
Cooperatively Catalyzed Activation of Thioglycosides with Iodine and Iron(III) Trifluoromethanesulfonate


Abstract
1. Introduction
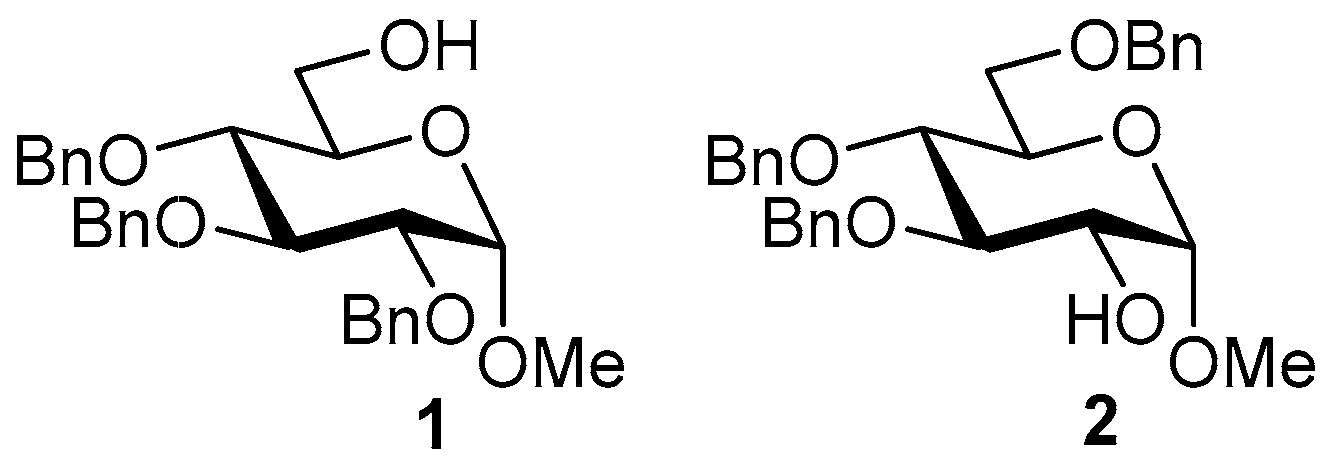
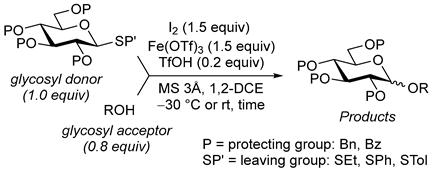
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. Synthesis of Building Blocks
3.3. Synthesis of Disaccharides
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Demchenko, A.V. Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Paulsen, H. Advances in selective chemical syntheses of complex oligosaccharides. Angew. Chem. Int. Ed. Engl. 1982, 21, 155–173. [Google Scholar] [CrossRef]
- Paulsen, H. Synthesis of complex oligosaccharide chains of glycoproteins. Chem. Soc. Rev. 1984, 13, 15–45. [Google Scholar] [CrossRef]
- Toshima, K.; Tatsuta, K. Recent progress in O-glycosylation methods and its application to natural-products synthesis. Chem. Rev. 1993, 93, 1503–1531. [Google Scholar] [CrossRef]
- Adero, P.O.; Amarasekara, H.; Wen, P.; Bohe, L.; Crich, D. The experimental evidence in support of glycosylation mechanisms at the SN1-SN2 Interface. Chem. Rev. 2018, 118, 8242–8284. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.S.; Wang, C.C.; Sabbavarapu, N.M.; Podilapu, A.R.; Liao, P.H.; Hung, S.C. “One-Pot” Protection, Glycosylation, and Protection-Glycosylation Strategies of Carbohydrates. Chem. Rev. 2018, 118, 8025–8104. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.M.; Pedersen, C.M. Catalytic Glycosylations in Oligosaccharide Synthesis. Chem. Rev. 2018, 118, 8285–8358. [Google Scholar] [CrossRef] [PubMed]
- Panza, M.; Pistorio, S.G.; Stine, K.J.; Demchenko, A.V. Automated chemical oligosaccharide synthesis: Novel approach to traditional challenges. Chem. Rev. 2018, 118, 8105–8150. [Google Scholar] [CrossRef] [PubMed]
- Michael, A. On the synthesis of helicin and phenolglucoside. Am. Chem. J. 1879, 1, 305–312. [Google Scholar]
- Fischer, E. Über die glucoside der alkohole. Ber. Dtsch. Chem. Ges. 1893, 26, 2400–2412. [Google Scholar] [CrossRef]
- Koenigs, W.; Knorr, E. Über einige derivate des traubenzuckers und der galactose. Ber. Dtsch. Chem. Ges. 1901, 34, 957–981. [Google Scholar] [CrossRef]
- Wulff, G.; Rohle, G. Results and problems of O-glycoside synthesis. Angew. Chem. Int. Ed. Engl. 1974, 13, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K. The Koenigs-Knorr reaction. Adv. Carbohydr. Chem. Biochem. 1977, 34, 243–283. [Google Scholar]
- Singh, Y.; Demchenko, A.V. Koenigs-Knorr glycosylation reaction catalyzed by trimethylsilyl trifluoromethanesulfonate. Chem. Eur. J. 2019, 25, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, A.V.; De Meo, C. The 4K reaction. Carbohydr. Res. 2024, 538, 109102. [Google Scholar] [CrossRef] [PubMed]
