1. Introduction
Among many smart tools for the colonization of human organs, pathogenic bacteria use a multilayer cell envelope [
1,
2], which is largely composed of glycans and their conjugates. The outermost thick layer found in the majority of Gram-negative and some Gram-positive bacteria is called a glycan capsule, the composition and structure of which depend on the particular bacterial species [
3]. Most capsular glycans are long-chain linear negatively charged CPSs or phosphoglycans. In the course of infection, this part of the bacterial outer shell first comes into contact with the components of innate and adaptive immunity. The anionic glycans of pathogenic bacteria shield them from the action of the components of the complement system, phagocytes and cationic antimicrobial peptides, which are secreted by immune and epithelial cells and destabilize the membrane of pathogenic bacteria [
4,
5]. Thus, it was established [
6] that the phosphoglycan capsule of
Hib prevents phagocyte attachment and the subsequent uptake of these bacteria. Moreover,
Hib can survive and multiply after having been engulfed by a phagocyte in the acidic medium of phagolysosomes [
7]. On the one hand, these biopolymers act as both protective shields and adhesive agents, enabling the bacterial evasion of host immunity [
8], and on the other hand, they represent a target for the host immune system.
It is known that a key step in the adaptive immune response is the production of antigen-specific antibodies, and the presence of IgG indicates the development of immunological memory to surface biopolymers [
9]. Immunological memory enables the body to quickly recognize the antigen on the surface of the pathogen during the second and subsequent contact and more effectively activate the body’s defenses. This phenomenon encouraged the development of a whole range of antibacterial prophylactic vaccines. To date, the most effective type of vaccines for the prevention of bacterial infections are conjugate vaccines [
10,
11,
12,
13,
14,
15,
16], in which a capsular glycan of the targeted pathogen is covalently linked to a protein carrier. The use of glycoconjugates helps circumvent the problem of the low immunogenicity of CPS and directs immunity along the T-dependent pathway [
17,
18,
19,
20].
The majority of commercial conjugate vaccines are currently manufactured on the basis of CPS produced in bacterial cell cultures. This approach has significant disadvantages, which are the laborious and operationally challenging steps of manufacturing, sophisticated quality control, and the high cost of producing pathogenic microbiological material. At present, an advanced approach to glycan conjugate vaccines is being developed, which employs synthetic OS antigens as an alternative to bacterial CPS [
21,
22,
23,
24,
25,
26].
In the first step of conjugate OS vaccine development, it is important to specify the particular bacterial glycoantigen to be mimicked. The probability factor speaks in favor of regular glycans, which have a repeating unit. These include the CPS of both Gram-positive and Gram-negative bacteria, lipopolysaccharide O-antigens of Gram-negative bacteria, and the cell-wall teichoic acids and lipoteichoic acids of Gram-positive bacteria. Today, commercial glycoconjugate vaccines incorporate bacterial glycan antigens exclusively.
In the second step, the optimum length of the OS, which is sufficient for the induction of protective immunity, has to be specified. On the one hand, the use of shorter OSs can significantly reduce production costs, and on the other hand, the OS chain has to be long enough to mimic the polymer and minimize the possible influence of the terminal monosaccharide residue. Identification of the minimum length of the OS antigen is performed in laboratory animals, which are immunized with the conjugates of synthetic oligosaccharides of different lengths, and the interaction of induced antibodies with bacterial antigens or directly with bacteria is investigated. It is commonly accepted [
27] that the minimum length of an effective glycoantigen is 3–4 repeating units. A reliable algorithm that can predict the optimum length of OS antigens has not yet been developed. Today, the identification of protective glycotopes for conjugate vaccine candidates can be defined by the screening of glycan arrays, which encompass a series of synthetic OSs related to capsular glycans [
28,
29] in combination with conformational studies [
29,
30]. Alternatively, the development of the anti-Hib vaccine Quimi-Hib
® (Centro de Ingeniería Genética y Biotecnología (CIGB), Republic of Cuba), which is a unique commercial synthetic OS-based conjugate vaccine, circumvented the problem of the choice of the optimum antigen length by the production of a protein-conjugated homologous phosphooligosaccharide obtained by oligomerization, with 7–8 repeating units as the average length of antigens [
31]. The choice of the optimum structure of an OS antigen for a glycovaccine is complicated by the possibility of structural changes in a synthetic antigen after the injection of the vaccine into the recipient’s body. Partial lysis or migration of acetyl groups can occur in lymphs (pH 7.4–9.0) or endolysosomes of follicular B-lymphocytes [
32,
33,
34], which are known to have an acidic environment.
In a number of human pathogens, a phosphodiester bond is involved in the formation of the main polymer chain. Phosphoglycans of this type were found, for example, in the cell wall of the human pathogens
Hia,
Hib,
Hic, and
Hif, the
S. pneumonia serotypes 6A, 6B, 11A, 17F, 19A, and 19F, the
N. meningitidis serogroups
A and
X, the
Campylobacter jejuni serotypes HS53 (strain RM1221) and HS1 (strain ATCC 43429), and
Escherichia coli K100 and K2 (
Figure 1) [
35,
36,
37,
38]. Today, eleven bacterial pathogens of this type (
Hib,
Men A, and the
S. pneumonia serotypes 6A, 6B, 10A, 11A, 15B, 18C, 19A, 19F, and 23F) are targeted by preventive vaccination, with commercial conjugate vaccines based on the corresponding capsular phosphooligosaccharides [
21].
In this review, we focus on the synthesis of spacer-armed mono-, di-, and oligomeric antigens structurally related to bacterial capsular phosphoglycans (
Figure 1), in which the repeating units are connected via a phosphodiester bond, and consider the preparation and immunogenicity of neoglycoconjugates on the basis of these antigens.
Phosphoglycans are the common glycocalyx components of pathogenic Gram-negative and Gram-positive bacteria [
35,
36,
39], yeast [
39], and protozoan parasites [
39,
40]. In a living cell, phosphodiester-linked carbohydrates are arranged via the transfer of a hexose 1-phosphate to a glycan acceptor. In the presence of enzymes, which belong to the Stealth enzyme family, a hydroxyl group of a glycan acceptor attacks the phosphoester group in the nucleotide donor, and finally, a phosphodiester interglycosidic bridge unit is formed. It was established [
41] (
Figure 2) that in
MenX bacteria, the hydroxyl group of
N-acetyl glucosamine acceptor attacks the P-atom of uridine-5′-diphosphate-
N-acetyl glucosamine to form an α-glycosyl phosphodiester, and the uridine monophosphate moiety serves as a leaving group.
A similar method of the establishment of a phosphodiester intersaccharide bridge is used in laboratory practice for the preparation of phosphooligosaccharides related to bacterial phosphoglycans. For the efficient and stereoselective formation of a phosphodiester linkage, a phosphodiester synthon is first introduced into one of the saccharide blocks, and the resulting product is reacted with a free hydroxyl group of another saccharide block.
As a rule, phosphodiesters, along with their diverse precursors (mono- and diphosphates, H-phosphonates, and phosphamidites;
Figure 3), decompose under the conditions of a glycosylation reaction. Therefore, the retrosynthetic analysis of phosphoglycans suggests the formation of phosphodiester bridges within the latest steps of the synthetic route after the glycosidic linkages are already established. Usually, the preparation of phosphooligosaccharides related to natural biopolymers is a multi-step synthesis, which can be performed following a linear, convergent, or oligomerization pathway via the formation of an O-P-O tether between selectively protected and activated saccharide blocks.
The conventional synthetic blocks for the preparation of glycosylphosphodiesters are monophosphates (
Figure 3A), diphosphates (
Figure 3B), H-phosphonates (
Figure 3C), and phosphoramidites (
Figure 3D). Previously developed methods of condensation involved phosphorus (V) chemistry (
Figure 3A,B) and were promoted by
N,
N′-dicyclohexylcarbodiimide and 1-(2,4,6-triisopropylbenzenesulfonyl)-3-nitro-1H-1,2,4-triazole. In the late 1980s, methods A and B gave way to fast, efficient, and convenient techniques that employed H-phosphonates (
Figure 3C) and phosphoramidites (
Figure 3D).
The H-phosphonate condensation general procedure [
42,
43] was first proposed in the 1950s for oligonucleotide synthesis by Todd et al. [
44], and it was further developed [
45,
46] and adapted for solid-phase synthesis [
47] and customized to the needs of phosphoglycan chemistry [
39,
48]. The phosphoramidite method was first proposed by van Boom [
49] and was later successfully applied in the solid-phase preparation of long-chain oligophosphodiesters [
50] and the P-modified analogs of glycosylphosphate oligomers [
51]. In addition to current methods, novel approaches are being actively developed, which are aimed at the preparation of glycosylphosphates with a predetermined anomeric configuration and the synthesis of the stabilized mimetics of phosphoglycans [
52,
53,
54,
55,
56,
57,
58].
One of the key features of bacterial phosphoglycans and synthetic phosphooligosaccharides is their susceptibility to degradation via hydrolytic cleavage, transesterification, and rearrangement. Thus, PRP (
Figure 1) was found to degrade spontaneously [
59,
60] in aqueous media. This molecule is destabilized by a hydroxyl group at C-2 (D-ribose), which is located in close proximity to the phosphodiester moiety and promotes the depolymerization and formation of cyclophosphate and phosphate monoester terminal groups [
61]. In vaccine production, the inherent tendency of PRP to autolyze results in the loss of manufactured phosphoglycan and conjugate preparations [
60]. Additionally, the low stability of PRP imposes significant limitations on the use of liquid Hib vaccines, especially in view of the acceleration of the degradation process in the presence of an alum adjuvant [
62]. Stability studies conducted by Cintra et al. [
60] showed that the rate of PRP depolymerization accelerates substantially with an increase in pH in the range 5.41–7.55 and with a rise in temperature in the interval of 28–40 °C. As a result, it may be assumed that in a host organism, PRP intensely degrades into fragments, which neutralize anti-PRP protective antibodies, thus hampering the immune response. In a similar way, partial PRP destruction after immunization with a conjugate
Hib vaccine may result in the loss of Hib epitopes and reduce the level of protective anti-Hib antibodies. Two more factors that affect the stability of PRP are the presence of Na
+ [
60] and Ca
2+ [
59] cations. Similar to PRP, the capsular phosphoglycan of
Hif (
Figure 1) was shown to decompose under mild conditions [
59].
MenA capsular phosphoglycan is especially susceptible to hydrolysis. It is assumed that the hydrolytic destruction of α-glycosylphosphodiester linkage occurs by two pathways. One of these includes the formation of an oxocarbenium ion, and within another pathway, the phosphodiester bond is cleaved with the assistance of the axial NAc group located at C-2 of the ManNAc, and a thermodynamically stable oxazoline is formed [
63].
2. Synthesis of Pseudo-Oligosaccharides Structurally Related to PRP
Highly effective infant immunization with conjugate
Hib vaccines, which are composed of a partially depolymerized
Hib capsular phosphoglycan and a protein carrier (see review [
15]), inspired researchers to develop conjugate vaccines using oligomer synthetic PRP fragments (
Figure 4). Several series of PRP-related oligomers were synthesized, which were equipped with different types and locations of amino-spacers for convenient attachment to a protein carrier (compounds
1–
15,
Figure 4).
In pseudo-oligosaccharide
13 with a
N-glycyl-aminopropyl spacer, which comprises two, three, or four residues of PRP repeating units [
64], as well as in hexamer
4 [
65] and oligomers
5–
8 [
66], the ω-aminospacer is linked to the D-ribose fragment via a phosphodiester bond. In pentamer
9 [
67], the spacer is attached immediately to C-1 of a D-ribose residue as an aglycone. In the oligomeric mixture
10 [
68] and in most of the representative oligomer series, which comprises the individual compounds
11–
15 [
69], the spacer is connected to a D-ribose via a phosphodiester bridge. In this review, basic synthetic blocks and strategies used in the preparation of compounds
1–
15 are briefly considered, with a focus on the arrangement of a phosphodiester linkage and the introduction of a ω-aminospacer. A more detailed consideration of the syntheses of pseudo-oligosaccharides
1–
15 was presented in our earlier review [
15].
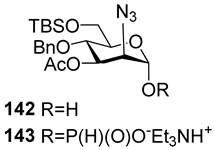
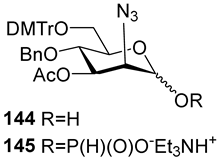
Between 1988 and 1992, van Boom and his research group at Leiden University synthesized spacer-armed PRP-related fragments for the first time. Dimer
1 [
64,
70], trimer
2 [
64,
70], and tetramer
3 [
71] (
Figure 4) were prepared by a sequential elongation of the oligomer chain from the non-reducing end, and the final equipment with an aminospacer. In brief, the phosphorylation of a free 3-OH group in the selectively protected riboside
16 with bis(benzotriazol-1-yl) (2-chlorophenyl) phosphate
17 produced the key activated phosphotriester building block
18 (
Scheme 1). The condensation of compound
18 with the selectively protected disaccharide
19 in the presence of
N-methylimidazole in Py, followed by the removal of the 1-
O-propenyl group, was carried out in a sequential manner, one, two, or three times, followed by the final capping with phosphotriester
20. Total deprotection afforded the series of conjugation-ready compounds
1–
3.
The spacer-armed trimer
2 and tetramer
3 were conjugated to TT (
Figure 5) to create the corresponding neoglycoconjugates
21 and
22. The antigenic properties of oligomers
2 and
3 as components of neoglycoconjugates
21 and
22 were examined [
71] in competitive inhibition ELISA experiments. A conjugate of bacterial PRP and tyramine was used as a coating antigen, and normal human serum and bacterial PRP were used as positive controls.
Total inhibition was observed when bacterial PRP and conjugate
22 (tetramer + TT,
Figure 5) at a concentration of 25 μg/mL were used as inhibitors, and conjugate
21 with the trimeric PRP–antigen was less effective. Compared to conjugates, the inhibitory capacity of the unconjugated oligomers
2 and
3 was much lower, and at a concentration of 25 μg/mL, it did not reach 40%.
The immunogenicity of glycoconjugates
21 and
22 was studied [
71] in mice at a dose of 1 µg of phosphoglycan per mouse. IgG antibodies in immune sera were analyzed in ELISA using a bacterial PRP–tyramine conjugate as a coating antigen and normal human serum as a positive control. Conjugate
21 with a trimeric antigen was found to be low immunogenic in a mouse model. In IgG ELISA, the mean optical density value for the sera of immunized mice was less than two times that of the corresponding value obtained for the sera of mice that received PBS.
For the immune sera from mice vaccinated with conjugate
22, which comprised tetrameric residues, the mean optical density value was three times that of the negative control, thus unambiguously evidencing the immunogenicity of conjugate
22. Immunization experiments were conducted using preparations formulated with AlPO
4 or without an adjuvant. It was shown that the presence of AlPO
4 had no effect on the result of immunization [
71].
To date, there is no animal model that reliably predicts the immunogenicity of glycoconjugate preparations in humans, and the choice of experimental animals for the examination of the immune activity of
Hib preparations is usually at the discretion of the researcher. In the blood sera of immunized mice and guinea pigs, the concentration of antibodies directed against
Hib antigens does not reach 1 mg/mL, and these animals are not recommended for laboratory studies of conjugate vaccines [
66].
As shown in
Scheme 1, the key reaction in the assembly of the synthetic
Hib phosphooligosaccharides
1–
3 is the arrangement of the O-P-O linkage, which can be applied iteratively. As no new chiral centers or other types of isomerism are created, this reaction sequence can be fulfilled using a solid-phase approach. This approach significantly decreases the number of laborious steps of the chromatographic separation of the product. In 1989, van Boom synthesized the spacer-armed hexamer
4 using controlled-pore glass as a solid support [
65]. First, the selectively protected pseudo-disaccharide
23 was phosphitylated with chlorophosphoramidite
24 (
Scheme 2), and the resulting phosphoramidite
25 was used for a stepwise extension of the oligomer chain starting from ribosylribitol
26, which was immobilized on a glass support. After each step of chain elongation, the phosphorus atom was oxidized with iodine in an acetonitrile/water/collidine mixture to obtain the corresponding phosphotriester, and the terminal DMTr protective group was removed to provide a free hydroxyl group for the next phosphitylation step. After the sequential attachment of five repeating units and spacer
27 and the removal of the protective groups, the target spacer-armed hexamer
4 was obtained.
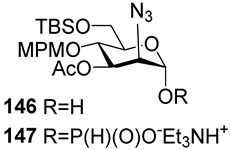
In 1992, Kandil et al. [
66] synthesized the series of spacer-armed Hib oligomers
5–
8 on a PEG support using a strategy similar to the synthesis of hexamer
4 (
Scheme 3). In this synthesis, the tert-butyldiphenylsilyl and benzyloxymethyl protective groups in the starting pseudo-disaccharide
28 were replaced with easily removable benzyl groups. Thus, the sequential condensation of phosphoramidite
29 with PEG-immobilized pseudo-disaccharide
30 and detritylation was repeated five times. Next, the pre-spacer
31 was attached, and finally, total deprotection resulted in the formation of hexamer
6. The latter was conjugated to synthetic peptides structurally related to the
Hib outer membrane protein, and one of these preparations showed immunogenicity comparable to that shown by the commercial
Hib vaccine [
72].
The same year, a group of Swedish researchers under the leadership of Norberg published [
67] the synthesis of the
Hib pentamer
9 on a polystyrene support (
Scheme 4). In this work, the H-phosphonate method was used for the establishment of a phosphodiester bond, which subsequently became the most popular in the preparation of oligophosphoglycans related to the glycocalyx components of bacteria and parasites [
39,
56]. The selectively protected disaccharide
32 was reacted with 5,5-dimethyl-2-chloro-1,3,2-dioxaphosphorinane 2-oxide (compound
33) in Py and the presence of phosphonic acid to obtain H-phosphonate
34 with an 84% yield.
The activation of H-phosphonates for subsequent attachment to alcohols can be achieved in the presence of the acyl chlorides of sterically hindered acids, e.g., PivCl or 1-adamantanecarbonyl chloride. In this reaction, the formation of by-products depends significantly on the order of the addition of reagents. For example, the activation of H-phosphonate by PivCl in Py in the absence of alcohol led to the formation of unwanted bis-acylated phosphites, and the use of a large excess of PivCl in solid-phase synthesis resulted in the pivaloylation of the support [
67]. The search for the optimum conditions for the attachment of H-phosphonate
34 to be selectively protected and immobilized on the solid support monomer
35 resulted in the discovery of the most efficient proportion—5 eq. of PivCl and 5 eq. of H-phosphonate
34—that provided the adduct with a 95% yield. The authors noted [
67] that the yield decreased with each chain extension step and dropped to 86% in the fourth cycle. The optimum sequence of reagent addition was found as follows: in the first step, H-phosphonate was activated by the addition of PivCl in Py, and then, the mixture was added to the immobilized alcohol. As a result, the yield of the pentamer increased to 96%. In the final step, the chain was capped with 2-(2-azidoethoxy) ethyl riboside
36, H-phosphonate was oxidized with iodine in 2% aqueous Py, and the protective groups were removed to obtain oligomer
9.
