron, sulfur-containing multi-resonance emitters with suppressed aggregation emission for narrowband OLEDs. Dyes Pigments 2023, 220, 111678. [Google Scholar] [CrossRef]
- Ye, K.; Li, G.; Li, F.; Shi, C.; Jiang, Z.; Zhang, F.; Li, Q.; Su, J.; Song, D.; Yuan, A. B-embedded disulfide-bridged p-conjugated compounds: Structures and optical tuning. Phys. Chem. Chem. Phys. 2024, 26, 2395–2401. [Google Scholar] [CrossRef] [PubMed]
- Krief, A. Synthesis of selenium and tellurium ylides and carbanions: Applications to organic synthesis. In The Chemistry of Organic Selenium and Tellurium Compound, 1st ed.; Patai, S., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 1987; Volume 2, pp. 677–764. [Google Scholar]
- Gilman, H.; Webb, F.J. The metalation of some sulfur-containing organic compounds. J. Am. Chem. Soc. 1949, 71, 4062–4066. [Google Scholar] [CrossRef]
- Dumont, W.; Bayet, P.; Krief, A. Cleavage of selenium compounds by butyllithium. A new, regiospecific, allyl alcohol synthon. Angew. Chem. Int. Ed. 1974, 13, 804–806. [Google Scholar] [CrossRef]
- Clarembeau, M.; Krief, A. Novel synthesis of benzyllithiums from benzylselenides. Tetrahedron Lett. 1985, 26, 1093–1096. [Google Scholar] [CrossRef]
- Clarembeau, M.; Krief, A. A novel method for the geminal dialkylation of the carbonyl group of aromatic aldehydes and ketones. Tetrahedron Lett. 1986, 27, 1719–1722. [Google Scholar] [CrossRef]
- Clarembeau, M.; Krief, A. Metallation of benzyl selenides and of α-aryl selenoacetals. Scope and limitations. Tetrahedron Lett. 1986, 27, 1723–1726. [Google Scholar] [CrossRef]
- Lide, D.R. Handbooks of Chemistry and Physics, 84th ed.; CRC Press: Boca Raton, FL, USA, 2003; pp. 9-65–9-71. [Google Scholar]
- Leroux, F.; Schlosser, M. The preparation of organolithium reagents and intermediates. In The Chemistry of Organolithium Compounds, 1st ed.; Rappoport, Z., Marek, I., Eds.; John Wiley & Sons, Ltd.: Hobooken, NJ, USA, 2004; pp. 435–493. [Google Scholar]
- Zender, E.; Karger, S.; Neubaur, R.; Virovets, A.; Lerner, H.-W.; Wagner, M. Green-Emitting extended B3,N2-doped polycyclic aromatic hydrocarbon with multiple resonance structure. Org. Lett. 2024, 26, 939–944. [Google Scholar] [CrossRef]
- Knöller, J.A.; Sönmez, B.; Matulaitis, T.; Gupta, A.K.; Zysman-Colman, E.; Laschat, S. A novel B,O,N-doped mesogen with narrowband MR-TADF emission. Chem. Commun. 2024, 60, 4459–4462. [Google Scholar] [CrossRef]
- Böser, R.; Haufe, L.C.; Freytag, M.; Jones, P.G.; Hörner, G.; Frank, R. Completing the series of boron-nucleophilic cyanoborates: Boryl anions of type NHC-B(CN)2−. Chem. Sci. 2017, 8, 6274–6280. [Google Scholar] [CrossRef]
- Böser, R.; Denker, L.; Frank, R. N-Heterocyclic carbene adducts of alkynyl functionalized 1,3,2-dithioborolanes. Molecules 2019, 24, 1690. [Google Scholar] [CrossRef]
- Böser, R.; Denker, L.; Frank, R. Benzyl borane NHC adducts: Beyond B−C bond scission. Chem. A Eur. J. 2019, 25, 10575–10579. [Google Scholar] [CrossRef] [PubMed]