- Lian, G.; Zhang, X.; Yu, B. Thioglycosides in Carbohydrate research. Carbohydr. Res. 2015, 403, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Fugedi, P.; Garegg, P.J. A novel promoter for the efficient construction of 1,2-trans linkages in glycoside synthesis, using thioglycosides as glycosyl donors. Carbohydr. Res. 1986, 149, c9–c12. [Google Scholar] [CrossRef]
- Dasgupta, F.; Garegg, P.J. Use of sulfenyl halides in carbohydrate reactions. Part I. Alkyl sulfenyl triflate as activator in the thioglycoside-mediated formation of beta-glycosidic linkages during oligosaccharide synthesis. Carbohydr. Res. 1988, 177, C13–C17. [Google Scholar] [CrossRef]
- Crich, D.; Smith, M. S-(4-Methoxyphenyl) benzenethiosulfinate(MPBT)/trifluoromethanesulfonic anhydride (Tf2O): A convenient system for the generation of glycosyl triflates from thioglycosides. Org. Lett. 2000, 2, 4067–4069. [Google Scholar] [CrossRef] [PubMed]
- Crich, D.; Smith, M. 1-Benzenesulfinyl piperidine/trifluoromethanesulfonic anhydride: A potent combination of shelf-stable reagents for the low-temperature conversion of thioglycosides to glycosyl triflates and for the formation of diverse glycosidic linkages. J. Am. Chem. Soc. 2001, 123, 9015–9020. [Google Scholar] [CrossRef] [PubMed]
- Codee, J.D.C.; Litjens, R.E.J.N.; Heeten, R.; Overkleeft, H.S.; van Boom, J.H.; van der Marel, G.A. Ph2SO/Tf2O: A powerful promotor system in chemoselective glycosylations using thioglycosides. Org. Lett. 2003, 5, 1519–1522. [Google Scholar] [CrossRef] [PubMed]
- Duron, S.G.; Polat, T.; Wong, C.H. N-(Phenylthio)-ε-caprolactam: A new promoter for the activation of thioglycosides. Org. Lett. 2004, 6, 839–841. [Google Scholar] [CrossRef] [PubMed]
- Veeneman, G.H.; van Leeuwen, S.H.; van Boom, J.H. Iodonium ion promoted reactions at the anomeric centre. II. An efficient thioglycoside mediated approach toward the formation of 1,2-trans linked glycosides and glycosidic esters. Tetrahedron Lett. 1990, 31, 1331–1334. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Seitz, S.P.; Papahatjis, D.P. A mild and general method for the synthesis of O-glycosides. J. Am. Chem. Soc. 1983, 105, 2430–2434. [Google Scholar] [CrossRef]
- Kihlberg, J.O.; Leigh, D.A.; Bundle, D.R. The in situ activation of thioglycosides with bromine: An improved glycosylation method. J. Org. Chem. 1990, 55, 2860–2863. [Google Scholar] [CrossRef]
- Kartha, K.P.M.; Aloui, M.; Field, R.A. Iodine: A versatile reagent in carbohydrate chemistry II. Efficient chemospecific activation of thiomethylglycosides. Tetrahedron Lett. 1996, 37, 5175–5178. [Google Scholar] [CrossRef]
- Burkart, M.D.; Zhang, Z.; Hung, S.-C.; Wong, C.-H. A new method for the synthesis of fluoro-carbohydrates and glycosides using selectfluor. J. Am. Chem. Soc. 1997, 119, 11743–11746. [Google Scholar] [CrossRef]
- Ercegovic, T.; Meijer, A.; Magnusson, G.; Ellervik, U. Iodine monochloride/silver trifluoromethanesulfonate (ICI/AgOTf) as a convenient promoter system for O-glycoside synthesis. Org. Lett. 2001, 3, 913–915. [Google Scholar] [CrossRef] [PubMed]