The multistep synthetic pathways discussed above are based on the linear sequential elongation of an oligomeric chain. As a rule, laboratory processes of this type cannot be scaled up to a commercial scale because of the high cost. For profitable manufacturing, a convergent synthetic scheme is needed. Under the leadership of Verez-Bencomo and Roy, a convergent synthesis of a mixture of homologous PRP oligomers (
Figure 4, compound
10) was developed and then scaled up to the commercial production of the anti-
Hib vaccine Quimi-Hib
® (Heber Biotec, S.A., Republic of Cuba). In this synthesis, polycondensation of a bifunctional monomer is employed for the establishment of O-P-O linkages as a key step of oligomer synthesis in the place of stepwise elongation (
Scheme 5).
In brief, the allyl group of the selectively protected pseudo-disaccharide
37 is isomerized into the 1-
O-propenyl group by the action of potassium tert-butoxide in DMSO. The resulting isomer is converted to H-phosphonate
38 by the action of PCl
3 and imidazole, and finally, the 1-
O-propenyl group is removed under acidic conditions to obtain the key heterobifunctional monomer
39. The interaction of
38 with the diethylene glycol spacer
40 in the presence of PivCl/Py, followed by oxidation with iodine in aqueous Py, and the further removal of the 1-
O-propenyl group, creates alcohol
41, which serves as a terminal unit in the polycondensation. The polycondensation of
39 and
41 is carried out in the presence of PivCl in Py, followed by oxidation of the mixture of H-phosphonates into the corresponding phosphodiester mixture, reduction of the azido groups,
N-acetylation, and total deprotection. The obtained mixture of the spacer-equipped oligomer
10 is activated by the action of SMP to obtain a conjugation-ready mixture of maleimide
42. The conjugation of the activated oligophosphodiesters with thiolated TT results in the production of a set of conjugate
43 with the average weight ratio PRP:TT of 1:2.6 [
31,
68].
On the basis of the mixture of conjugate
43, an anti-
Hib vaccine, Quimi-Hib
®, was developed, which successfully passed all the required preclinical [
73] and clinical trials [
31,
74]. The antigenicity of synthetic PRP oligomer
10 was compared to the antigenicity of bacterial PRP in ELISA experiments [
31]. A conjugate of a mixture of the spacer-equipped oligomer
10 with HSA (
10-HSA), prepared similarly to conjugate
43, and a conjugate of partially depolymerized bacterial PRP (PRPDp30) with HSA were used as coating antigens. The PRPDp30-HSA conjugate was obtained by the reductive amination of the mixture of bacterial PRP depolymerized via periodate oxidation to a length of ~30 monomeric units and conjugation with HSA [
68]. Standard rabbit anti-PRP antibodies were obtained by the immunization of animals with two licensed commercial conjugate vaccines based on bacterial PRP. These were Hiberix
® (PRP-TT), which contains PRP activated by cyanogen bromide and conjugated to TT via an adipic acid dihydrazide linker, and Vaxem-Hib
® (PRP-CRM197), which is a product of the conjugation of partially depolymerized bacterial PRP with an avDP10 repeating unit and the protein carrier CRM197 via an adipic acid dihydrazide spacer. The correlation coefficient for the titers obtained in the ELISA experiments with standard rabbit sera and the coating antigens 10-HSA and PRPDp30-HSA was in the high-value range (0.972–0.978), indicating the similarity of the antigenic properties of the synthetic oligosaccharide
10 and
Hib CPS [
75].
For the preliminary evaluation of the immunological activity of the conjugate
43 in vivo, rats, mice, and rabbits were chosen as experimental animals. Rats were immunized subcutaneously twice at an interval of 4 weeks with a dose of conjugate
43 containing 2 µg of the ligand
10 [
75]. Mice were immunized intraperitoneally three times at an interval of 2 weeks with a dose of conjugate
43 containing 2.5 µg of ligand
10. Rabbits were immunized with conjugate
43 using both two-step and three-step regimens at a dose of 5 µg of ligand
10 per animal. The efficiency of immunization was assessed by the evaluation of IgG titers, which were calculated as the logarithm of the highest dilution at which the light absorbance of the diluted serum sample is twice as high as that of the negative control. For the negative control, the animals were injected with PBS. A PRP-HSA conjugate was used as a coating antigen in the ELISA experiments. The study of the PRP-specific antibodies showed that the immune response to conjugate
43 in rodents was weak and inconsistent. In contrast, the immunization of rabbits with conjugate
43 efficiently evoked PRP-specific antibodies, as shown in the inhibition ELISA experiments with bacterial PRP as the inhibitor [
75].
In a phase 1 clinical trial [
31], more than 100 children aged 4 to 5 years were immunized with a single dose of a vaccine preparation formulated on the basis of conjugate
43 without an adjuvant. Comparative studies of the immunogenicity of synthetic antigens in conjugate
43 and partly depolymerized bacterial PRP in Vaxem-Hib
® showed that the average anti-PRP IgG titers were similar. It was found that PRP-specific IgG antibodies induced in children by immunization with conjugate
43 had bactericidal activity comparable to that stimulated by the action of Vaxem-Hib
®. In the course of phase 2 clinical trials [
31], 1141 infants were immunized three times at 2, 4, and 6 months of age with or without AlPO
4, and the positive control group was immunized with Vaxem-Hib
®. Antibody tests showed that 99.7% of infants had serum anti-PRP IgG concentrations >1 µg/mL, which is known to provide prolonged protection against Hib infection [
76], and the mean concentration of anti-PRP IgG was as high as 27.4 µg/mL.
It is noteworthy that in Quimi-Hib®, different-sized oligomeric antigens were present, thus circumventing the problem of the elucidation of the optimal length of an oligomeric PRP antigen. The success of the development and application of the cost-effective vaccine Quimi-Hib® with a synthetic Hib-antigen inspired scientists to search for the shortest protective Hib-antigen, which is likely to be in the range of chain lengths that are obtained during the polycondensation of 39.
One of the novel and efficient approaches to the preparation of PRP-related oligomeric phosphoglycans suggested by Seeberger et al. [
69] involved the elongation of the chain by two PRP repeating units at once (
Scheme 6). The applied strategy effectively shortened the task of the oligomer assembly and raised the degree of convergence.
For the preparation of a selectively protected and activated bifunctional dimeric key unit, the universal precursor
44 was deallylated, and the resulting diol was regioselectively 4,4′-dimethoxytritylated at the primary hydroxyl group to obtain compound
45, which was then converted to H-phosphonate
46 by the action of PCl
3, Et
3N, and imidazole. For the preparation of alcohol
47, the universal precursor
44 was levulinoylated and deallylated. Alcohol
47 was reacted with H-phosphonate
46 in PivCl/Py to obtain the key dimeric precursor
48 with readily removable orthogonal levulinoyl and dimethoxytrityl protective groups. The de-4,4′-dimethoxytritylation of pseudo-tetrasaccharide
48 in the presence of trichloroacetic acid afforded alcohol
49 with a free terminal hydroxyl group in the ribitol residue. The delevulinoylation of universal precursor
48 by the action of hydrazinium acetate resulted in the formation of alcohol
50, which was converted into H-phosphonate
51. The combination of H-phosphonate
51 and primary alcohol
49, followed by oxidation, yielded a tetramer, which was, in turn, de-4,4′-dimethoxytritylated and reacted with H-phosphonate
46 for the elongation of the chain or with the 5-azidopentyl derivative
52 for chain termination. Using this elegant strategy, PRP-related tetramer (85%), hexamer (85%), octamer (83%), and decamer (80%) were prepared, which were subjected to sequential detritylation, the attachment of a 5-azidopentyl spacer under the action of H-phosphonate
52, the transformation of the azido group into an amino group, and total deprotection to obtain the spacer-armed oligomers
12–
15 (
Figure 4). After activation, oligomers
12–
15 were reacted with TT to obtain conjugates
53–
56 [
69].
The antigenicity of oligomers
11–
15 (
Figure 4) was examined using a glycan microarray. Compounds
11–
15 were immobilized on a
N-hydroxysuccinimide hydrogel surface on glass slides and reacted with polyclonal anti-
Hib hyperimmune rabbit sera or standard human sera, which were mixed with different concentrations of the WHO PRP standard as an inhibitor. The subsequent addition of a fluorescent secondary antibody revealed the dependence of the fluorescence intensity on the PRP concentration, thus proving the presence of antibodies specific to the synthetic PRP-oligomers
11–
15. Hexamer
13, octamer
14, and decamer
15 interacted with standard human sera in a similar way, binding with tetramer
12 was weaker, and in the case of dimer
11, the adsorption of the antibodies was even less effective. In contrast, rabbit hyperimmune sera interacted with all five oligomers equally well [
69].
The immunogenicity of conjugates
53–
56 was studied using an animal model. Rabbits were immunized with these conjugates at a dose of 5 μg of an oligomer without an adjuvant, and the positive control group received ActHib
® (PRP-TT). Bacterial PRP was used as a coating antigen. Immunization with conjugates
53 and
55, based on tetramer
12 and octamer
14 ligands, respectively, resulted in a high level of PRP-specific antibodies. Conjugates
54 and
56, which comprised hexamer
13 and decamer
15, induced substantially lower levels of PRP-specific antibodies [
69]. Therefore, in a series of conjugates equipped with antigens with an even number of repeating units, the tetramer was the most promising synthetic antigen candidate for the design of an anti-
Hib vaccine.
In a continuation of this work, Seeberger et al. synthesized [
77] a series of mimetics of synthetic PRP-related dimer, tetramer, hexamer, and octamer compounds (
11–
14), which comprised 2-deoxy-, 2-deoxy-2-fluoro-, 2-deoxy-2-(
N,
N-dimethyl)-carbamoyloxy-, and 2-O-methylated ribose residues in different combinations. This research was aimed at the preparation of analogs of PRP antigens with higher hydrolytic stability. The study of the conjugates of these antigens with the protein carrier CRM197 showed that the most promising are mimetics, in which ribose residues are methylated at the position O-2. Conjugates of these mimetics with CRM197 stimulated Hib-specific immune responses in both animals and humans, which confirms the possibility of their commercial use in anti-
Hib vaccines. The replacement of the 2-OH group, which promotes the hydrolysis of the phosphodiester bond, with a methoxy group allowed for a significant increase in the integrity of the antigen [
77].
3. Synthesis of Oligomers Structurally Related to Capsular Phosphoglycans of Hia, Hic, and Hif
Hib bacteria are the common and most dangerous serotype of the
H. influenzae species, which comprises at least five more encapsulated serotypes and numerous non-capsulated (non-typeable) variants. The introduction of anti-
Hib conjugate vaccines [
13,
15,
21,
23] in national immunization schedules in a number of European countries, North and South America countries, Australia, and the South African Republic significantly contributed to a reduction in invasive diseases caused by
Hib-infection [
78,
79]. At the same time, along with the expansion of anti-
Hib vaccination programs, the spread of other serotypes of
H. influenzae was reported [
80,
81,
82,
83,
84,
85,
86,
87,
88,
89]. As a consequence, after more than thirty years’ use of anti-Hib conjugate vaccines, an “antigenic shift” is observed, which is expressed in the increased incidence of invasive diseases, including meningitis, meningoencephalitis, and septicemia caused by encapsulated
H. influenzae serotypes different from b [
82,
90].
In European countries, the number of invasive diseases caused by
Hia is also growing. For example, in England, in 2022, Hia was responsible for 19% of all invasive disease cases caused by encapsulated
H. influenzae. Before 2017, only sporadic cases occurred [
80], and the contribution of
Hif and
Hie is also growing [
90]. In South Africa, the rise of
Hie is observed [
82]. In American countries,
H. influenzae diseases are often caused by
Hif [
82,
91]. Also, an increase in invasive diseases caused by
Hia [
85,
89,
92,
93] and
Hic [
91] was reported.
In the early 1990s, in the northern regions of Canada and Alaska, a routine anti-
Hib immunization schedule was introduced, and in the late 1990s, a growing number of cases of
Hia infection were registered. The majority of cases (more than 60%) were reported in children under five years of age. As a result, special research is being conducted in Canada aimed at the development of a vaccine for the prevention of diseases caused by
Hia [
84]. Considering that invasive diseases caused by encapsulated
H. influenzae serotypes different from serotype b remain relatively rare and are often localized in specific areas, corresponding vaccines will have limited use according to epidemic indications. In this situation, conjugate vaccines with synthetic antigens structurally related to
H. influenzae capsular glycans may be considered as a drug of choice.
Inspired by the success of the Quimi-Hib
® vaccine, which comprises synthetic oligomeric phosphoglycans structurally related to
Hib capsular phosphoglycan, researchers synthesized [
94,
95] the series of oligomers
57–
61 with one, two, three, four, and five repeating units of
Hia capsular phosphoglycans and 3-aminopropyl linkers (
Figure 6). In another recent study, the series of fragments of
Hia capsular phosphoglycans
62–
64 equipped with a
66 diethyleneglycol linker was obtained [
96] (
Figure 6). Syntheses of aminoalkyl glycosides related to the
Hic phosphoglycan (compounds
65 and
66) and related to the
Hif phosphoglycan (compounds
67 and
68) were performed by Oscarson et al. [
97].
In the two series of the spacer-armed oligomers
57–
61 [
94] and
62–
64 [
96] structurally related to
Hia capsular phosphoglycans, the linker is connected to the glycan antigen via a phosphodiester bridge. Compounds in both series were prepared using a convergent synthetic strategy with iterative elongation of the linear structure starting from the non-reducing end, and finally, the spacer was added (
Scheme 7 and
Scheme 8). The major difference between the syntheses of these two series is the type of universal, selectively protected bifunctional synthon. In the preparation of oligomers
58–
61 [
94], a monomer block with a phosphoramide group at C-4 (Glc) was used, and for compounds
63–
64 [
96], the synthetic pathway involved the use of a non-phosphorus monomer.
For the choice of the optimum strategy for the assembly of oligomers
57–
61 (
Scheme 7) [
95], two alternative ways were considered. The first strategy involved pseudo-disaccharide
69 with a free hydroxyl group in the ribitol residue, which was reacted with 2-cyanoethyl
N,
N,
N′,
N′-tetraisopropylphosphordiamidite (compound
70) in the presence of diispropylammonium tetrazolide to obtain the selectively protected phosphoramidite
71 as a universal precursor with a 95% yield. The following condensation of phosphoramidite
71 and glycoside
72 with a free hydroxyl group at C-4 (Glc), oxidation of a phosphite into a phosphodiester, and detritylation created the conjugation-ready dimer
73 with a 73% yield over three steps.
Another way of preparing phosphotriester
73 (
Scheme 7) employed phosphoramidite
74 with a pro-phosphodiester group located at C-4 (Glc). The sequential combination of phosphoramidite
74 with alcohol
69, oxidation, and detritylation resulted in the formation of the phosphodiester block
73 with a 79% yield over three steps [
94,
95]. Therefore, both assembly strategies were equally effective. Starting with phosphoramidite
74, protected pseudo-oligosaccharides
75–
77 were obtained and then converted to ligands
58–
61 by interaction with the pre-spacer phosphoramidite
78 and total deprotection. The authors pointed out that in comparison to the H-phosphonate approach, the phosphoramidite protocol was more efficient for the construction of longer phosphodiester-linked chains. Oligomers
57–
61 were
N-acylated with a di(
N-hydroxysuccinimidyl) adipate linker, and the activated esters were conjugated to CRM197. The resulting conjugates contained 13–25 copies of the oligomeric antigen per CRM197 unit. Rats were immunized with three doses, which contained 2 μg of the glycan antigen, and the IgG antibodies were analyzed in the sera using ELISA. Conjugates of trimer
59 and pentamer
61 with HSA were used as coating antigens. All the conjugates were immunogenic and induced similar levels of IgG antibodies, which recognized immobilized synthetic antigens. The researchers noted that the immunogenicity data for the CRM197-based conjugates of oligomers
57–
61 was independent of the length of the oligomeric antigen [
95].
Compounds
62–
64 [
96] were prepared (
Scheme 8) starting from the universal precursor pseudo-disaccharide
79. The acetylation of compound
79 resulted in acetate
80, which was phosphitylated to obtain key monomeric H-phosphonate
81 with a 97% yield. The condensation of H-phosphonate
81 with a pre-spacer
N-carbamoyl aminopropanol, as shown in
Scheme 8, created phosphodiester
82 (yield: 34%), which, after the removal of the protective groups, was transformed into the spacer-armed target monomer
62 (
Figure 6). To obtain dimer
83, alcohol
79 was condensed with H-phosphonate
81 with a 77% yield and then oxidized. Phosphodiester
83 was, in turn, converted into H-phosphonate
84. The interaction of H-phosphonate
84 with alcohol
79 produced phosphodiester
85, which was converted into H-phosphonate
86. Spacer-armed derivatives were obtained by the interaction of H-phosphonates
84 and
86 with
N-carbamoyl aminopropanol to obtain, after total deprotection, the spacer-armed dimer
63 and trimer
64, respectively (
Figure 6). It is interesting to note that the efficiency of condensation with N-protected aminopropanol increased in the series
81 >
84 >
86 with the rise in the number of phosphodiester fragments present in these H-phosphonates, whereas numerous experimental data suggest the opposite [
39].
In 2001, Oscarson et al. synthesized the amino spacer-armed oligomeric fragments of
Hic (compounds
65 and
66) and
Hif (compounds
67 and
68) phosphoglycans [
97] (
Figure 6), in which the repeating disaccharide units are linked via a phosphodiester bridge. Similar to the block syntheses of
Hib and
Hia fragments described above, the establishment of a phosphodiester linkage between selectively protected oligosaccharide blocks was used for the chain elongation. However, unlike
Hib and
Hia, in
Hic and
Hif capsular phosphoglycans, the anomeric carbons in hexoses are involved in the formation of a phosphodiester bridge. Therefore, the development of a synthetic strategy for the preparation of
Hic and
Hif fragments has to be developed with respect to the possibility of the formation of an unwanted anomer. At the step of chain assembly, it can be particularly difficult to provide stereocontrol in the formation of a C-1-O-P bond. Instability of the anomeric phosphodiester linkage also has to be considered, which makes it possible to use anomeric phosphodiesters as glycosyl donors in the glycosylation reaction. A convenient strategy for the preparation of the desired anomer includes the initial stereocontrolled glycosylation of a phosphodiester synthon, which provides an intermediate product with the target anomeric configuration, and the subsequent coupling of this compound in mild conditions for the prevention of anomerization. These conditions are met in the H-phosphonate protocol, as hexose hemiacetals retain the anomeric configuration, while they are transformed into H-phosphonates upon the action of triimidazolyl phosphine prepared in situ from PCl
3 and imidazole in the presence of Et
3N [
39].
For the preparation of the spacer-armed oligomers
65 and
66 (
Figure 6) [
97], which are structurally related to
Hic phosphoglycans, hemiacetal
87 with an axial hydroxyl group at C-1 was converted into the key α-H-phosphonate 88 (yield: 89%), which was condensed with the selectively protected alcohol
89 and oxidized with I
2 (
Scheme 9A) to provide phosphodiester
90 (yield: 71%). The desilylation of phosphodiester
90 produced alcohol
91, which was readily condensed with α-H-phosphonate
88, and the phosphite group was oxidized to produce phosphate
92 in a moderate yield (36%). After total deprotection, the reduction of the azido group, and the N-acetylation of compounds
91 and
92, the target spacer-armed phosphooligosaccharides
65 and
66 were obtained.