- Dolati, H.; Haufe, L.C.; Denker, L.; Lorbach, A.; Grotjahn, R.; Hörner, G.; Frank, R. Two π-electrons make the difference—From BODIPY to BODIIM switchable fluorescent dyes. Chem. A Eur. J. 2020, 26, 1422–1428. [Google Scholar] [CrossRef] [PubMed]
- Dolati, H.; Denker, L.; Trzaskowski, B.; Frank, R. Superseding β-diketiminato ligands: An amido imidazoline-2-imine ligand stabilizes the exhaustive series of B = X boranes (X = O, S, Se, Te). Angew. Chem. Int. Ed. 2021, 60, 4633–4639. [Google Scholar] [CrossRef] [PubMed]
- Güven, Z.; Denker, L.; Wullschläger, D.; Martínez, J.P.; Trzaskowski, B.; Frank, R. Reductive Al−B σ-bond formation in alumaboranes: Facile scission of polar multiple bonds. Angew. Chem. Int. Ed. 2022, 61, e202209502. [Google Scholar] [CrossRef] [PubMed]
- Denker, L.; Wullschläger, D.; Martínez, J.P.; Świerczewski, S.; Trzaskowski, B.; Tamm, M.; Frank, R. Cobalt(I)-catalyzed transformation of Si–H bonds: H/D exchange in hydrosilanes and hydrosilylation of olefins. ACS Catal. 2023, 13, 2586–2600. [Google Scholar] [CrossRef]
- Reshi, N.U.D.; Bockfeld, D.; Baabe, D.; Denker, L.; Martínez, J.P.; Trzaskowski, B.; Frank, R.; Tamm, M. Iron(I) and iron(II) amido-imidazolin-2-imine complexes as catalysts for H/D exchange in hydrosilanes. ACS Catal. 2024, 14, 1759–1772. [Google Scholar] [CrossRef]
- Denker, L.; Dolati, H.; Barthen, M.; Frank, R. Amino imidazolin-2-imine ligands in magnesium complexes: Approaches towards low-valent Mg(I) species. Z. Anorg. Allg. Chem. 2024, 650, e202300247. [Google Scholar] [CrossRef]
- Dolati, H.; Denker, L.; Martínez, J.P.; Trzaskowski, B.; Frank, R. Iminoboranes with parent B = NH entity: Imino group metathesis, nucleophilic reactivity and N−N coupling. Chem. Eur. J. 2023, 29, e202302494. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction. CrysAlisPRO Softw. Syst. version 1.171.39.46; Rigaku Corporation: Oxford, UK, 2018. [Google Scholar]
- Sheldrick, G.M. SHELXT-Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Güven, Z.; Dolati, H.; Wessel, L.; Frank, R. Facile Synthetic Access Towards Sulfur- and Selenium-Functionalized Boron-Based Multiresonance TADF Emitters. Molecules 2024, 29, 5819. https://doi.org/10.3390/molecules29245819
Güven Z, Dolati H, Wessel L, Frank R. Facile Synthetic Access Towards Sulfur- and Selenium-Functionalized Boron-Based Multiresonance TADF Emitters. Molecules. 2024; 29(24):5819. https://doi.org/10.3390/molecules29245819
Chicago/Turabian StyleGüven, Zeynep, Hadi Dolati, Leo Wessel, and René Frank. 2024. "Facile Synthetic Access Towards Sulfur- and Selenium-Functionalized Boron-Based Multiresonance TADF Emitters" Molecules 29, no. 24: 5819. https://doi.org/10.3390/molecules29245819
APA StyleGüven, Z., Dolati, H., Wessel, L., & Frank, R. (2024). Facile Synthetic Access Towards Sulfur- and Selenium-Functionalized Boron-Based Multiresonance TADF Emitters. Molecules, 29(24), 5819. https://doi.org/10.3390/molecules29245819