- Mao, R.-Z.; Guo, F.; Xiong, D.-C.; Li, Q.; Duan, J.; Ye, X.-S. Photoinduced C-S Bond Cleavage of Thioglycosides and Glycosylation. Org. Lett. 2015, 17, 5606–5609. [Google Scholar] [CrossRef] [PubMed]
- Wever, W.J.; Cinelli, M.A.; Bowers, A.A. Visible Light Mediated Activation and O-Glycosylation of Thioglycosides. Org. Lett. 2013, 15, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Spell, M.L.; Deveaux, K.; Bresnahan, C.G.; Bernard, B.L.; Sheffield, W.; Kumar, R.; Ragains, J.R. A Visible-Light-Promoted O-Glycosylation with a Thioglycoside Donor. Angew. Chem. Int. Ed. 2016, 55, 6515–6519. [Google Scholar] [CrossRef] [PubMed]
- Mao, R.-Z.; Xiong, D.-C.; Guo, F.; Li, Q.; Duan, J.; Ye, X.-S. Light-driven highly efficient glycosylation reactions. Org. Chem. Front. 2016, 3, 737–743. [Google Scholar] [CrossRef]
- Escopy, S.; Demchenko, A.V. Transition-Metal-Mediated Glycosylation with Thioglycosides. Chem. Eur. J. 2022, 28, e202103747. [Google Scholar] [CrossRef] [PubMed]
- Ferrier, R.J.; Hay, R.W.; Vethaviyasar, N. A potentially versatile synthesis of glycosides. Carbohydr. Res. 1973, 27, 55–61. [Google Scholar] [CrossRef]
- Goswami, M.; Ellern, A.; Pohl, N.L. Bismuth(V)-mediated thioglycoside activation. Angew. Chem. Int. Ed. Engl. 2013, 52, 8441–8445. [Google Scholar] [CrossRef] [PubMed]
- Goswami, M.; Ashley, D.C.; Baik, M.H.; Pohl, N.L. Mechanistic Studies of Bismuth(V)-Mediated Thioglycoside Activation Reveal Differential Reactivity of Anomers. J. Org. Chem. 2016, 81, 5949–5962. [Google Scholar] [CrossRef] [PubMed]
- Vibhute, A.M.; Dhaka, A.; Athiyarath, V.; Sureshan, K.M. A versatile glycosylation strategy via Au (III) catalyzed activation of thioglycoside donors. Chem. Sci. 2016, 7, 4259–4263. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Schmidt, R.R. New principles for glycoside-bond formation. Angew. Chem. Int. Ed. 2009, 48, 1900–1934. [Google Scholar] [CrossRef] [PubMed]
- Escopy, S.; Singh, Y.; Demchenko, A.V. Palladium(II)-assisted activation of thioglycosides. Org. Biomol. Chem. 2021, 19, 2044–2054. [Google Scholar] [CrossRef] [PubMed]
- Pooladian, F.; Escopy, S.; Demchenko, A.V. Activation of thioglycosides with copper(II) bromide. Molecules 2022, 27, 7354. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, L.M.; Das, A.; Shadrick, M.L.; Demchenko, A.V. Ferric Chloride Promoted Glycosidation of Alkyl Thioglycosides. Molecules 2024, 29, 4845. [Google Scholar] [CrossRef] [PubMed]
- Dent, A.; Escopy, S.; Demchenko, A.V. Cooperatively Catalyzed Activation of Thioglycosides That Bypasses Intermediacy of Glycosyl Halides. Chem. Eur. J. 2023, 29, e202300873. [Google Scholar] [CrossRef] [PubMed]