Analogously, the favorable α-configuration of the GalNAc hemiacetal residue in disaccharide
93 (
Scheme 9B) paved the way to the preparation of the spacer-armed phosphooligosaccharides
67 and
68 related to
Hif phosphoglycans [
97]. Upon action with PCl
3 and imidazole, hemiacetal
93 was transformed into α-H-phosphonate
94 with a retention of configuration (yield: 87%). The condensation of α-H-phosphonate
94 with disaccharide
95 and the subsequent oxidation resulted in the efficient formation of phosphodiester
96 (yield: 81%), which was desilylated to obtain alcohol
97. Similar to the condensation of compounds
91 and
88, the interaction of
97 and
94 was less efficient, and phosphodiester
98 was obtained with a 37% yield. Most likely, low yield in this reaction is associated with the high lability of the phosphodiester group in acceptors
91 and
97 in the conditions of oxidation with I
2 [
39]. The authors [
97] observed the formation of a significant amount (up to 40%) of 3′-O-phosphate
99, which indicates the lability of a glycosyl-O-phosphodiester tether under oxidation conditions. The low yields of attachment of the second glycosyl-phosphodiester residue for variants A and B, shown in
Scheme 9, suggest that the stability of a phosphodiester bond is largely determined by the condensation and oxidation conditions and is less dependent on the structure of disaccharide residues. The target spacer-armed phosphooligosaccharides
67 and
68 were obtained after complete deblocking of the compounds and the reduction of the nitrophenyl residue into an aniline residue.
In view of the development of an anti-Hia vaccine, two aspects have to be addressed. Similar immunogenic properties of synthetic Hia antigens from monomer to pentamer can be connected with the low stability of phosphodiester linkages in aqueous solutions. In this case, the design of a vaccine may require a mimetic structure for the antigen. Also, it is advisable to use a phosphoramidite-based strategy for the preparation of phosphooligosaccharide antigens for industrial production, as the considered examples demonstrate the advantage of the phosphoramidite-based method over the H-phosphonate procedure. Meanwhile, anti-Hic and anti-Hif conjugate vaccines are not considered to be of high importance in contemporary glycoscience, as researchers have not referred to this topic for more than 20 years.
4. Synthesis of Phosphooligomers Related to Capsular Phosphoglycan of MenA
In 2010, a conjugate polysaccharide meningococcal monovalent vaccine, MenAfriVac
® (MenA-TT), was licensed [
98,
99]. In contrast to meningococcal polysaccharide vaccines, the conjugate preparation was found to be highly effective. For example, the results of mass vaccination campaigns in African meningitis belt countries that were carried out in 2010–2015 showed more than a 99% decrease in the incidence of
MenA-associated meningitis [
100]. Today,
MenA phosphoglycan is a component of a number of polyvalent conjugate meningococcal vaccines, including Menactra
® (A-meningococcal component MenA-diphtheria toxoid), Menveo
® (A-meningococcal component MenA-CRM197), and Nimerix
® (A-meningococcal component MenA-TT) [
14].
Polymer chains of
MenA phosphoglycan are highly labile in aqueous media [
63]. This intrinsic property of
MenA phosphoglycan creates the need for the cold-chain transport of conjugate anti-
MenA vaccines [
101], which significantly increases the cost per dose and, in some cases, is an insurmountable obstacle to the use of this type of preparation [
102]. Also, this feature imposes additional requirements on the production of both monovalent vaccines and polyvalent vaccines in a convenient liquid form. In this regard, considerable research efforts have been directed towards the synthesis of oligomeric antigens structurally related to
MenA CPS fragments or corresponding mimetics with a view to preparing commercial conjugate vaccines. The use of anti-
MenA conjugate preparations with a synthetic oligoside antigen, the structure of which fully corresponds to
MenA CPS fragments, could allow the monitoring of the structural integrity of this preparation during storage and transportation, and the design of an antigen using ManNAc phosphate mimetics obtained by replacing the oxygen atom with the methylene group in the pyranose ring with (carba-analogs) or by replacing the anomeric oxygen atom with a methylene group (C-phosphonates) will make the antigen structure resistant to hydrolysis.
It is important to emphasize that in
MenA bacterial phosphoglycans, ~80% of 3-OH groups and ~10% of 4-OH groups in ManNAc residues are acetylated [
103]. A study of phosphoglycan structures present on the surface of a living
MenA bacterial cell [
104], which was conducted using high-resolution magic-angle spinning, showed the presence of acetyl substituents at 50–60% of 3-OH groups and 25–30% of 4-OH groups. The acetylation of
MenA phosphoglycans is known to be an important antigenicity factor [
105,
106].
Spacer-armed synthetic antigens structurally related to the
MenA capsular phosphoglycan and the corresponding phosphono- and carba-analogs are attractive synthetic compounds considering their potential use as ligands in marketed meningococcal vaccines. Since the first time a phosphodiester bridge was arranged between two αManNAc residues (
Figure 7, compounds
100 and
101) in 1993 by the Shibaev group [
107], a vast number of
MenA-related mono-, di-, and oligosaccharides
102–
109 (
Figure 7) have been synthesized [
108,
109,
110,
111,
112]. However, the preparation of αManNAc anomeric phosphodiesters remains a challenge, as these compounds comprise a highly labile linkage between C-1 and O-1 of αManNAc, which is, in addition, destabilized by the presence of the NHAc group at C-2 of αManNAc. As a result, researchers have focused significant efforts on the design of hydrolytically stable phosphono-mimetics (compounds
111–
115) [
113,
114,
115] and carba-mimetics (compounds
116–
127) [
116,
117,
118]. However, new challenges are emerging, which are associated with the establishment of C-C bonds and the stereoselective formation of new chiral centers. The majority of publications that describe the preparation of oligomeric antigens related to
MenA phosphoglycans concern nonacetylated molecules, whereas bacterial
MenA phosphoglycans comprise the acetyl groups at O-3/O-4 (
Figure 6), which are important for the antigenic properties of the polymer [
105,
106]. Additionally, a number of synthetic
MenA-related antigens were reported (compounds
107–
109 and
127), which are totally or partially acetylated at O-3 of αManNAc, with a view to bringing their antigenic properties closer to those of the bacterial antigen. Synthetic fragments of
MenA phosphoglycans with 3,4-di-O-acetylated αGManNAc residues are not considered in this review.
As the capsular MenA glycan is an αManNAc(1→(-PO
3)→6) polymer, the H-phosphonate method is applicable, provided that 1-OH has the axial orientation in the selectively protected H-phosphonate ManNAc or 2-deoxy-2-azido mannose precursor, as described above for oligomers related to
Hic and
Hif phosphoglycans (
Scheme 9). A number of convenient and efficient procedures have been developed for the preparation of this type of compound. The common protocol suggests phosphitylation with tri(1-imidazolyl) phosphine (
Figure 8, compound
128), which is obtained in situ by the interaction of PCl
3 and imidazole in the presence of Et
3N and salicylchlorophosphite
129 (2-chloro-4H-1,3,2-benzodioxaphosphin-4-one;
Figure 8). Alternatively, within a phosphoramidite protocol, chloroanhydrides
130 and
131 (
Figure 8) were especially designed for automated synthesis to produce stable phosphoramidites as synthons of α-mannosamine phosphodiesters [
108].
For the PCl
3/imidazole protocol, the key prerequisite for the preparation of ManNAc or 2-deoxy-2-azido mannose α-H-phosphonates is the α-configuration of the 1-OH group in the corresponding selectively protected hemiacetal. This feature imposes certain restrictions on the use of the H-phosphonate protocol. Hemiacetal
132 (
Table 1) with a 5:1 ratio of α/β anomers was quantitatively converted into the 5:1 mixture of α- and β-isomeric H-phosphonate
133 upon the action of PCl
3 and imidazole in acetonitrile at 0 °C, followed by hydrolysis with an aqueous solution of Et
3NH∙HCO
3 at 20 °C (
Table 1, entry 1) [
107]. In a similar way, phosphitylation of the mixture of hemiacetal
134 with a 3.5:1 ratio of α/β anomers in analogous conditions proceeded with the retention of the configuration of the anomeric center and resulted in the formation of the mixture of H-phosphonate
135, with the α:β ratio 3.5:1 and an 88% yield (
Table 1, entry 2) [
110]. In similar conditions, the interaction of the α-anomer of hemiacetal
136 (
Table 1, entry 3) [
111] afforded α-H-phosphonate
137 with a 97% yield, and hemiacetal
138 with a dibenzyl phosphate group at C-6 was converted into α-H-phosphonate
139 (
Table 1, entry 4) [
111]. The phosphitylation of hemiacetal
140 (
Table 1) with an α-configuration of the anomeric center upon the action of (PhO)
2P(O)H in Py, followed by hydrolysis, resulted in a mixture of α- and β-isomer
141 in a ratio of 93:3 (
Table 1, entry 5) [
112]. Similarly, the phosphitylation of α-hemiacetal
142 in these conditions formed α-H-phosphonate
143 with a 78% yield (
Table 1, entry 6) [
109]. Anomeric mixtures of hemiacetals, which contain considerable quantities of β-isomers, can be efficiently converted into α-H-phosphonates with chlorophosphite
129 (
Figure 8) [
110,
119]. For example, the phosphitylation of the anomeric mixture
144, with a ratio of α- and β-anomers of 4.5:1, was transformed into α-H-phosphonate
145 by the action of chlorophosphite
129 in a mixture of dioxane and triethylamine with an 88% yield (
Table 1, entry 8) [
110]. Similarly, in these conditions, hemiacetals
142 and
146 were converted into the corresponding α-H-phosphonates
143 and
147 at 91 and 90% yields, respectively (
Table 1, entries 7 and 9) [
110,
119]. Unlike H-phosphonates, phosphoramidites are rarely used as synthetic blocks for the establishment of phosphodiester bonds between α-ManNAc residues because of their utmost lability. Two successful examples of ManNAc phosphoramidites are compounds
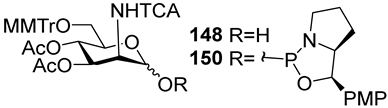
149 and
150, which are stabilized by rigid oxazaphospholidine substituents. These compounds were obtained from the anomeric mixture of hemiacetal
148 by the action of tricyclic chlorophosphoroamidites
130 and
131 at yields of 36 and 39% (
Table 1, entries 10 and 11), respectively [
108]. The generation of α-H-phosphonates and the anomerization of the α/β mixtures of hemiacetals or the stereoselective cleavage of the unwanted β-isomer in the presence of H
3PO
3 [
107] or AgOTf [
110] were ineffective.
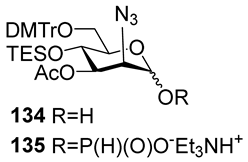
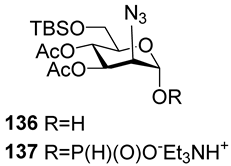
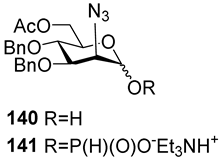
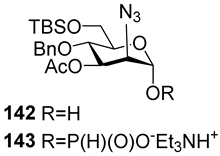
The majority of syntheses of the spacer-armed oligomers related to
MenA phosphoglycans (
Figure 7) were performed using a straightforward and efficient protocol suggested by Van Boom [
120], which constitutes the condensation of H-phosphonates with alcohols, with the subsequent oxidation of phosphites into phosphodiesters. In the first step, the dissolved alcohol is activated by the addition of sterically hindered chloroanhydride, e.g., PivCl, followed by the addition of H-phosphonate in Py and stirring for 5–30 min. In the second step, the intermediate products are subjected to oxidation with a 0.5 M solution of I
2 in a Py:H
2O mixture within the temperature interval from −40 °C to 0 °C for 30 min. The first syntheses of the non-acetylated fragments of
MenA phosphoglycans as methyl glycoside
100 and nitrophenyl ether
101 were accomplished by the Shibaev group [
107] using the H-phosphonate protocol (
Figure 7). Then, monomers
102 and
103, dimer
104, and trimer
105 (
Figure 7), related to
MenA phosphoglycans and equipped with a spacer carrying a primary amino group for conjugation with protein carriers, were synthesized by Pozsgay et al. [
112]. The condensation of H-phosphonate
141 with alcohol
151, with subsequent oxidation, resulted in the formation of phosphodiester
152 with a yield of 95% over two steps (
Scheme 10). The successive reduction of an azido group into the amino group and N-acetylation yielded mannosaminyl phosphodiester
153, which was converted into alcohol
154 by chemoselective 6-O′-deacetylation. The condensation of H-phosphonate
141 and alcohol
154 was less effective (a yield of 74% over two steps) in connection with the destruction of the phosphodiester bond in the reaction conditions. The authors noted that attempts at further elongation of the chain were unsuccessful. Phosphoester
102 and phosphodiesters
103,
104, and
105 were converted into conjugates with HSA. The antigenic properties of the conjugates were confirmed by a double immunodiffusion assay with the
MenA phosphoglycan as a positive reference and chemically modified HSA as a negative reference, and anti-
MenA horse serum [
112].
In 2005, the non-acetylated spacer-armed
MenA phosphoglycan-related oligomer
105 (
Figure 7), which comprises three ManNAc residues connected by phosphodiester bridges and trimer
106 (
Figure 7), composed of three non-acetylated
MenA repeating units, was synthesized by the Oscarson group [
111]. In contrast to the abovementioned synthetic strategy designed by the Pozsgay group, which suggested the reduction of the azido group and N-acetylation after each chain elongation step (
Scheme 10), 2-azido glycoside
158 was used as a starting unit for further chain elongation, and 2-azido H-phosphonate
137 (
Table 1, entry 3) was applied as a repeating unit synthon without intermediate N
3→NHAc transformation (
Scheme 11). The condensation of H-phosphonate
137 and acceptor
158 with a free hydroxyl group at C-6, followed by oxidation, resulted in the efficient formation of phosphodiester
159 (yield: 96%), which was then 6′-O-desilylated to create a new active site in acceptor
160. However, the attachment of the next monomer unit
137 to phosphodiester
160 was less effective and, after oxidation, formed the corresponding phosphodiester
161 with a 62% yield. With a view to synthesizing trimer
106 with a phosphoester substituent at C-6, acceptor
160 was reacted with phosphodiester
139 (
Table 1, entry 4) and a dibenzylphosphate group at C-6 to obtain the trimeric precursor
162 with a 59% yield. A reduction in all azido groups, total N-acetylation, and deprotection in compounds
161 and
162 afforded the spacer-armed conjugation-ready antigens
105 and
106 [
111].
3-O-Acetylated dimer
107 and trimer
108 related to
MenA capsular phosphoglycans were synthesized as methyl glycosides (
Figure 7) [
110]. The phosphitylation of acceptors
163,
164, and
165, which comprised one, two, or three 2-deoxy-2-azido mannose residues with H-phosphonate
145, followed by oxidation and detritylation, afforded phosphodiesters
164,
165, and
166 with 88%, 74%, and 68% yields, respectively. The alcohols obtained were subjected to phosphitylation with H-phosphonate
167 and subsequent oxidation (
Scheme 12). The connection of H-phosphonate
167 to the 6-OH of the terminal monosaccharide was effective only for the shorter alcohols
164 and
165 (88% over two steps) and resulted in the preparation of protected di- and trimers
168 and
169 (
Scheme 12). The transformation of azido groups into amino groups and the total N-acetylation and debenzylation of compounds
168 and
169 formed the spacer-armed dimer
107 and trimer
108, which carry acetyl groups at O-3 of each ManNAc residue, as they do in bacterial
MenA phosphoglycans. In the homologous series
164–
166, attempts to phosphitylate the longest alcohol
166 with H-phosphonate
145 in order to obtain a tetramer or with the pre-spacer H-phosphonate
167 were not successful, in contrast to compounds
164 and
165.
In general, the examples of the synthesis of oligomers related to
MenA capsular phosphoglycans discussed above are in line with the tendency outlined in an earlier review [
39], which discussed the decrease in the efficiency of phosphitylation with H-phosphonates with the increase in the number of already formed phosphodiester bonds and suggested the lability of phosphodiester fragments in the conditions of the H-phosphonate protocol. However, in 2017, a group of Indian researchers synthesized an aminohexyl glycoside of oligomer
109 related to capsular phosphoglycans, which contained four 3-O-acetylated ManNAc residues, using a linear, synthetic strategy (
Scheme 13) [
109]. In a sequence of phosphitylation steps, H-phosphonate
143 was used as a universal monomer (
Table 1, entry 6). The establishment of the first phosphodiester linkage between alcohol
170 and H-phosphonate
143 afforded compound
171 (yield: 92%), which was, in turn, desilylated to form acceptor
172. The phosphitylation of alcohol
172 with H-phosphonate
143, followed by oxidation, resulted in the formation of compound
173 with two phosphodiester bridges (yield: 89%). The desilylation of compound
173 formed acceptor
174, which was phosphitylated with H-phosphonate
143 and, after oxidation, compound
175 with three phosphodiester bridges was obtained with a 76% yield.
After the transformation of azido groups into NHAc groups and the desilylation, debenzylation, and deprotection of the spacer amino group, the spacer-armed ligand
109 was conjugated to TT. The antigenic properties of oligomer
109 were evaluated in a competitive ELISA experiment. Both oligomer
109 and its conjugate with TT in the concentration range 12.5–400 μg glycan/mL were found to neutralize anti-MenA rabbit antiserum and inhibit the binding of antibodies to the bacterial
MenA phosphoglycan used as a coating antigen. In comparison to the conjugate, oligomer
109 showed lower inhibition [
109]. It can be concluded that the improvement of synthetic protocols made it possible to obtain oligomeric antigens related to
MenA capsular phosphoglycans, which contain up to four ManNAc residues. However, for the efficient application of antigens of this type in immunodiagnostic tests and vaccine production, the hydrolytic lability issue has to be addressed.
5. Synthesis of Glycomimetics of MenA Capsular Phosphoglycans
Today, all anti-MenA conjugate vaccines include a lyophilized bacterial MenA component except for the fully liquid commercial vaccine preparation Menactra®. The presence of the MenA antigen in the dissolved form substantially shortens the shelf life for Menactra® to 18 months at 2–8 °C compared to the 4-year shelf life of MenQuadfi® and the 3-year shelf life of Menjugate® and MenAfriVac® in these conditions. With a view to preparing hydrolytically stable MenA antigens, a number of oligomeric analogs were designed, in which NHAc groups were replaced with trichloroacetamide groups as they are not likely to undergo transformation into oxazolines. Another type of mimetics is compounds with the isosteric replacement of one of the hemiacetal oxygens with a methylene group (phosphono- and carba-analogs).
A comparative conformational analysis of a hexapyranose ring in 2-deoxy-2-acetamido mannohexapyranosyl phosphate
176 and its phosphono-analog
177 and carba-analog
178 (
Figure 9) in a study of conformations in combination with NMR experiments showed that for compound
176, 4C1 was almost the only populated conformer, whereas for phosphonate
177, the proportion of pyranose ring conformers other than 4C1 was 4%, and for the carba-analog
178, this proportion rose to 7% [
121]. The most populated 4C1 conformer for compound
178 was confirmed by quantum mechanics and molecular dynamics calculations [
122]. The molecular dynamics calculations performed for a decamer of a
MenA capsular phosphoglycan repeating unit and its carba-analog showed that, despite a number of conformational and dynamic variations, the carba-mimetics of the fragments of
MenA capsular phosphoglycans can be considered candidate antigens for the construction of anti-
MenA vaccine preparations [
123].
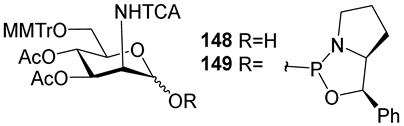
As mentioned previously, the axial NHAc group at C-2 of the ManNAc residue contributes largely to the lability of
MenA capsular phosphoglycans and their fragments via neighboring group participation. One of the possible ways to circumvent this obstacle is the replacement [
55] of the acetamide group at C-2 with the trichloroacetamide group, which is not likely to form oxazolines. To study the possibility of the preparation of the N-trichloroacetamide mimetics of ManNHAc, oxazaphospholidines
149 and
150 (
Table 1) were obtained in a solid-phase synthesis. Bis(2-hydroxyethyl) hydroquinone
179 (
Scheme 14) [
108] was immobilized on a glass support and phosphitylated with phosphoramidites
149 and
150. The phosphitylation of hydroquinone
179 with phosphoramidite
149 in the presence of 1-(cyanomethyl) pyrrolidinium trifluoromethanesulfonate (compound
180) resulted in the formation of phosphite
181, which carried a 2-phenylpyrrolydine residue as a result of the cycle opening. Phosphotriester
181 was subjected to mild oxidation with DCSO (compound
182) and desilylated with TFA/TES to produce phosphotriester 183, with a free 6-OH group for the following elongation of the oligomeric chain. Finally, the cleavage of the 2-phenylpyrrolydine ether was fulfilled in the presence of a base, followed by deacetylation and removal of the solid support by the action of MeONa/MeOH.
However, researchers faced considerable difficulties at the step of the cleavage of the 2-phenylpyrrolydine ether. After the support was removed, analysis of the products showed that the use of DBU for the cleavage of the ether did not provide target phosphodiester 110, and the identified products 184–186 did not contain a ManNAc residue. When the weaker bases of Et3N or 2,6-lutidine were used as basic catalysts, the target product 110 was obtained with a moderate yield (17%) and low conversion.
In order to replace the step of the basic hydrolysis of the O(P)-protective group for acidic hydrolysis, phosphoramidite
150 with a p-MeO-Ph residue in the place of the Ph residue, as in compound
149, was used. Phosphoramidite
150 was attached to a solid support, and phosphite
187 was oxidized with DCSO (compound
182) to yield phosphotriester
188, followed by the simultaneous acid hydrolysis of the trityl group and removal of the 2-(p-methoxybenzyl) pyrrolidine moiety. However, after deacetylation and removal of the solid support with 50 mM NaOMe/MeOH, the unwanted products
186 and
187 were detected. Without taking into account inefficient deprotection, this method provided the preparation of tetramer
189, which was O-acetylated and N-trichloroacetylated. The authors noted that the developed method in the current state is not suitable for the synthesis of oligomers [
108].
In 2005, the preparation of the first isosteric hydrolysis-resistant phosphono-analog
111 structurally related to
MenA phosphoglycans was published by Lay et al. [
115]. The interaction of iodide
190, in which the iodomethylene group is arranged axially, with trimethylphosphite (
Scheme 15) afforded phosphonodiester
191, which was further converted to ester
192 by the action of triethylamine and thiophenol.
Similarly, 6-O-acetylated phosphonodiester
193 was converted to the corresponding ester
194 by partial hydrolysis. Mitsunobu reaction conditions were used for the condensation of ester
192 with the selectively protected ManNAc
195, which formed phosphonodiester
196 (yield: 97%) [
113]. Partial hydrolysis of phosphonodiester
196 into phosphonoester
197 was also efficient. 6′-O-Acetylated phosphonodiester
198 was synthesized by the condensation of
195 with phosphonoester
194, with a 90% yield. By deacetylation, phosphonodiester 198 was converted into alcohol
199 and condensed with phosphonoester
194 to form compound
200 with two phosphonodiester bridges and an 83% yield. Chemoselective hydrolysis of the methyl phosphonate moieties in compound
200 formed phosphonoester
201. Total deprotection of phosphonoester
197 with two ManNAc residues and
201 with three ManNAc residues resulted in phosphono-analogs
111 and
112 (
Figure 6) [
113].
A similar strategy in combination with Mitsunobu reaction modification, where Ph
3P is replaced with tris(4-chlorophenyl) phosphine, was used for the synthesis of the series of phosphono-analogs
113–
115 of
MenA phosphoglycans (
Scheme 16) [
114]. The 6″-O-deacetylation of phosphonodiester
202 resulted in alcohol
203, which was then reacted with universal monosaccharide block
194 in modified Mitsunobu conditions to obtain the phosphono-analog
204 of
MenA phosphoglycans with three phosphonodiester-bridged fragments (yield: 87%). Partial hydrolysis of diester moieties afforded compound
205 (yield: 75%), and after total deprotection, phosphono-analog
115 was obtained with three pseudo-ManNAc residues.
The antigenic properties of ligands
111 and
112 [
113] were studied in competitive ELISA experiments with anti-
MenA human antisera. Native
MenA phosphoglycans were used as a coating antigen and positive control, and
MenY phosphoglycans were used as a negative control. For both ligands, the EC50 was about 10
−3 mg/mL, which is three orders higher than the EC50 of 6.6 × 10
−6 mg/mL for
MenA phosphoglycans.
Aminopropyl glycosides
111 and
112 and 3-aminopropyl β-D-ManNAc were transformed into conjugates with HSA using the squarate procedure [
124] (
Figure 10). One series consisted of conjugates
206–
208 (
Figure 10) with the maximum saccharide/protein molar ratio, and another series of conjugates was composed of compounds
209–
211 (
Figure 10) with the saccharide/protein molar ratio being half of the value achieved for conjugates
206–
208. Competitive ELISA experiments with the mouse polyclonal anti-
MenA antisera and
MenA capsular phosphoglycans as a coating antigen showed that at an inhibitor concentration of 1 mg/mL, inhibition using the fully loaded conjugate
206 was 55%. For conjugates
207 and
208, it reached 65%, whereas
MenA capsular phosphoglycans provided 100% inhibition. Inhibition with the half-loaded conjugates
209–
211 was 5–15% lower than for the fully loaded conjugates with the same antigen type [
124]. Conjugates
206–
211 were used for the immunization of mice at a dose of 2 μg/mouse and efficiently evoked IgG antibodies, which is a reliable marker of the induction of thymus-dependent immune responses. Quantitative elucidation of the level of anti-
MenA IgG antibodies developed against conjugates
206–
211 showed that immunization with the half-loaded conjugates
209–
211 was more efficient compared to the fully loaded conjugates
206–
208. It is important to note that the level of induced anti-
MenA antibodies was similar for the half-loaded conjugates
209–
211, regardless of the length of the pseudo-oligosaccharide antigen. The authors concluded that this result indicated the antibodies’ recognition of the ManNAc epitope [
124].
In contrast to C-phosphono mimetics, in which the methylene group stands in the place of anomeric oxygen, in carba-analogs, the decrease in electrophilicity of the carbonyl carbon, along with the increase in the stability of the phosphodiester linkage, is achieved by the replacement of the ring hemiacetal oxygen of ManNAc with a methylene group [
125].
Synthesis of the series of carba-analogs
116–
118 (
Figure 7) of aminopropyl glycosides of a monomer, a dimer, and a trimer of
MenA capsular phosphoglycans, and the advanced series of carba-analogs
119–
126, from monomers to octamers, as aminohexyl glycosides, was performed by the Lay group [
116,
117,
118]. Carba-analogs
116–
118 [
118] were obtained by the sequential elongation of a pseudo-oligosaccharide chain, starting from the spacer-equipped monomer (
Scheme 17). For the preparation of monomer
116, the universal orthogonally protected precursor
212 was transformed into alcohol
213, which was phosphitylated with H-phosphonate
214 and oxidized to obtain the spacer-armed phosphodiester
215 with an 81% yield.
For the preparation of oligomers
117 and
118, the universal precursor
212 was desilylated and converted into H-phosphonate
216, which was used as a universal monomer block for chain elongation. The interaction of H-phosphonate
216 with alcohol
213 and the subsequent oxidation formed phosphodiester
217 (yield: 82%), which was deacetylated to obtain alcohol
218. The condensation of alcohol
218 with H-phosphonate
216 resulted in pseudo-trisaccharide
219 (yield: 81%), which was deacetylated to produce alcohol
220. Finally, pre-spacer
214 was introduced into alcohols
218 and
220 to obtain the spacer-armed, protected dimers
221 and
222 with yields of 85% and 57%, respectively. The high efficiency of the phosphitylation of alcohol
218, which already includes a phosphodiester bridge with H-phosphonates
214 and
216, evidences the increase in stability of the phosphodiester linkage in protected carba-analogs compared to phosphodiester-linked oligosaccharides. However, the phosphitylation of
220, which already comprised two phosphodiester moieties, was less efficient, indicating the limitations of the application of the H-phosphonate procedure to the linear synthesis of longer oligomers. The target carba-analogs
116–
118 of monomers, dimers, and trimers related to
MenA phosphoglycans were obtained after the total deprotection of compounds
215,
221, and
222 [
118].
For the synthesis of the advanced series of carba-analogs
119–
126, from monomers to octamers, the Lay group used the strategy of a step-by-step chain extension using the spacer-equipped monomer as a starting compound and phosphitylation with a selectively protected monomeric phosphoramidite as the key step (
Scheme 18) [
116]. Alcohol
223 was converted into the universal phosphoramidite block
224 under the action of chlorophosphoroamidite
24. The interaction of alcohol
223 with the phosphoramidite pre-spacer
225 in the presence of DCI, followed by oxidation with DCSO and detritylation, yielded phosphotriester
226 with a free hydroxyl group for further phosphitylation, which was used as a starting compound for chain elongation. The sequential execution of phosphitylation with the universal monomer block
224, oxidation, and detritylation afforded the protected compounds
227–
233 with excellent yields. After total deblocking, the spacer-armed carba-analogs
119–
126, structurally related to MenA capsular phosphoglycans, were obtained. For better resemblance of the octamer antigen to the natural structure, octamer
126 was N-Boc-protected, subjected to random monoacetylation, and N-deblocked, and the mixture of oligoacetates
234 was obtained, which contained a certain amount of oligomer
127 related to MenA capsular phosphoglycans [
116].
The stability of compound
126 was studied using an accelerated stability test and fragments of natural and deacetylated
MenA phosphoglycans, with avDP15 as a reference compound. During four weeks of keeping these samples in buffered 5 mM sodium acetate at pH 7 and 37 °C, a drastic decrease in chain length for the deacetylated bacterial oligosaccharide was observed, and the natural sample with acetyl groups was subjected to partial depolymerization, and for compound
126, no trace of decomposition was detected [
116].
Competitive ELISA experiments with polyclonal immune mouse anti-
MenA sera and
MenA capsular phosphoglycans as a coating antigen were used for the assessment of the antigenic properties of the mono-, di-, and trimeric pseudo-oligosaccharides
116–
118 [
118].
MenA capsular phosphoglycans and their derivatives
MenA avDP15 and
MenA avDP3 were used as positive controls. The highest inhibition rate was found for
MenA capsular phosphoglycans and
MenA avDP15, with an IC50 of 5.15 × 10
−6 and 4.3 × 10
−3 mM, respectively. For the carba-dimer
117, the IC50 was about 0.16–0.091 mM, which is less than the IC50 (4.3 × 10
−2) found for
MenA avDP3. For the carba-monomer
116 and carba-trimer
118, only 30% inhibition was achieved [
118]. The synthetic carba-analogs
116–
118 were converted into conjugates with CRM197 (compounds
235–
237,
Figure 11) and HSA (compounds
238–
240,
Figure 11). Mice were immunized three times with conjugates
235–
237,
241, and
242, which comprised
MenA avDP5 and
MenA avDP15 antigens, respectively, at a dose of 2 μg of antigen per mouse. The induced antibodies were analyzed in ELISA experiments using the HSA-based conjugates
238–
240 as coating antigens and
MenA capsular phosphoglycans. The immunogenicity of conjugates
235,
236, and
237 was evaluated using the HSA-conjugated coating antigens
238,
239, and
240, respectively. In this experiment, the immunogenicity of conjugate
238, which comprised the monomeric antigen
116, was somewhat lower compared to conjugates
239 and
240 with dimeric and trimeric antigens, respectively. In ELISA experiments with
MenA capsular phosphoglycans as a coating antigen, it was found that only conjugate
240 with a trimeric antigen evoked antibodies specific to the natural phosphoglycans, and conjugates
238 and
239 did not induce anti-
MenA immunity. However, the level of IgG antibodies induced by immunization with
240 was 2–3 orders lower than that induced by conjugates
241 and
242, which contained fragments of natural phosphoglycans with intact acetyl substituents. These data are in line with the results of an in vitro bactericidal assay, which showed the intensity of the complement-mediated lysis of
N. meningitidis [
118].
The carbocyclic hexamer
124 and the non-acetylated and acetylated octamers
126 and
234 were conjugated to CRM197 to form compounds
243,
244, and
245, respectively (
Figure 11). Conjugates
243–
245 were used for the immunization of mice, and another group of mice was immunized with a conjugate composed of partially depolymerized
MenA phosphoglycans as a positive control. ELISA analysis of IgG antibodies on plates coated with
MenA capsular phosphoglycans showed that immunization with conjugate
245, based on the acetylated carba-octamer, efficiently induced anti-
MenA immunity. For this conjugate, the IgG titers were comparable to the titers registered for conjugate
242 based on fragmented bacterial antigens, whereas the titers measured in animals immunized with conjugates
243 and
244, which carried non-acetylated synthetic antigens, were two orders lower [
116].
The urgency of the development of anti-MenA conjugate vaccines is evidenced by the considerable efforts of several research groups aimed toward antigenic and non-hydrolyzable synthetic phosphooligosaccharides. However, the antigenicity studies of phosphono- and carba-mimetics show that these oligomers do not induce anti-MenA immunity and, therefore, are unable to prevent MenA infection. Another obstacle in the way of the design of the semisynthetic anti-MenA vaccine is the importance of acetyl groups at the O3/O4 of ManNAc for the protective properties of the vaccines. The introduction of these substituents increases production costs.
To date, synthetic MenA antigens cannot compete with multiple conjugate anti-MenA vaccine preparations with a bacterial antigen. However, the repeating unit of MenA phosphoglycans is a simple monosaccharide, and it is a suitable target for production on automated synthesis platforms. We expect that the development of a stable mimetic with high antigenicity, in combination with automated synthesis technology, will help create an affordable anti-MenA semisynthetic conjugate vaccine.
6. Synthesis of Phosphooligomers Related to MenX Capsular Phosphoglycans
High anti-
MenA vaccination coverage in the African meningitis belt in 2010–2015 resulted in a significant reduction of
MenA-associated invasive diseases [
100,
126]. At the same time, outbreaks of invasive diseases caused by
MenX were registered in Sub-Saharan Africa [
127,
128]. The clinical and epidemiological characteristics of diseases associated with
MenX are similar to those of
MenA. Over 90% of cases are registered during the dry season, the median age of patients is 9.2 years, and the mortality rate is 11.9% [
129]. As a result, the development of a conjugated anti-
MenX vaccine became an urgent issue [
130].
For the elucidation of the minimal immunogenic epitope of
MenX capsular phosphoglycans, a bactericidal mAb,
MenX.01, was obtained, which induced bactericidal killing at a physiological concentration of 1 μg/mL in the first step. The interaction of this antibody with fragments of the natural
MenX capsular phosphoglycans of different lengths showed that the minimal immunogenic epitope comprises 5–6 repeating units [
131].
With a view to developing a conjugated anti-
MenX glycovaccine with a fully synthetic glycan epitope, phosphodiester
246 [
132] and the series of spacer-armed mono-, di-, and trimeric phosphoglycans
247–
249 [
129,
133], were synthesized. In addition, a mixture of oligomeric phosphoglycan
250, with an average length of 12 repeating units, was prepared using a combined chemo-enzymatic protocol, and pseudo-tetrasaccharide
251 was used as a glycoside with an aminohexyl spacer attached as an aglycon (
Figure 12) [
134].
The preparation of the synthetic oligosaccharide fragments of MenX capsular phosphoglycans was most commonly performed using an H-phosphonate procedure for the establishment of the phosphodiester linkage. In MenX phosphoglycans, the GlcNAc residue was incorporated in the polymer chain as an α-anomer. In synthetic fragments, the anomeric configuration of GlcNAc was determined with the configuration of the H-phosphonate, which, in turn, depends on the configuration of C-1 in the starting hemiacetal.
In 1991, pseudo-disaccharide
246 was synthesized for the first time by the Shibaev group (
Scheme 19) [
132]. Hemiacetal
252 with the NHAc group at C-2 was transformed into α-H-phosphonate
253 with a 95% yield under the action of in situ generated tris(imidazol-1-yl) phosphine. The condensation of H-phosphonate
253 and alcohol
254, followed by oxidation, afforded phosphodiester
255 (yield: 72%), which was deblocked to obtain pseudo-disaccharide
246 (
Figure 12).
In 2013, the series of spacer-armed oligomers
247 –
250 was synthesized by the Lay group [
133]. Unsuccessfully, 2-deoxy-2 azido hemiacetals
256 and
257, intended for the preparation of the corresponding H-phosphonates
258 and
259, were obtained as anomeric mixtures, thus challenging the preparation of H-phosphonates
258 and
259 as α-anomers. In order to provide the α-stereoselectivity of phosphonylation, the reaction of hemiacetals
256 and
257 with chlorophosphite
129 was conducted in the presence of phosphoric acid, which is known to destroy the β-anomer [
39] (
Scheme 20). The reaction took more than a week and afforded α-H-phosphonates
258 and
259 with 41% and 52% yields, respectively.
The phosphitylation of benzyl N-(3-hydroxypropyl) carbamate with benzylidenated H-phosphonate 258, followed by oxidation, resulted in phosphodiester
260 (yield: 62%), and the condensation of this alcohol with 4-O-acetylated H-phosphonate
259 formed phosphodiester
262 (yield: 64%), which was deacetylated to afford acceptor
262. The condensation of acceptor
262 with 4,6-O-benzylidenated H-phosphonate
258 and 4-O-acetylated H-phosphonate
259 was conducted with a moderate yield of phosphodiester
263 (45%) and
264 (40%). After deacetylation, pseudo-disaccharide
264 was converted into alcohol
265, which was reacted with 4,6-O-benzylidenated H-phosphonate
258 to afford trimer
266 with a 33% yield. The authors attribute the low efficiency of the phosphodiester linkage formation in trimer
266 to the presence of the azido group at C-2 of the hexapyranose ring. Total deprotection of compounds
260,
263, and
266 and transformation of the azido group into NHAc produced the target spacer-armed mono-, di-, and trimeric phosphodiesters
247,
248, and
249, structurally related to MenX capsular phosphooligoglycan [
133].
Unlike 2-deoxy-2-azido hemiacetals
256 and
257, hemiacetal
267 with an NHAc group at C-2 exists as an α-anomer and is readily converted into α-H-phosphonate
268 under the action of chlorophosphite
129 (
Scheme 20). Further improvement was achieved when the process was conducted in a microreactor, which provided a reduction in the reaction time to 30 min. and an increase in efficiency of the formation of α-H-phosphonate 268 to 76% [
129].
In the synthesis of a mixture of the spacer-armed oligomer
250 with avDP12, the trimeric phosphoglycan
249 was extended using an enzymatic method. The mixture of trimer
249 and a nucleotide GlcNAc-UDP was passed through a HiTrap
® column with immobilized recombinant capsule polymerase CsxA [
135], followed by fractionation of the resulting oligomers.