- Dent, A.R.; Demchenko, A.V. Halophilic Metal Salts for the Cooperatively Catalyzed Activation of Thioglycosides. J. Org. Chem. 2025, 90, 6478–6490. [Google Scholar] [CrossRef] [PubMed]
- Bandara, M.D.; Yasomanee, J.P.; Demchenko, A.V. Application of armed, disarmed, superarmed and superdisarmed building blocks in stereocontrolled glycosylation and expeditious oligosaccharide synthesis. In Selective Glycosylations: Synthetic Methods and Catalysts; Bennett, C.S., Ed.; Wiley: Weinheim, Germany, 2017; pp. 29–58. [Google Scholar]
- Shadrick, M.; Singh, Y.; Demchenko, A.V. Stereocontrolled α-galactosylation under cooperative catalysis. J. Org. Chem. 2020, 85, 15936–15944. [Google Scholar] [CrossRef] [PubMed]
- Shadrick, M.; Stine, K.J.; Demchenko, A.V. Expanding the scope of stereoselective α-galactosylation using glycosyl chlorides. Bioorg. Med. Chem. 2022, 73, 117031. [Google Scholar] [CrossRef] [PubMed]
- Premathilake, H.D.; Mydock, L.K.; Demchenko, A.V. Superarming common glycosyl donors by simple 2-O-benzoyl-3,4,6-tri-O-benzyl protection. J. Org. Chem. 2010, 75, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Yasomanee, J.P.; Demchenko, A.V. The effect of remote picolinyl and picoloyl substituents on the stereoselectivity of chemical glycosylation. J. Am. Chem. Soc. 2012, 134, 20097–20102. [Google Scholar] [CrossRef] [PubMed]
- Geringer, S.A.; Mannino, M.P.; Bandara, M.D.; Demchenko, A.V. Picoloyl protecting group in synthesis: Focus on a highly chemoselective catalytic removal. Org. Biomol. Chem. 2020, 18, 4863–4871. [Google Scholar] [CrossRef] [PubMed]
- Ranade, S.C.; Kaeothip, S.; Demchenko, A.V. Glycosyl alkoxythioimidates as complementary building blocks for chemical glycosylation. Org. Lett. 2010, 12, 5628–5631. [Google Scholar] [CrossRef] [PubMed]
- Sail, D.; Kovac, P. Benzoylated ethyl 1-thioglycosides: Direct preparation from per-O-benzoylated sugars. Carbohydr. Res. 2012, 357, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Ferrier, R.J.; Furneaux, R.H. 1,2-trans-1-Thioglycosides. In Methods in Carbohydrate Chemistry; Whistler, R.L., BeMiller, J.N., Eds.; Academic Press: Cambridge, MA, USA, 1980; Volume 8, pp. 251–253. [Google Scholar]
- France, R.R.; Rees, N.V.; Wadhawman, J.D.; Fairbanks, A.J.; Compton, R.G. Selective activation of glycosyl donors using electrochemical techniques: A study of the thermodynamic oxidation potentials of a range of chalcoglycosides. Org. Biomol. Chem. 2004, 2, 2188–2194. [Google Scholar] [CrossRef] [PubMed]
- Andersson, F.; Fugedi, P.; Garegg, P.J.; Nashed, M. Synthesis of 1,2-cis-linked glycosides using dimethyl(methylthio) sulfonium triflate as promoter and thioglycosides as glycosyl donors. Tetrahedron Lett. 1986, 27, 3919–3922. [Google Scholar] [CrossRef]
- Weïwer, M.; Sherwood, T.; Linhardt, R.J. Synthesis of Floridoside. J. Carbohydr. Chem. 2008, 27, 420–427. [Google Scholar] [CrossRef]