An advanced method of phosphonylation [
134] of an anomeric mixture of hemiacetal
259 with chlorophosphite
129 was used by Indian researchers for the preparation of pseudo-tetrasaccharide
251 as an aminohexyl glycoside (
Figure 12). Interaction of the hemiacetal mixture
259 with a phosphonylating agent (PhO)
2POH in Py in the presence of Et
3N, followed by treatment with H
3PO
3 for 4 days, afforded the desired α-H-phosphonate
261 with a 65% yield [
134].
In the first step of the assembly of pseudo-tetrasaccharide
251, the selectively protected aminohexyl glycoside
269 was condensed with H-phosphonate
259, and after oxidation, the pseudo-tetrasaccharide
270 was formed. By sequential deacetylation, condensation with H-phosphonate
259, and oxidation, the protected pseudo-tetrasaccharide
271 was obtained and further deblocked to yield the target pseudo-tetrasaccharide
271 (
Scheme 21). In the course of the synthesis, the efficiency of the formation of the phosphodiester linkage decreased in each subsequent step (yields of 86% > 60% > 53%) [
134], reflecting the tendency of the phosphodiester bond to be destroyed in the conditions of the H-phosphonate procedure.
The spacer-armed pseudo-saccharides
247–
251 (
Figure 12) were conjugated to protein carriers, and the immunogenic and antigenic properties of the resulting neoglycoconjugates were studied. Oligomers
247–
249 were attached to CRM197 to yield conjugates
273–
275 (
Figure 13) and the corresponding conjugates
277–
279 with HSA (
Figure 13). Conjugates
273–
275 were used for the immunization of mice at a dose of 0.3 μg of glycan per mouse. In addition, conjugate
275 was used at a dose of 1 μg per mouse. For positive controls, a group of mice was immunized with a conjugate of the partially fragmented
MenX phosphoglycan avDP15 with CRM197 (
MenXDP15-CRM197) at a dose of 0.3 μg and 1 μg of phosphoglycans, and bacterial MenX phosphoglycans were used as a coating antigen in ELISA experiments.
Conjugates
273 and
274 did not induce anti-
MenX IgG antibodies. Immunization with conjugate
275 with trimeric antigens and
MenXDP15-CRM197 efficiently elicited anti-
MenX IgG antibodies, yet the intensity of the specific immune response for conjugate
275 was significantly weaker. For these conjugates, the immune response did not depend on the dose [
133].
In the ELISA experiments, the immunogenic capacity of conjugates
273–
275 was assessed using HSA conjugates
277–
279 as coating antigens. It was found that the hyperimmune antisera of mice immunized with conjugates
273 (monomeric ligand) and
274 (dimeric ligand) contained negligible amounts of IgG antibodies to synthetic antigens. In contrast, immunization with conjugate
279 elicited anti-trimer IgG antibodies. An assessment of bactericidal activity in the rSBA tests showed that the pooled mouse sera obtained from animals immunized with conjugates
273 and
274 revealed that these conjugates did not elicit bactericidal antibodies. For conjugate
276, rSBA titers were 16 times lower compared to
MenXDP15-CRM197. Therefore, the length of the synthetic oligomers
247–
250 (
Figure 12), which are incorporated into conjugates
273–
275, was too small to effectively induce an anti-
MenX immune response [
133].
The mixture of the spacer-armed oligomer
250 with avDP12 was conjugated with the protein carrier CRM197. The resulting conjugate
276 was used for the immunization of mice, and the group of mice immunized with
MenXDP15-CRM197 was used as a positive control. The antigenicity of conjugates
276 and
MenXDP15-CRM197 was assessed in ELISA experiments with
MenX capsular phosphoglycans as a coating antigen. It was found that conjugates
276 and
MenXDP15-CRM197 demonstrate similar antigenic properties. However, SBA titers evidence the slightly greater bactericidal activity of antibodies elicited by
MenXDP15-CRM197 [
135].
7. Synthesis of Pseudo-Oligosaccharides Structurally Related to Phosphoglycans of S. Pneumonia
On a global scale, one in five infants under 1 year of age has had pneumonia, and ¾ of these pneumonia cases are caused by bacteria belonging to the
S. pneumoniae species [
136]. The prevention of community-acquired diseases associated with the most virulent S. pneumoniae serotypes is provided by a number of polyvalent conjugated pneumococcal vaccines, which comprise a number of pneumococcal capsular glycans conjugated to protein carriers.
Today, the pharmaceutical industry provides a whole range of anti-pneumococcal conjugated vaccines, including PCV7 (the
S. pneumoniae serotypes 4, 6B, 9V, 14, 19F, 18C, and 23F), PCV10 (the
S. pneumoniae serotypes 1, 4, 5, 6B, 7F, 9V, 14, 18C, 19F, and 23F) and (the
S. pneumoniae serotypes 1, 5, 6A, 6B, 7F, 9V, 14, 19A, 19F, and 23F), PCV13 (the
S. pneumoniae serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 19A, 19F, 18C, and 23F), PCV15 (the
S. pneumoniae serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F, 22F, 23F, and 33F), and PCV20 (
S. pneumoniae 1, 3, 4, 5, 6A, 6B, 7F, 8, 9V, 10A, 11A, 12F, 14, 15B, 18C, 19A, 19F, 22F, 23F, and 33F). Among the capsular glycan antigens used in these vaccines, five biopolymers, in particular
S. pneumoniae 6A,
S. pneumoniae 6B,
S. pneumoniae 10A,
S. pneumoniae 19A, and
S. pneumoniae 19F, comprise phosphodiester bonds in the main chain.
S. pneumoniae 6A and
S. pneumoniae 6B phosphoglycans are built of pseudo-tetrasaccharide repeating units connected by phosphodiester linkage. The repeating unit of
S. pneumoniae 10A phosphoglycans is a pseudo-heptaglycosyl phosphate, and the phosphoglycans of
S. pneumoniae 19A and
S. pneumoniae 19F are composed of trisaccharide phosphate and hexasaccharide phosphate repeating units [
137].
Although the anti-pneumococcal vaccines for the prevention of
S. pneumoniae 6A,
S. pneumoniae 6B,
S. pneumoniae 19A, and
S. pneumoniae 19F have been available for more than 10 years, these serotypes are still frequently identified as the causative agents in pneumococcal pneumonia [
136] and meningitis [
138] and colonize the upper respiratory tract as opportunistic pathogens [
139]. One of the factors hindering the broad coverage of people with anti-pneumococcal vaccination programs is high costs [
9], which can be reduced by the replacement of bacterial antigens with their synthetic analogs [
22].
The
S. pneumoniae strains of serotype 6 are the common causative agents of invasive disease. The group is subdivided into serotypes 6A and 6B, which both have a capsule composed of phosphoglycans with Rha–ribitol–Gal–Glu subunits, which differ in the type of Rha–ribitol linkage. In serotype 6A, it is Rha1→3ribitol, and in serotype 6B, it is Rha1→4ribitol. It was reported that point mutation between
S. pneumoniae 6A and
S. pneumoniae 6B, as well as recombination, can compromise the linkage specificity and mediate serotype change [
140]. As a result, it is reasonable to include both glycan antigens in combined vaccines. The surveillance of invasive pneumococcal disease in different countries evidences the immense importance of pneumococcal conjugated vaccines in the prevention of
S. pneumoniae 6a- and
S. pneumoniae 6b-associated invasive diseases [
139,
141,
142]. In contrast, in regions with low immunization coverage [
143], this pathogen makes a substantial contribution to the burden of invasive pneumonia.
Recently, syntheses of the spacer-armed pseudo-oligosaccharides
280–
286 [
144,
145] structurally related to
S. pneumoniae 6A phosphoglycans (
Figure 14) have been reported. For the construction of a phosphodiester bond in compound
280, Nifantiev et al. [
144] used the H-phosphonate procedure. Glycoside
287 (
Scheme 22) was converted into H-phosphonate
288 by treatment with H
3PO
3 in Py and the presence of PivCl. The condensation of H-phosphonate
288 with the primary alcohol
289, followed by oxidation, afforded phosphodiester
290, which, after removal of the protective groups, was converted into aminoethyl glycoside
280.
Pseudo-oligosaccharides
281–
286 were assembled by Taiwan scientists [
145] using a phosphoramidite procedure for the establishment of phosphodiester linkage (
Scheme 23). The selectively protected pseudo-tetrasaccharide
291 with a primary hydroxyl group in the ribitol residue was transformed into phosphoramidite
292 by the action of diamidite
70 and diisopropylammonium tetrazolide. In similar conditions, pseudo-disaccharide
293 was converted into phosphoramidite
294.
The interaction of the key phosphoramidite
292 with 5-azidopentanol (compound
295) in the presence of 1H-tetrazole, followed by oxidation with mCPBA, the removal of the protective groups, and the reduction of the azido group, resulted in pseudo-tetrasaccharide
281 (
Scheme 23). The condensation of phosphoramidite
292 with alcohols
296–
299 was carried out in the presence of 5-ethylthio-1H-tetrazole, followed by oxidation of a phosphite group to a phosphotriester with I
2 in THF. The efficiency of the formation of the phosphotriester linkage decreased in the sequence of monosaccharide
296 (94%), disaccharide
297 (71%), trisaccharide
298 (66%), and pseudo-tetrasaccharide
299 (62%), along with an increase in the steric hindrance of the hydroxyl group involved in condensation. The reduction and deblocking of the obtained phosphotriesters afforded the amino-spacered derivatives
282–
285. The condensation of phosphoramidite
294 with disaccharide
297, followed by oxidation, the reduction of the azido group, and deblocking, formed pseudo-tetrasaccharide
286 [
145].
Due to interest in the development of vaccine preparations based on the synthetic phosphooligosaccharides related to
S. pneumoniae 6B, extensive libraries of the spacer-armed pseudo-oligosaccharides
300–
314 [
145,
146,
147,
148] (
Figure 15) related to
S. pneumoniae 6B CPS fragments have been created. Despite their structural diversity, these compounds comprise only one phosphodiester linkage that is connected with the synthetic complexity of the target molecules.
The assembly of the spacer-armed pseudo-disaccharides
300 and
301 and pseudo-trisaccharides
302 and
303 was carried out by Vliegenthart [
146] using the H-phosphonate method. For the preparation of pseudo-disaccharide
300, the starting pseudo-disaccharide
315 was converted to the key H-phosphonate
316 by the action of chlorophosphite
129 (
Scheme 24). H-phosphonate
316 was condensed with N-protected aminopropanol in the presence of PivCl in Py, the product was oxidized, and a phosphodiester
317 was obtained with a 43% yield. The deprotection of phosphodiester
317 yielded the target spacer-armed pseudo-disaccharide
300.
Under similar conditions, alcohol
318 (
Scheme 25) was transformed into H-phosphonate
319, and the phosphonylation of alcohol
320 produced H-phosphonate
321 with an excellent yield. Phosphodiester
322 was prepared via two alternative routes. The condensation of the secondary hydroxyl group in galactoside
318 with H-phosphonate
321, followed by oxidation, formed
322 with a 40% yield, and the interaction of a primary hydroxyl group in the selectively protected ribitol
320 with H-phosphonate
319 and oxidation of the intermediate phosphonate afforded pseudo-disaccharide
322 with a 38% yield. By the removal of the benzyl and Cbz groups, compound
322 was converted into the target pseudo-disaccharide
301 (
Figure 15) [
146].
Unlike the synthesis of pseudo-disaccharide
322, the phosphitylation of alcohol
315 with H-phosphonate
319 was more efficient and readily formed pseudo-trisaccharide
323 (yield: 89%) [
147], which was deprotected to yield the target spacer-armed pseudo-trisaccharide
302 (
Scheme 24) [
146]. The substantial difference in the yields for pseudo-disaccharide
322 (from
319 and
320) and pseudo-trisaccharide
323 may be related to the easier accessibility of a less sterically hindered phosphodiester bond in pseudo-disaccharide
322 for the I
2-induced rapid rupture of the bridging P-O bonds. The total deprotection of pseudo-trisaccharide
323 resulted in the formation of the target spacer-armed pseudo-trisaccharide
302 (
Figure 15) [
147].
The condensation of alcohol
324 with H-phosphonate
321 (
Scheme 24), followed by oxidation, afforded pseudo-tetrasaccharide
325 with a 77% yield [
147]. Similar to the phosphitylation of alcohols
315 and
320 with H-phosphonate
319, the improved efficiency of preparation of more sterically hindered pseudo-tetrasaccharide
325 compared to
322 (from
318 and
321, 40%) [
146] can be attributed to the easier destruction with I
2 during the oxidation step. By deprotection, pseudo-trisaccharide
325 was converted into the target spacer-armed pseudo-trisaccharide
303 (
Figure 15) [
147].
The interaction of H-phosphonate
321 with the trisaccharide alcohol
326 (
Scheme 24), followed by oxidation, formed pseudo-tetrasaccharide
327 (78%) with a phosphodiester bond between a bulky trisaccharide and a flexible ribitol residue. On the contrary, the efficiency of the formation of pseudo-tetrasaccharide
328 (from disaccharide
324) and pseudo-tetrasaccharide
330 (from pseudo-tetrasaccharide
329), which comprise bulky protected glycoside residues on both sides of a phosphate moiety, was much lower, and the yields did not exceed 50%. The total deprotection of pseudo-tetrasaccharides
328 and
329 resulted in the target spacer-armed compounds
304 and
305, respectively [
147].
The condensation of pseudo-tetrasaccharide
331 with H-phosphonate
214, followed by oxidation, provided the introduction of a pre-spacer group, which is connected to the pseudo-tetrasaccharide via a phosphodiester bridge. The moderate yield of compound
332 (53%) can be attributed to the conformational flexibility of the pre-spacer group and the easier availability of the phosphate group for the action of I
2. Compounds
331 and
332 were deblocked to yield the target spacer-armed pseudo-tetrasaccharides
307 and
309, respectively (
Scheme 24) [
147].
For the assembly of pseudo-tetrasaccharide
308, Nifantiev et al. [
148] investigated two alternative pathways according to the schemes [3 + 1], which differed in the position of the H-phosphonate group on one or the other condensing part (
Scheme 25). For this purpose, alcohol
333 was converted into H-phosphonate
334 by the action of chlorophosphite
129, and galactoside
335 was converted into H-phosphonate
336 under similar conditions. The condensation of H-phosphonate
336 with the primary hydroxyl group of pseudo-tetrasaccharide acceptor
333, followed by oxidation, produced phosphodiester
337 in a high yield (85%), whereas the interaction of H-phosphonate
334 with the secondary hydroxyl group in galactoside
335 proceeded with low efficiency (the yield of pseudo-tetrasaccharide
337 was 22%).
For the preparation of pseudo-oligosaccharides
306 and
310–
314 [145], Taiwan researchers used the universal pseudo-tetrasaccharide block
338 (
Scheme 26), which was readily converted into the corresponding phosphoramidite
339 by the action of diamidite
70 in the presence of diisopropylammonium tetrazolide. In similar conditions, pseudo-disaccharide
340 was converted into phosphoramidite
341. The condensation of alcohols
295–
298 and
342 with compound
339 added a repeating unit to the chains. Thus, the phosphitylation of the primary hydroxyl group in alcohol
295 with phosphoramidite
339 in the presence of 1H-tetrazole, followed by the oxidation of phosphite to phosphate and the removal of the 2-naphthylmethyl protective group, afforded alcohol
342, and the total deprotection and reduction of the azido group resulted in the target spacer-armed pseudo-tetrasaccharide
310, with the spacer group connected to the pseudo-tetrasaccharide by a phosphodiester bridge.
In a similar way, phosphoramidite
339 readily phosphitylated secondary hydroxyl groups in monosaccharide
296, disaccharide
297, and trisaccharide
298 (
Scheme 26). The resulting phosphites were oxidized and deprotected to obtain target compounds
311–
313. The condensation of two pseudo-tetrasaccharides
342 and
339, followed by oxidation, afforded a pseudo-octasaccharide (87%), with two bulky fragments connected by a phosphodiester bridge. After the deprotection and reduction of the azido group, the spacer-armed pseudo-octasaccharide
314 was obtained. Disaccharide
297 was phosphitylated with phosphoramidite
341. The oxidation of the intermediate phosphite, removal of the protective groups, and reduction of the azido group afforded pseudo-tetrasaccharide
306. It can be concluded that the use of phosphoramidites as phosphitylating agents in the preparation of compounds
306 and
310–
314 was efficient regardless of the steric demands of the secondary hydroxyl group in the acceptor molecule. This result shows that the phosphoramidite method is preferred for the synthesis of larger structures.
The phosphoglycan structure of
S. pneumoniae 6C differs from
S. pneumoniae 6A in the orientation of a single hydroxyl group in one of the monosaccharide blocks (Glc in
S. pneumoniae 6C vs. Gal in
S. pneumoniae 6A), and a similar structural difference is observed between
S. pneumoniae 6D and
S. pneumoniae 6B (
Figure 16). It is generally accepted that a close structural resemblance gives rise to similar antigenic properties, and one could expect cross-reactivity between all serotypes in group
S. pneumoniae 6. However, surveillance data and clinical efficacy studies for subtypes of
S. pneumoniae 6 obtained in connection with the use of pneumococcal vaccines are controversial. Generally, cross-protection was observed from the
S. pneumoniae 6B conjugate in PCV7 and PCV10 against
S. pneumoniae 6A but not against
S. pneumoniae 6C and
S. pneumoniae 6D. However, it was shown that PCV13, which comprises the
S. pneumoniae 6A antigen, was effective against serotypes 6C and 6D invasive pneumococcal disease and microbial carriage [
149,
150]. The challenges associated with the assessment of the cross-reactivity and cross-protection of
S. pneumoniae group 6 could be met via the identification of a glycotope for each
S. pneumoniae 6 subtype. To this end, the series of oligosaccharides
343 and
344, structurally related to
S. pneumoniae 6C, and
345 and
346, structurally related to
S. pneumoniae 6D, were synthesized (
Figure 16) [
151]. The preparation of pseudo-tetrasaccharides
343 and
344, which comprise phosphodiester linkages, was performed using phosphoramidite chemistry. To obtain pseudo-oligosaccharide
343 with an interglycosidic phosphodiester bridge, disaccharide
347 was reacted with phosphoramidite
294 in acetonitrile in the presence of 5-ethylthio-1H-tetrazole (
Scheme 27). The reaction mixture was oxidized under the action of iodine in THF with the addition of water, the protective groups were removed, and the N
3 group was reduced to NH
2. The phosphitylation of the primary OH-group in pseudo-tetrasaccharide
348 with diamidite
70 resulted in the efficient formation of phosphoramidite
349. The condensation of phosphoramidite
349 with alcohol
295, followed by the reduction of the N
3 group and total deprotection, afforded pseudo-tetrasaccharide
344, in which a phosphodiester linkage connects the ribitol part and a spacer [
151].
In a similar way, pseudo-tetrasaccharide 345 with an interglycosidic phosphodiester bridge was obtained by the condensation of disaccharide 247 and phosphoramidite 341, oxidation, reduction of the N3 group, and deprotection. For the preparation of phosphodiester 346, pseudo-tetrasaccharide 350 was transformed into phosphoramidite 351, which was then condensed with alcohol 295. After oxidation, the condensation product was reduced and deprotected to yield pseudo-tetrasaccharide 346, with a phosphodiester bridge between ribitol and a spacer. Notably, in the preparation of compounds 343–346, the corresponding protected phosphodiesters were obtained in moderate yields regardless of the position of the phosphodiester bridge in the molecule. It can be concluded that the used method is not sensitive to steric factors and can be applied for the condensation of bulky counterparts.
The cross-reactivity of
S. pneumoniae 6A and
S. pneumoniae 6B was evaluated in an immunological study of pseudo-tetrasaccharides
280 [
144] related to
S. pneumoniae 6A (
Figure 14) and
308 [
148] (
Figure 15) related to
S. pneumoniae 6B. Conjugates of these compounds with BSA (
280-BSA and
308-BSA, respectively) were used for the immunization of mice at a dose of 20 μg/mouse on days 0 and 14. The hyperimmune serum obtained after immunization with
280-BSA and
308-BSA was analyzed in ELISA experiments using the corresponding N-biotinylated conjugates
352 and
353 on streptavidin-coated plates. It was shown that the antibodies induced by the
S. pneumoniae 6A-related conjugate
280-BSA recognized not only the
S. pneumoniae 6A-related N-biotinylated conjugate
352 but also the
S. pneumoniae 6B-related N-biotinylated conjugate
353. Inversely, the specificity of the serum obtained from mice immunized with the
S. pneumoniae 6B-related conjugate
308-BSA to the
S. pneumoniae 6A-related conjugate
352 was detected. These results evidence the cross-reactivity of pseudo-tetrasaccharides
280 and
308 and indicate the presence of a common epitope in these antigens.
The immunogenic properties of synthetic phosphooligosaccharides
285 and
286 related to
S. pneumoniae 6A phosphoglycans (
Figure 14) and phosphooligosaccharides
306 and
314 related to
S. pneumoniae 6B phosphoglycans were studied. The conjugates of these compounds with CRM197 were obtained and used for the immunization of mice at a dose of 2.2 μg of glycan per mouse, with three shots in two-week intervals. The glycan-specific IgG in the serum was analyzed using a microarray with a number of synthetic glycan antigens [
145]. Mice in the negative control group were immunized with CRM197. It was found that conjugates
286-CRM197 and
306-CRM197, which contain pseudo-tetrasaccharides, effectively induced IgG antibodies against the majority of other synthetic antigens. On the contrary, conjugates
285-CRM197 and
314-CRM197 showed low immunogenicity [
145].
The antigenic properties of
S. pneumoniae 6B-related pseudo-disaccharide
301, pseudo-trisaccharide
303, and pseudo-tetrasaccharide
305 were investigated using their conjugates
301-KLH–
305-KLH with the highly immunogenic protein KLH (
Figure 17) [
152]. The neoglycoconjugates
301-KLH–
305-KLH and a conjugate obtained by the condensation of bacterial
S. pneumoniae 6B phosphoglycans and KLH (
S. pneumoniae 6B-KLH) were used for the immunization of mice at a dose of 2.5 μg and rabbits at a dose of 10 μg. The hyperimmune sera were analyzed in ELISA experiments with bacterial
S. pneumoniae 6B and
S. pneumoniae 6A phosphoglycans. Human serum samples were obtained by pooling infant antisera obtained by vaccination with PCV7-CRM197 at a dose of 4 mg of 6B PS.
It was shown that in mice conjugates, 301-KLH with pseudo-disaccharide antigens and 303-KLH with pseudo-trisaccharide antigens were poorly immunogenic. On the contrary, the conjugate 305-KLH was more immunogenic than S. pneumoniae 6B-KLH. ELISA analysis of the rabbit hyperimmune antisera using bacterial S. pneumoniae 6A phosphoglycans as a coating antigen showed that conjugates 303-KLH and 305-KLH were able to induce anti-S. pneumoniae 6A antibodies. Antibody specificity was confirmed in ELISA inhibition and phagocytosis experiments. The binding of human PCV7-CRM197 antisera to the conjugates 301-KLH–305-KLH was inhibited by bacterial S. pneumoniae 6A and S. pneumoniae 6B phosphooligosaccharides.
S. pneumoniae 19F is one of the most virulent pneumococcal serotypes. Similarly to
S. pneumoniae 6A and
S. pneumoniae 6B, the diseases caused by
S. pneumoniae 19F infection are associated with increased mortality and morbidity [
153] despite the fact that the
S. pneumoniae 19F capsular glycan is a component of many pneumococcal conjugated commercial vaccines. In light of this, it could be useful to develop an anti-
S. pneumoniae 19F conjugate vaccine with a synthetic glycan antigen structurally related to the capsular glycan of
S. pneumoniae 19F. N-trifuoroacetamidopropyl glycoside
357, which comprises a pseudo-hexasaccharide fragment of the