- Wang, C.; Sanders, B.; Baker, D.C. Synthesis of a glycodendrimer incorporating multiple mannosides on a glucoside core. Can. J. Chem. 2011, 89, 959–963. [Google Scholar] [CrossRef]
- Elie, C.; Verduyn, R.; Dreef, C.; Brounts, D.; van der Marel, G.; van Boom, J. Synthesis of 6-0-(α-D-mannopyranosyl)-D-myo-inositol: A fragment from mycobacteria phospholipids. Tetrahedron 1990, 46, 8243–8254. [Google Scholar] [CrossRef]
- Garcia, B.A.; Gin, D.Y. Dehydrative glycosylation with activated diphenyl sulfonium reagents. Scope, mode of C(1)-hemiacetal activation, and detection of reactive glycosyl intermediates. J. Am. Chem. Soc. 2000, 122, 4269–4279. [Google Scholar] [CrossRef]
- Nigudkar, S.S.; Parameswar, A.R.; Pornsuriyasak, P.; Stine, K.J.; Demchenko, A.V. O-Benzoxazolyl imidates as versatile glycosyl donors for chemical glycosylation. Org. Biomol. Chem. 2013, 11, 4068–4076. [Google Scholar] [CrossRef] [PubMed]
- Codee, J.D.C.; Van den Bos, L.J.; Litjens, R.E.J.N.; Overkleeft, H.S.; Van Boeckel, C.A.A.; Van Boom, J.H.; Van der Marel, G.A. Chemoselective glycosylations using sulfonium triflate activator systems. Tetrahedron 2004, 60, 1057–1064. [Google Scholar] [CrossRef]
- Hasty, S.J.; Kleine, M.A.; Demchenko, A.V. S-Benzimidazolyl glycosides as a platform for oligosaccharide synthesis by an active-latent strategy. Angew. Chem. Int. Ed. 2011, 50, 4197–4201. [Google Scholar] [CrossRef] [PubMed]
- Vankar, Y.D.; Vankar, P.S.; Behrendt, M.; Schmidt, R.R. Synthesis of β-O-glycosides using enol ether and imidate derived leaving groups. Emphasis on the use of nitriles as a solvent. Tetrahedron 1991, 47, 9985–9992. [Google Scholar] [CrossRef]
- Premathilake, H.D.; Demchenko, A.V. 2-Allylphenyl glycosides as complementary building blocks for oligosaccharide and glycoconjugate synthesis. Beilstein J. Org. Chem. 2012, 8, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Pornsuriyasak, P.; Demchenko, A.V. S-Thiazolinyl (STaz) glycosides as versatile building blocks for convergent selective, chemoselective, and orthogonal oligosaccharide synthesis. Chem. Eur. J. 2006, 12, 6630–6646. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.; Wang, T.; Geringer, S.A.; Stine, K.J.; Demchenko, A.V. Regenerative glycosylation. J. Org. Chem. 2018, 83, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Neralkar, M.; Hotha, S. Stable alkynyl glycosyl carbonates: Catalytic anomeric activation and synthesis of a tridecasaccharide reminiscent of Mycobacterium tuberculosis cell wall lipoarabinomannan. Angew. Chem. Int. Ed. 2016, 128, 7917–7922. [Google Scholar] [CrossRef]
- Geringer, S.A.; Demchenko, A.V. Iron(III) chloride-catalyzed activation of glycosyl chlorides. Org. Biomol. Chem. 2018, 16, 9133–9137. [Google Scholar] [CrossRef] [PubMed]