S. pneumoniae 19F capsular polysaccharide, was obtained as a mixture of isomers. In the first step, the selectively protected trisaccharide
354 with a free hydroxyl group at C-4 (ManNAc) was converted into H-phosphonate
355 under the action of chlorophosphite
129 in Py [
154] (
Scheme 28). The condensation of H-phosphonate
355 with hemiacetal
356, followed by oxidation and deprotection, resulted in the formation of a diastereomeric mixture of pseudo-hexasaccharides, which contained the spacer-armed phosphooligosaccharide
357.
The structure of the repeating unit of
S. pneumoniae 19A is close to that of 19F, with the only difference being the type of linkage between Glc and Rha monosaccharide residues, which is α-D-Glc-(1→3)-Rha in
S. pneumoniae 19A phosphoglycans and α-D-Glc-(1→2)-Rha in
S. pneumoniae 19F phosphoglycans. Whereas
S. pneumoniae 19F phosphoglycans are a component of all commercial PCV compositions,
S. pneumoniae 19A phosphoglycans were not included in PCV7 and PCV10. After the introduction of PCV7, serotype replacement resulted in an increase in the number of invasive pneumococcal diseases caused by S. pneumoniae 19A [
155,
156]. This observation shows the absence of cross-protection between serotypes 19A and 19F. Nevertheless, the synthetic complexity of the repeating units of
S. pneumoniae 19A and
S. pneumoniae 19F phosphoglycans, which share a common structure, stimulated attempts to develop a universal synthetic glycoantigen for the induction of protective antibodies to both serotypes.
With a view to investigating the ability of a neoglycoconjugate with a chimeric synthetic antigen to induce a protective immune response, the Seeberger group prepared [
157] phosphohexasaccharide
358, which is a combination of
S. pneumoniae 19A and S. pneumoniae 19F phosphoglycan repeating units in one molecule and the spacer-armed phosphotrisaccharides
359 and
360 related to repeating units of
S. pneumoniae 19F and
S. pneumoniae 19A, respectively (
Figure 18).
Hemiacetal
361 was readily transformed into the corresponding α-H-phosphonate
362 under the action of chlorophosphite
129 and H
3PO
3 in Py (
Scheme 29). The authors suggest that α-stereoselectivity was attained due to thermodynamic reaction control, which favored α-H-phosphonate
362 in the conditions of S
N2 displacement with H
3PO
3 at the anomeric center. In a similar manner, hemiacetal
363 was stereoselectively converted into α-H-phosphonate
364 as a precursor of the
S. pneumoniae 19A repeating unit. α-H-Phosphonates
362 and
364 were coupled with alcohol
295, oxidized, reduced, and deprotected to yield the spacer-armed glycoantigens
359 and
360, which correspond to repeating units of
S. pneumoniae 19F and
S. pneumoniae 19A phosphoglycans [
157].
For the preparation of the hybrid antigen 358, another precursor of the
S. pneumoniae 19A repeating unit, hemiacetal
365 with an acetyl protecting group at O-4 (MaNAc) was phosphonylated to obtain α-H-phosphonate
366, which was then condensed with alcohol
295 and oxidized. The following 4-O-deacetylation and phosphitylation with α-H-phosphonate
362 and oxidation yielded the protected phosphodiester
367. The deblocking and reduction of the azido group in phosphodiester
367 afforded the conjugation-ready chimeric phosphodiester 358 [
157]. Phosphooligosaccharides
358–
361 were conjugated to CRM197 via a succinimidyl adipate linker, and the resulting conjugates
358-CRM197 (average glycan loading: 5),
359-CRM197 (average glycan loading: 5), and
360-CRM197 (average glycan loading: 7) were adsorbed on alum and used for the vaccination of rabbits on day 0 and boosted on days 14, 28, and 133. IgG antibodies were raised in response to immunization with the semisynthetic neoglycoconjugates
358-CRM197,
359-CRM197,
360-CRM197, and Prevnar 13
® (PCV13).
Oligosaccharide-specific antibody titers were analyzed by glycan microarray with specific immobilized oligosaccharides. Chimeric
358-CRM197 generated antibodies that recognized trisaccharide antigens
359 and
360 in quantities exceeding those generated by Prevnar 13
®. At the same time, ELISA experiments showed that antibodies produced by vaccination with Prevnar 13
® more efficiently recognized
S. pneumoniae 19A and