- Ravidà, A.; Liu, X.; Kovacs, L.; Seeberger, P.H. Synthesis of glycosyl phosphates from 1,2-orthoesters and application to in situ glycosylation reactions. Org. Lett. 2006, 8, 1815–1818. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
 | |||
|---|---|---|---|
| Entry | Donor | Acceptor | Product, Time, Yield, Ratio α/β |
| 1 | 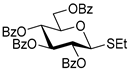 3 | 1 | 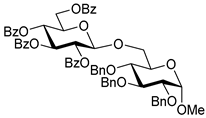 4, 16 h, 96%, β-only |
| 2 | 3 | 2 | 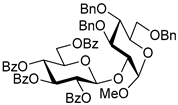 5, 24 h, 87%, β-only |
| 3 | 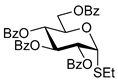 6 | 1 | 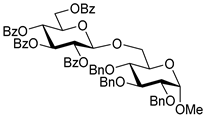 4, 30 h, 92%, β-only |
| 4 |  7 | 1 | 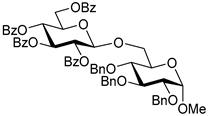 4, 30 h, 90%, β-only |
| 5 | 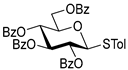 8 | 1 | 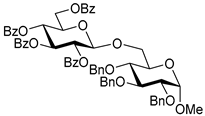 4, 30 h, 95%, β-only |
| 6 | 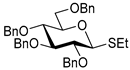 9 | 1 | 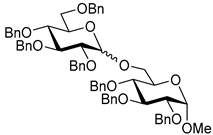 10, 16 h, 86%, α:β = 1:1.5 |
| 7 | 9 | 2 |  11, 16 h, 73%, α:β = 1:1 |
| 8 | 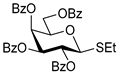 12 | 1 | 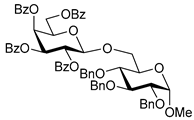 13, 16 h, 96%, β-only |
| 9 | 12 | 2 | 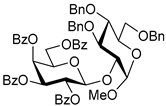 14, 24 h, 89%, β-only |
| 10 | 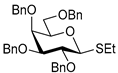 15 | 1 | 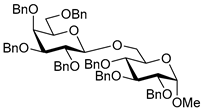 16, 16 h, 92%, β-only |
| 11 | 15 | 2 | 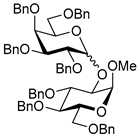 17, 16 h, 79%, α:β = 1.6:1 |
| 12 | 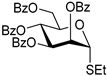 18 | 1 | 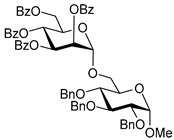 19, 24 h, 84%, α-only |
| 13 | 18 | 2 | 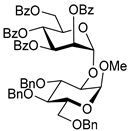 20, 24 h, 85%, α-only |
| 14 | 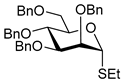 21 | 1 | 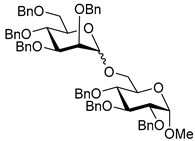 22, 16 h, 81%, α:β = 1:4 |
| 15 | 21 | 2 | 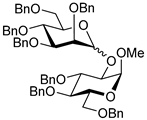 23, 16 h, 70%, α:β = 1:1.2 |
| 16 |  24 | 1 |  25, 16 h, 78%, α-only |
| 17 | 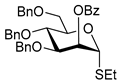 26 | 1 |  27, 16 h, 79%, α-only |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dent, A.R.; DeSpain, A.M.; Demchenko, A.V. Cooperatively Catalyzed Activation of Thioglycosides with Iodine and Iron(III) Trifluoromethanesulfonate. Molecules 2025, 30, 3058. https://doi.org/10.3390/molecules30153058
Dent AR, DeSpain AM, Demchenko AV. Cooperatively Catalyzed Activation of Thioglycosides with Iodine and Iron(III) Trifluoromethanesulfonate. Molecules. 2025; 30(15):3058. https://doi.org/10.3390/molecules30153058
Chicago/Turabian StyleDent, Ashley R., Aidan M. DeSpain, and Alexei V. Demchenko. 2025. "Cooperatively Catalyzed Activation of Thioglycosides with Iodine and Iron(III) Trifluoromethanesulfonate" Molecules 30, no. 15: 3058. https://doi.org/10.3390/molecules30153058
APA StyleDent, A. R., DeSpain, A. M., & Demchenko, A. V. (2025). Cooperatively Catalyzed Activation of Thioglycosides with Iodine and Iron(III) Trifluoromethanesulfonate. Molecules, 30(15), 3058. https://doi.org/10.3390/molecules30153058