S. pneumoniae 19F phosphoglycans compared to vaccine preparations with synthetic antigens. This observation reveals that upon immunization with Prevnar 13
®, the antibodies are elicited to larger glycotopes. Immunization with chimeric
358-CRM197 produced antibodies that efficiently recognized phosphotrisaccharide antigens
359 and
360 and
S. pneumoniae 19A and
S. pneumoniae 19F phosphoglycans, whereas antibodies elicited by
359-CRM197 and
360-CRM197 did not recognize
S. pneumoniae 19A phosphoglycans, and only moderate interaction of
359-CRM197-induced antibodies and S. pneumoniae 19F phosphoglycans was detected. Bactericidal properties of the obtained hyperimmune sera were studied in the opsonophagocytic killing assay. Chimeric
358-CRM197-induced opsonic antibodies were able to kill both
S. pneumoniae 19A and
S. pneumoniae 19F. The opsonic activity of
359-CRM197-induced antibodies was very weak, and antibodies induced by the ST19A conjugate were not bactericidal [
157].
As previously mentioned, clinically significant cross-protection between
S. pneumoniae 19A and
S. pneumoniae 19F serotypes has not been reported. One of the possible reasons for weak antibody cross-protection is the conformational difference between these two phosphoglycans [
158]. In shorter oligosaccharides, the chain exists as another ensemble of conformers, which may be able to induce antibodies to phosphoglycans of both serotypes. The capsular phosphoglycans
S. pneumoniae 19A and
S. pneumoniae 19F share a common disaccharide structural element: P(1→4)ManNAc-β-(1→4)-Glc. With a view to studying the universal glycotope for these serotypes, a series of short glycans
368–
370 (
Figure 19) were prepared, which comprise the basic structural elements of the common disaccharide.
The phosphoramidite approach was applied for the construction of the phosphodiester bridge in pseudotrisaccharide
370 [
159]. The selectively protected glycoside
371 with a free hydroxyl group at C-4 (ManNAc) was transformed into phosphamidite
372 under the action of chlorophosphoramidite
24 (
Scheme 30). The condensation of phosphamidite
372 with hemiacetal
373 resulted in the formation of a mixture of α- and β-glucopyranosides (α:β 55:45). The α-isomer was oxidized, decyanoethylated, and deprotected to obtain phosphodiester
370.
Compounds 368–370 were printed on epoxysilane-coated slides together with bacterial S. pneumoniae 19A and S. pneumoniae 19F phosphoglycans as controls. A glycan microarray was arranged, and the glycan antigens interacted with the hyperimmune sera of rabbits immunized with whole bacteria. The group’s 19 sera were bound to S. pneumoniae 19A and S. pneumoniae 19F and phosphodisaccharide 368, and the interaction with disaccharide 369 and the pseudotrisaccharide was weak. The interaction of synthetic and bacterial glycans was studied with factor reference antisera obtained by vaccination with whole cell bacteria S. pneumoniae 19A or S. pneumoniae 19F and cleared of the antibodies recognizing the common epitopes. Reference antisera to S. pneumoniae 19A readily bound to phosphate 368 and did not interact with compounds 369 and 370. Reference antisera to S. pneumoniae 19F showed moderate binding to compounds 368–370.
Two considered approaches to the search for a common epitope in S. pneumoniae 19A and S. pneumoniae 19F phosphoglycans open new horizons in the development of semisynthetic conjugated glycovaccines. The design of a universal glycotope for introduction in neoglycoconjugate vaccines is aimed at a decrease in protein loading of the vaccination dose and reduction of the production costs.




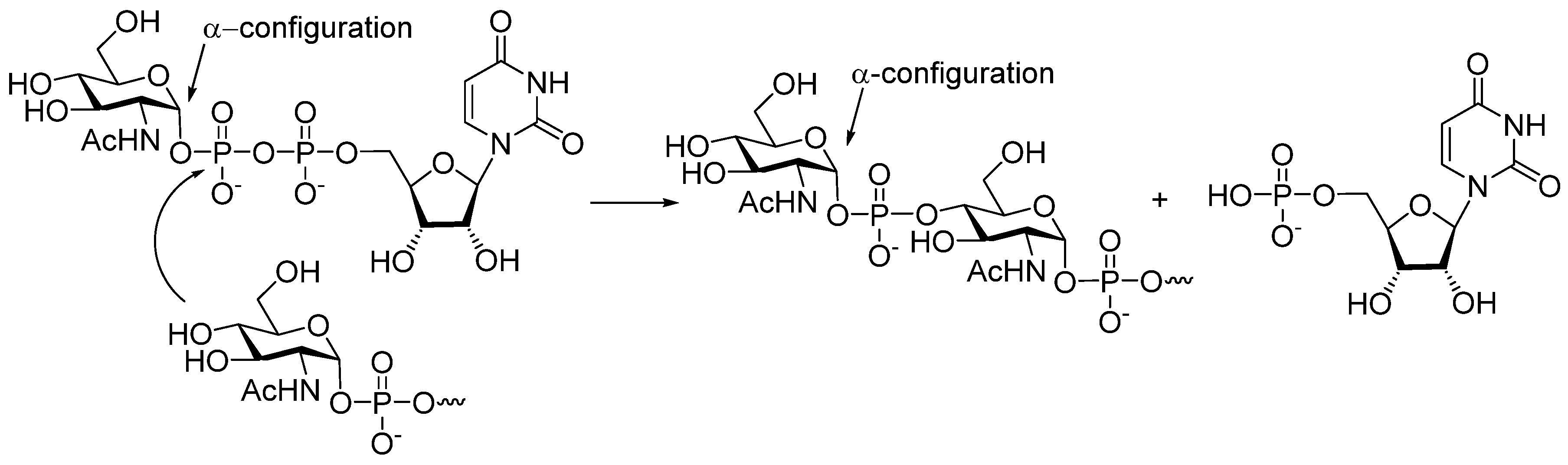

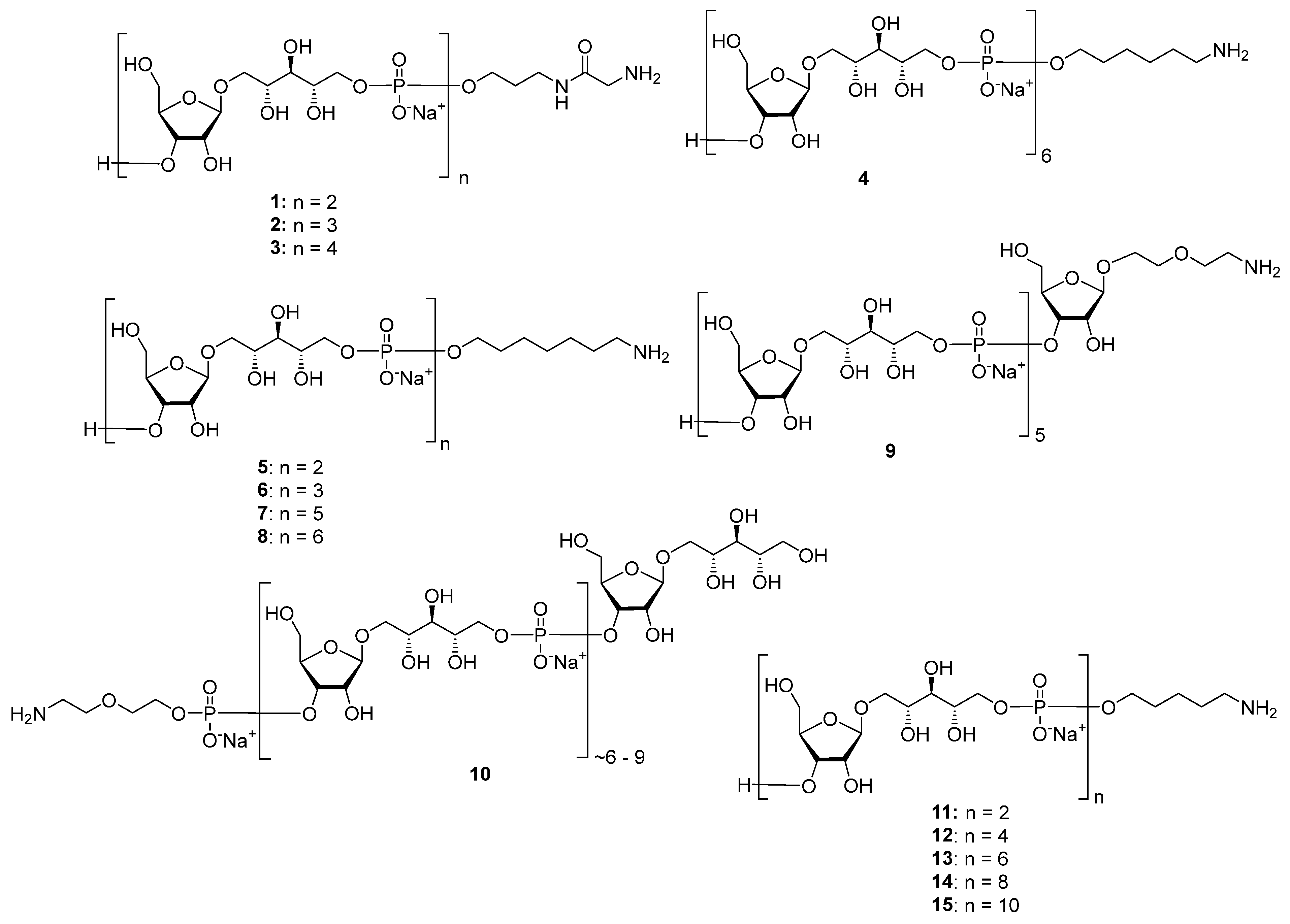
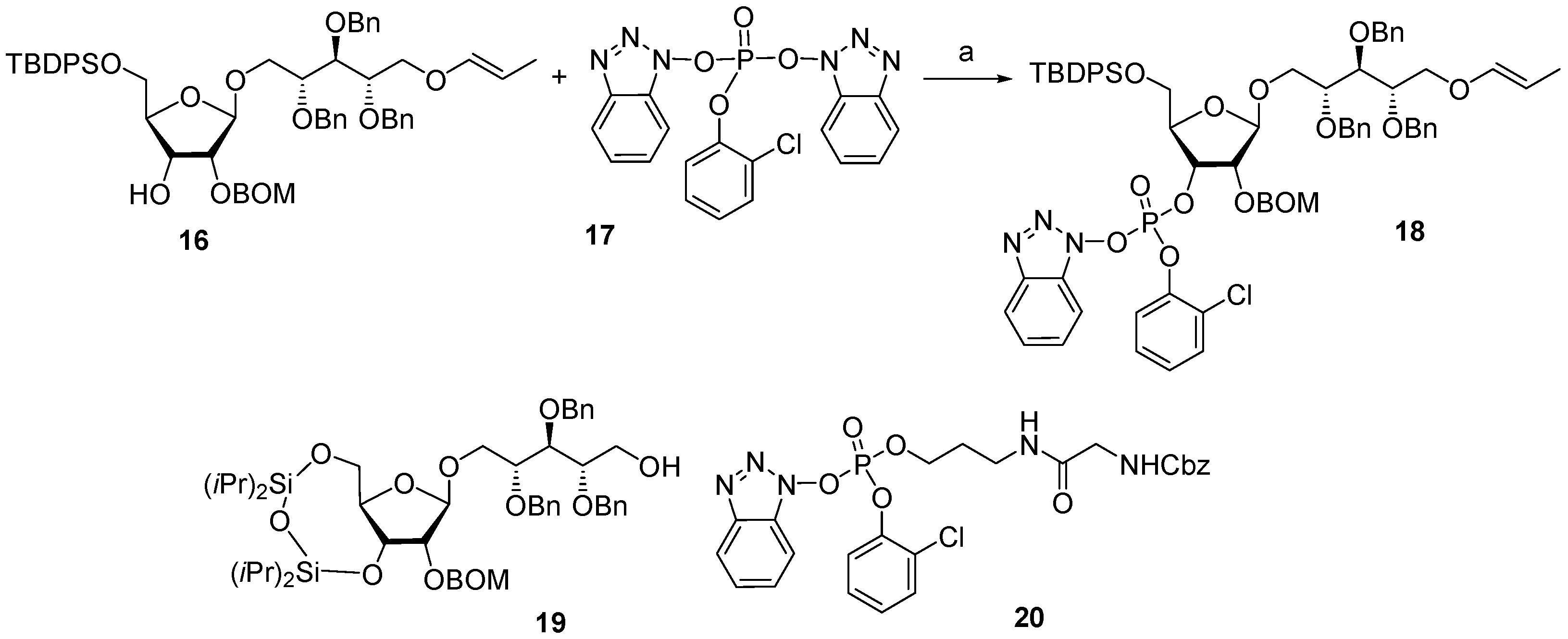

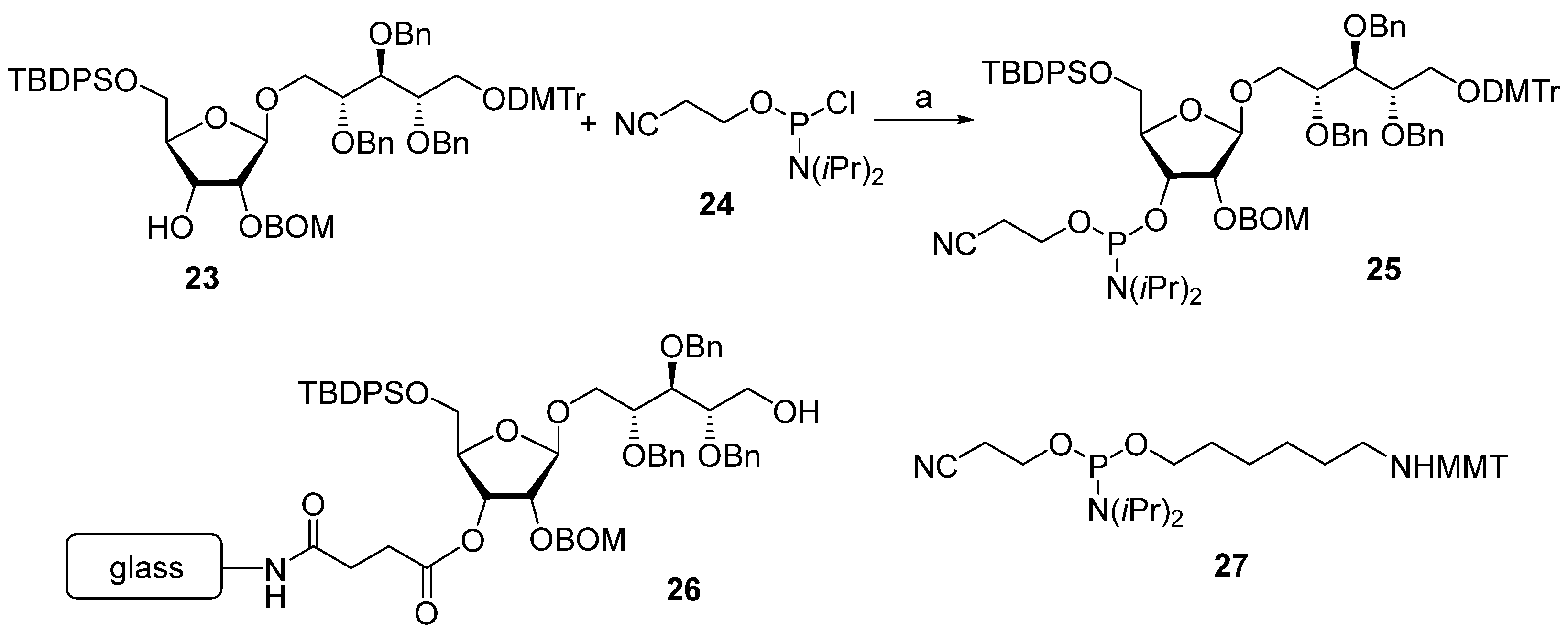
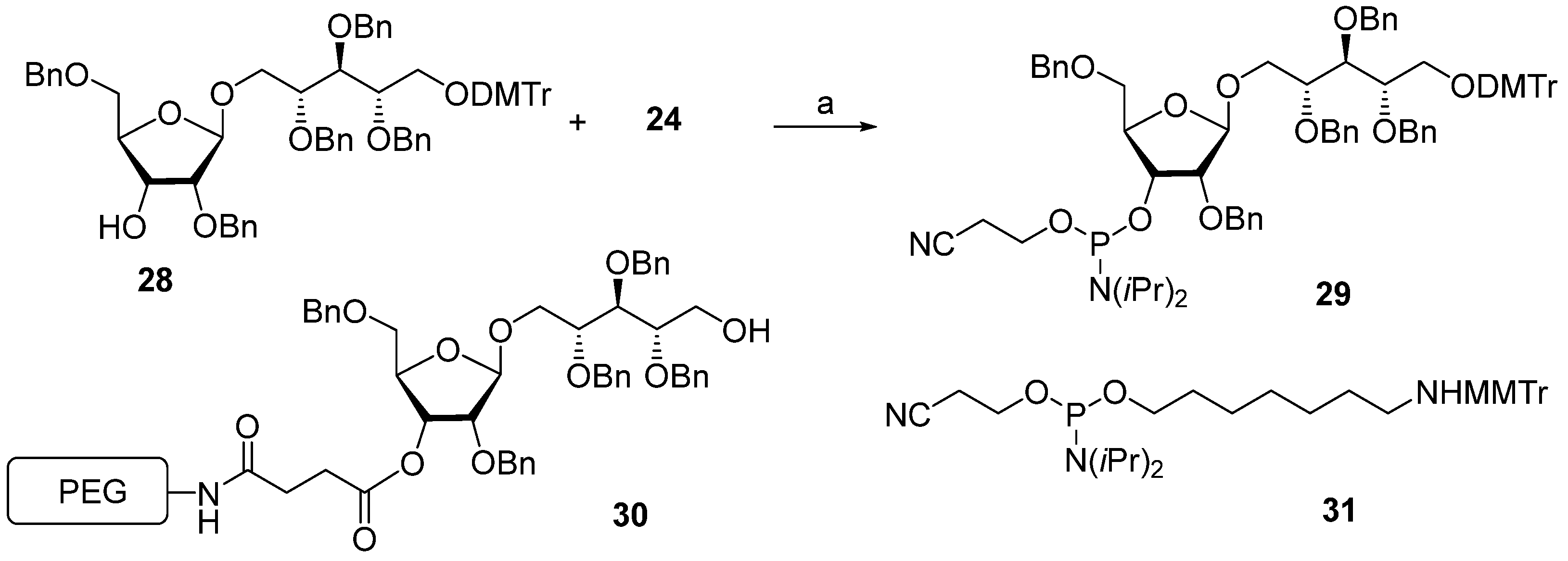
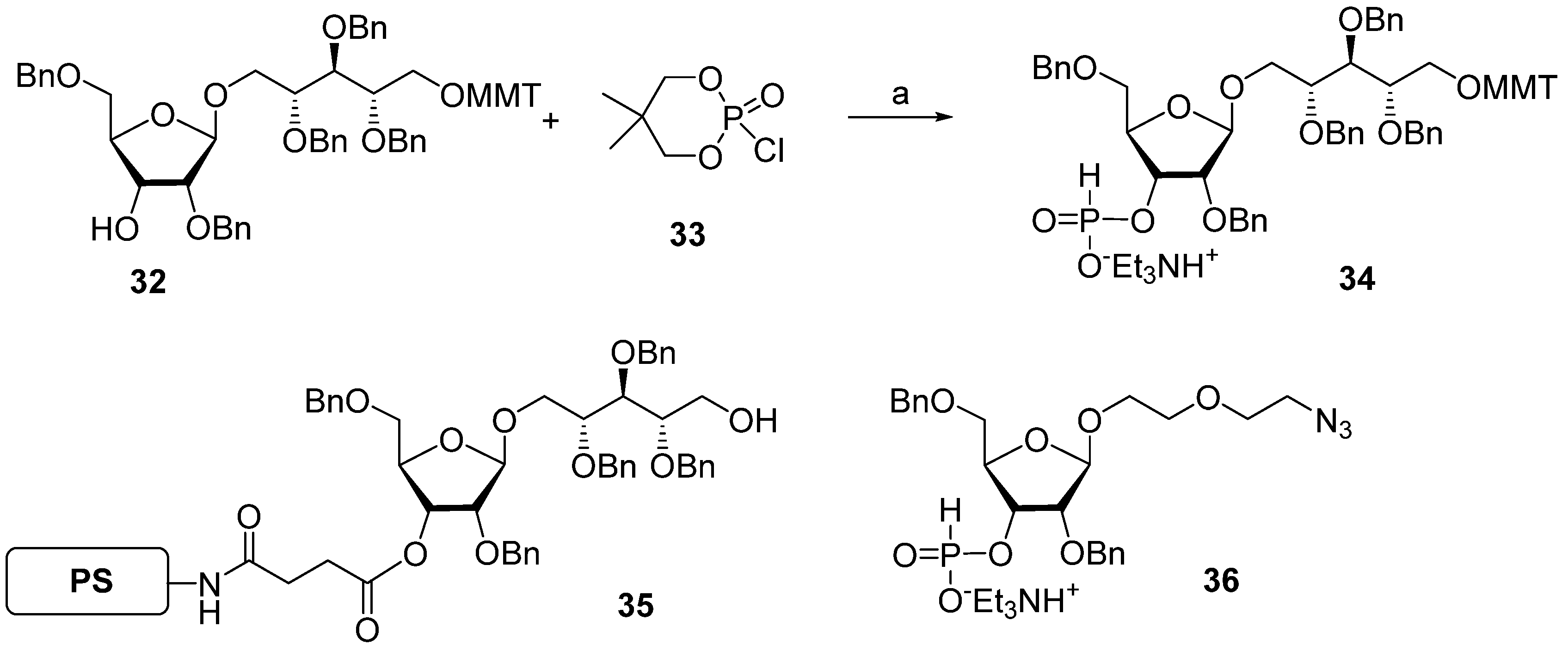
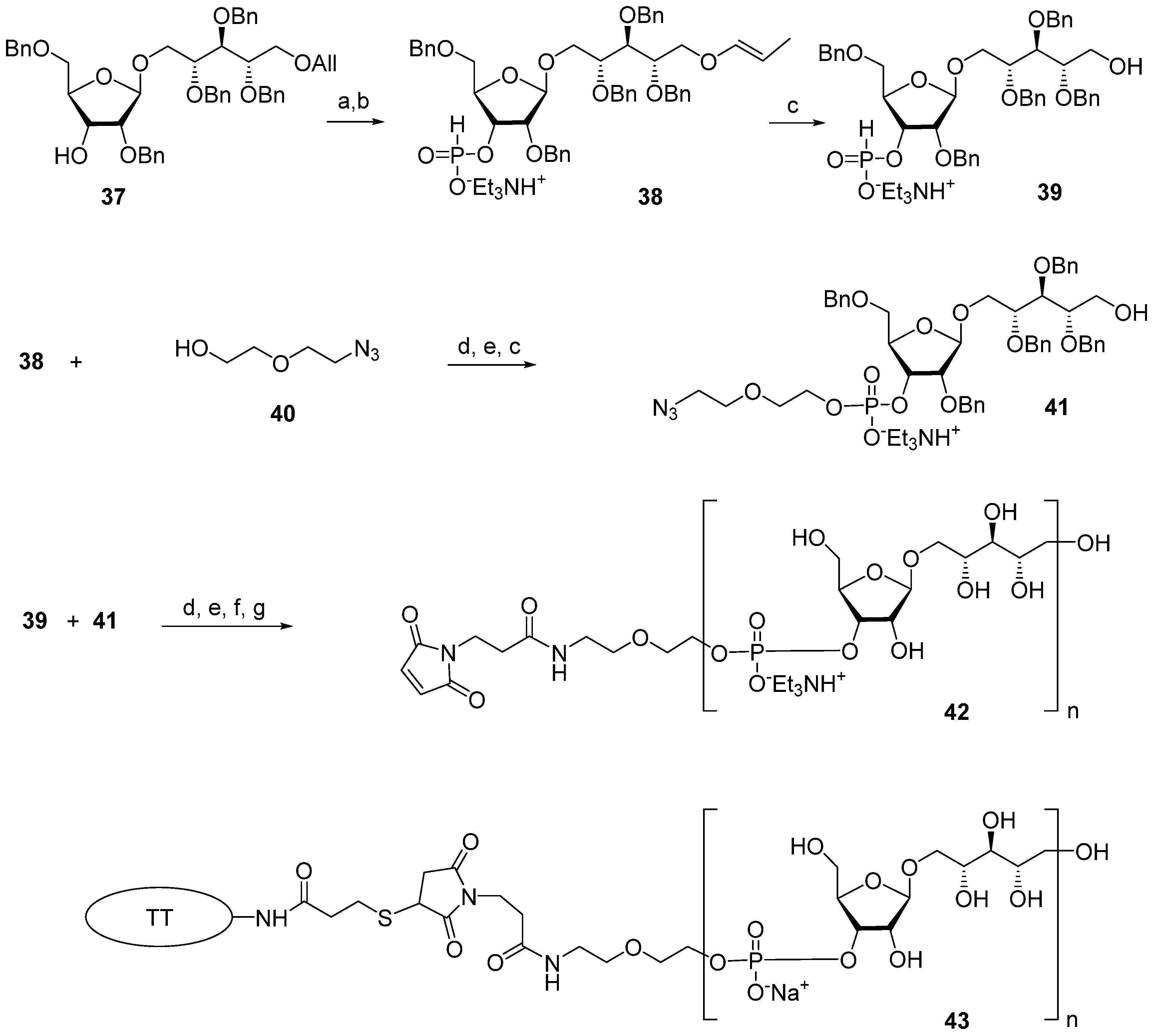
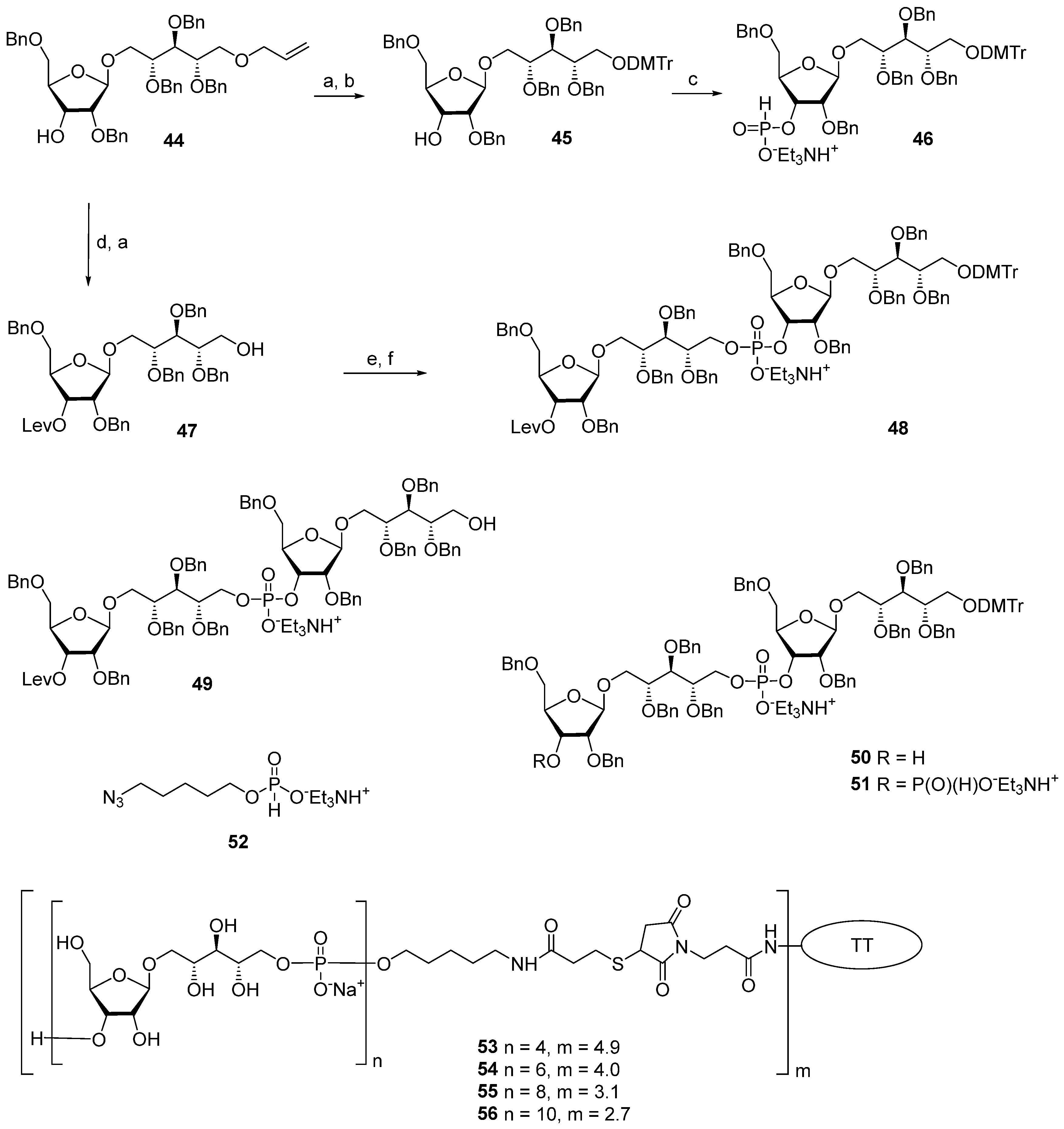
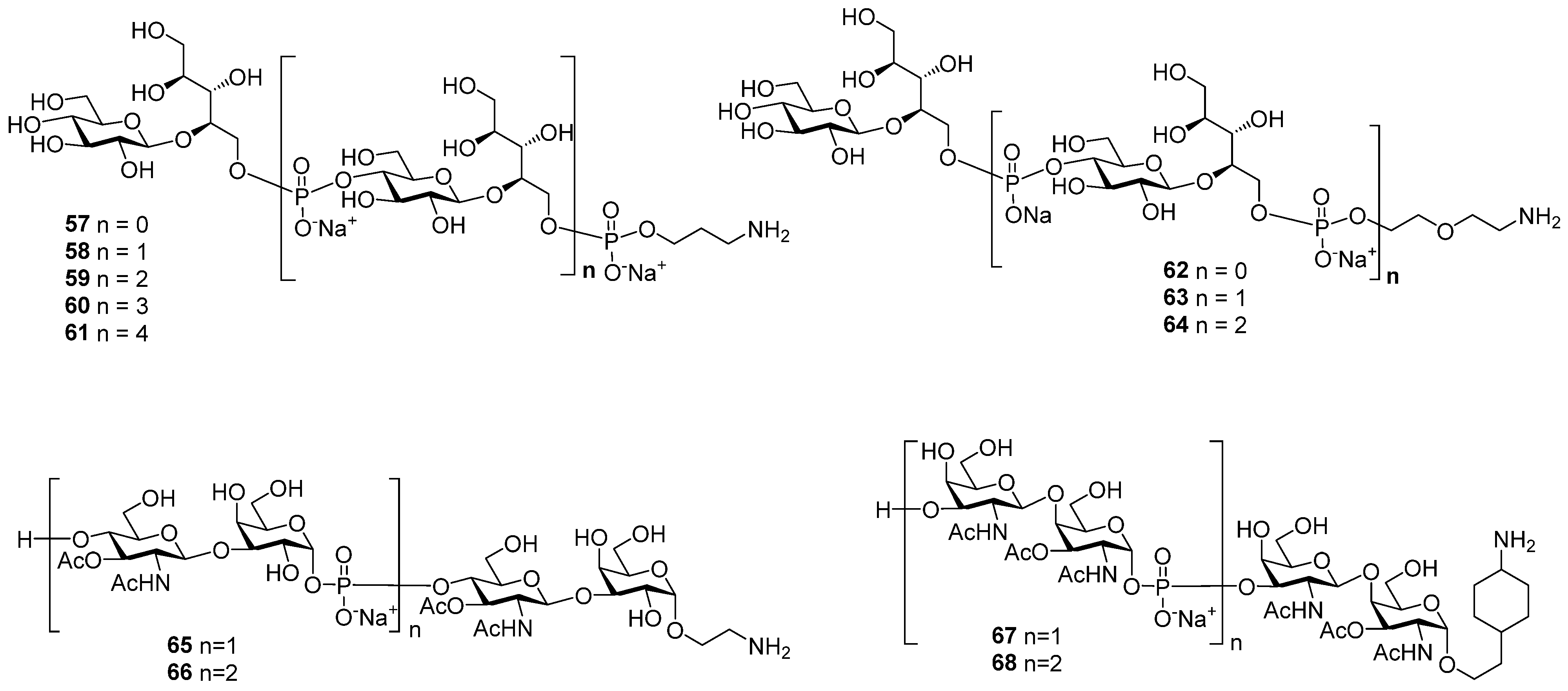
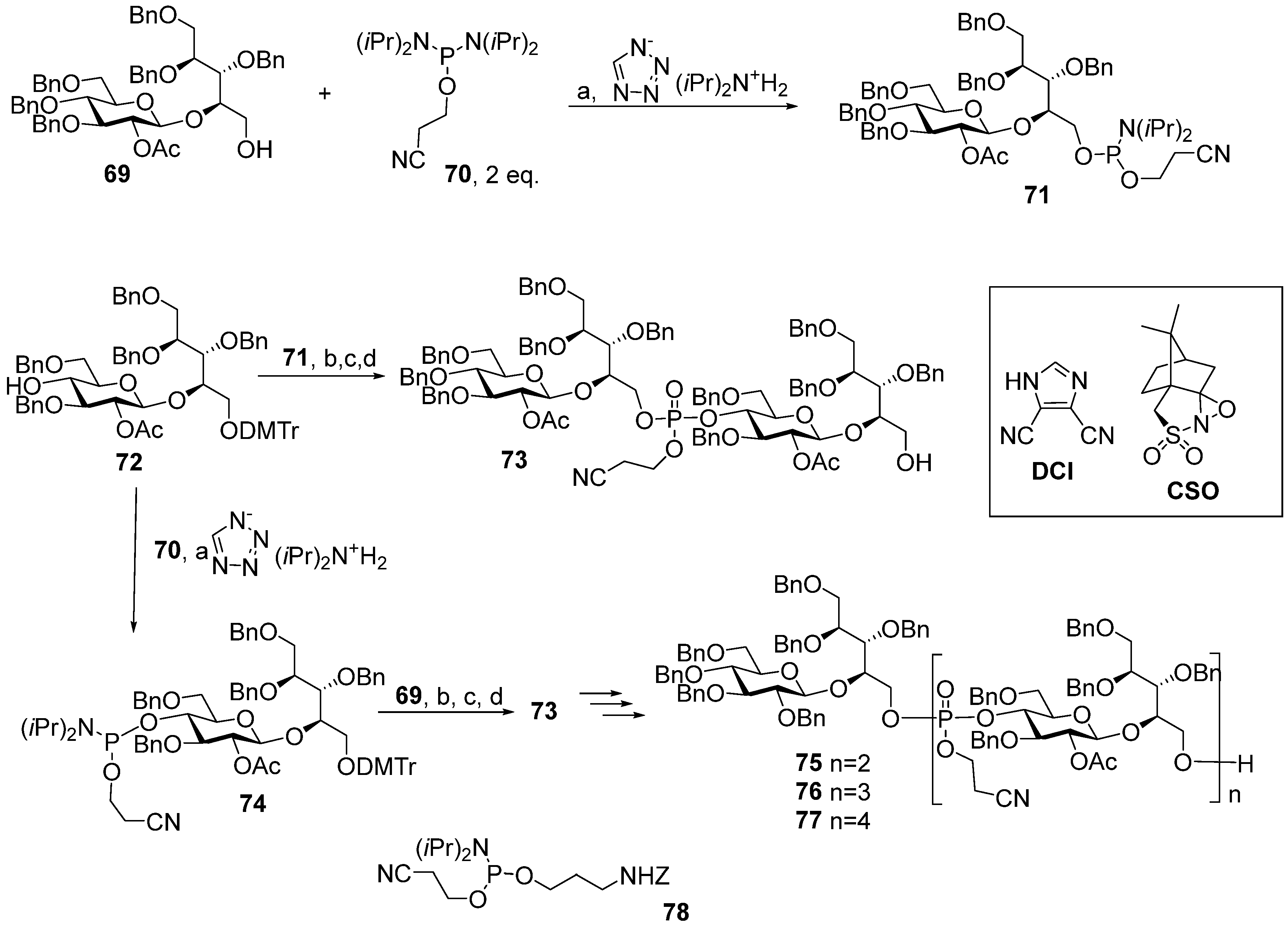
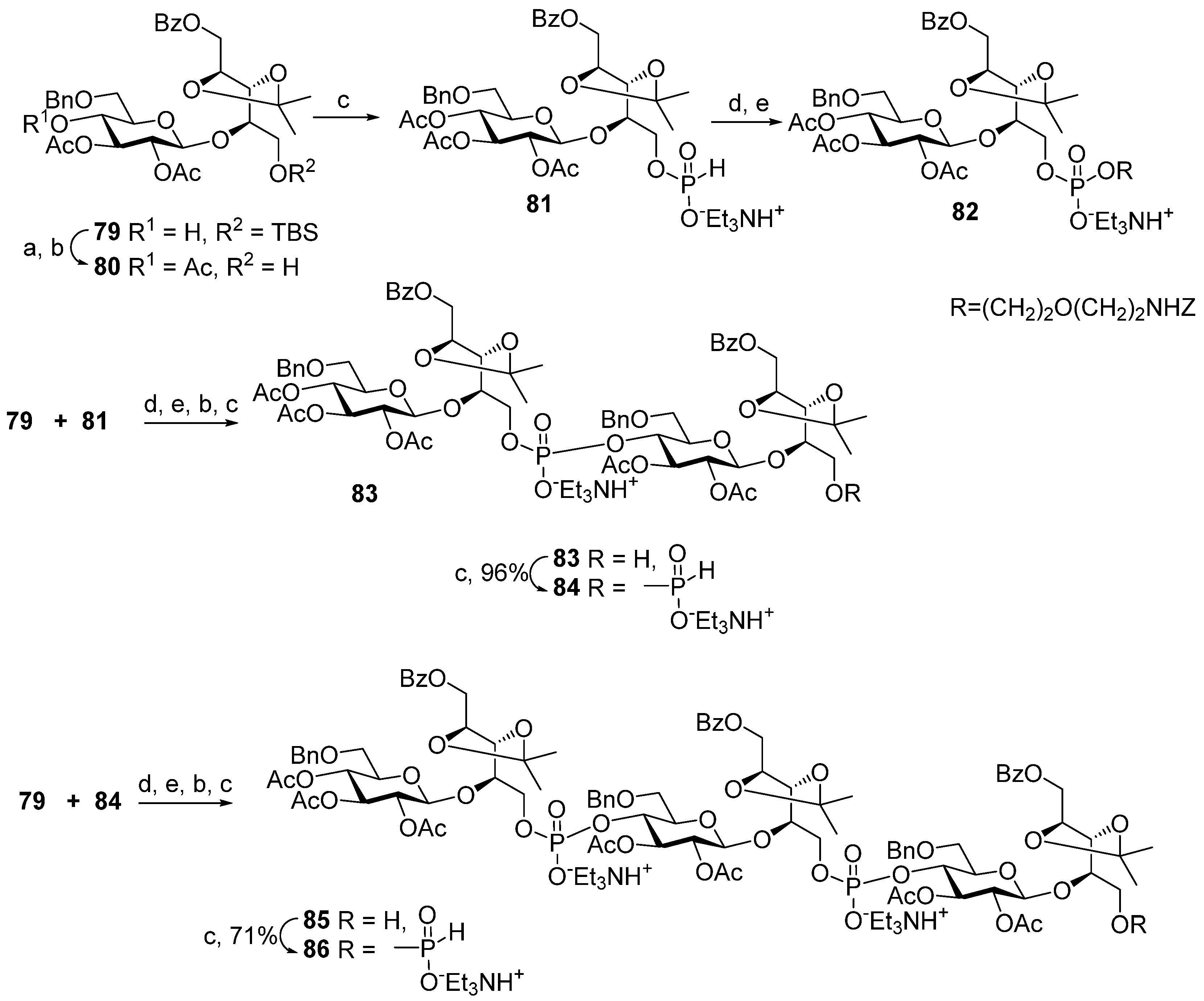



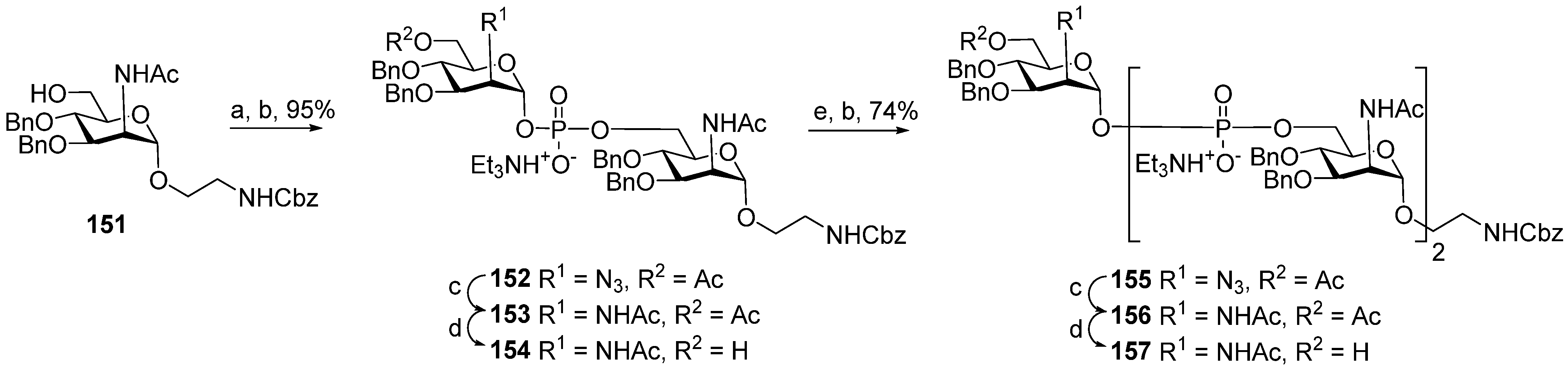
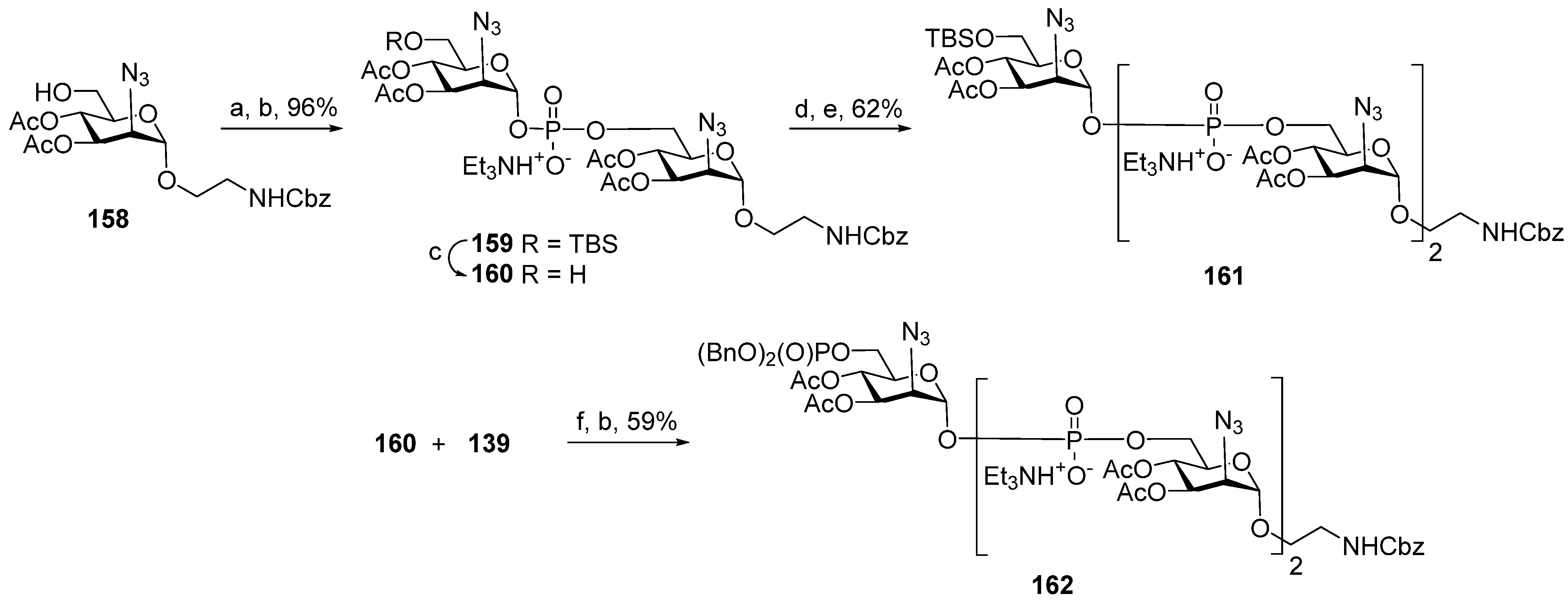
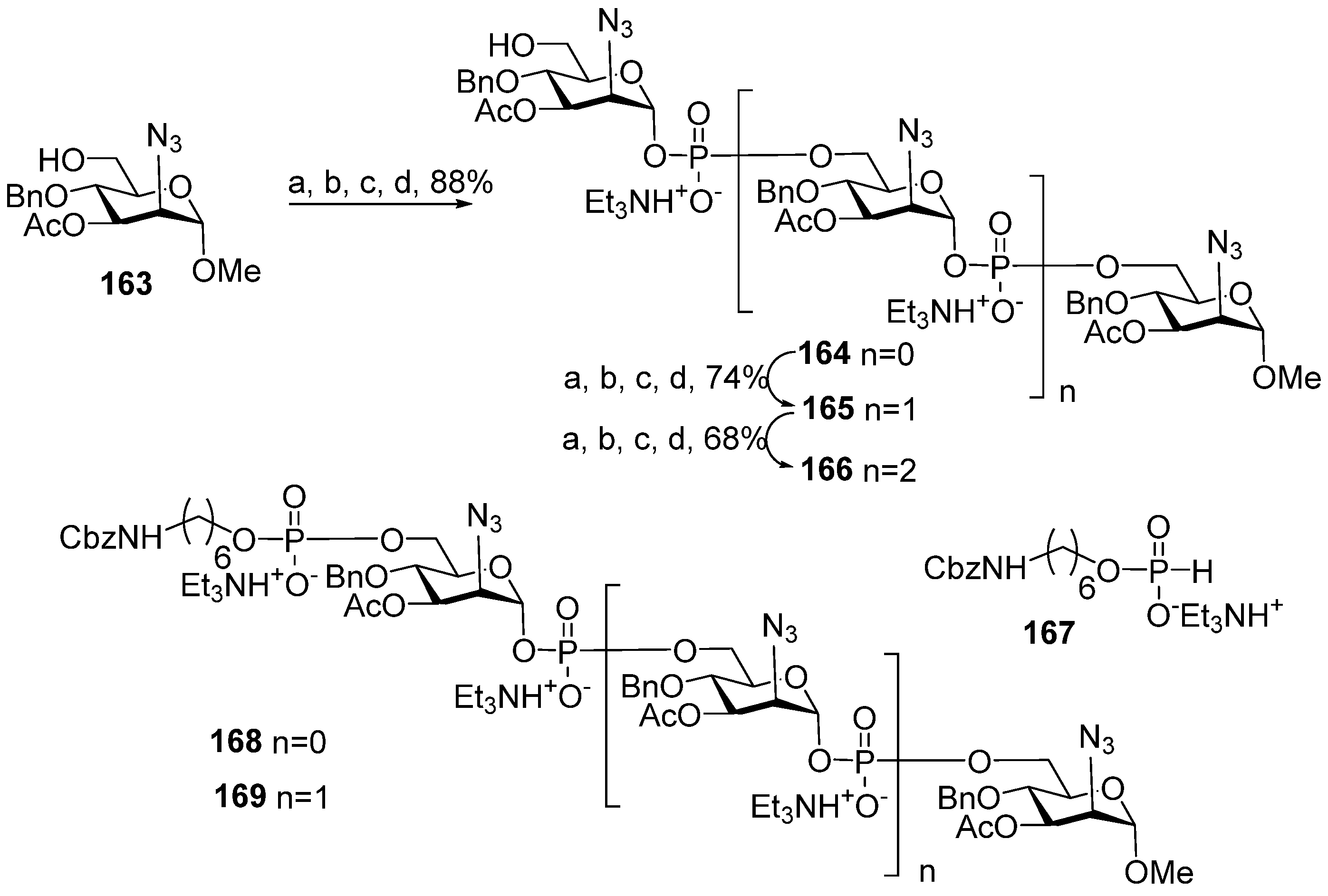
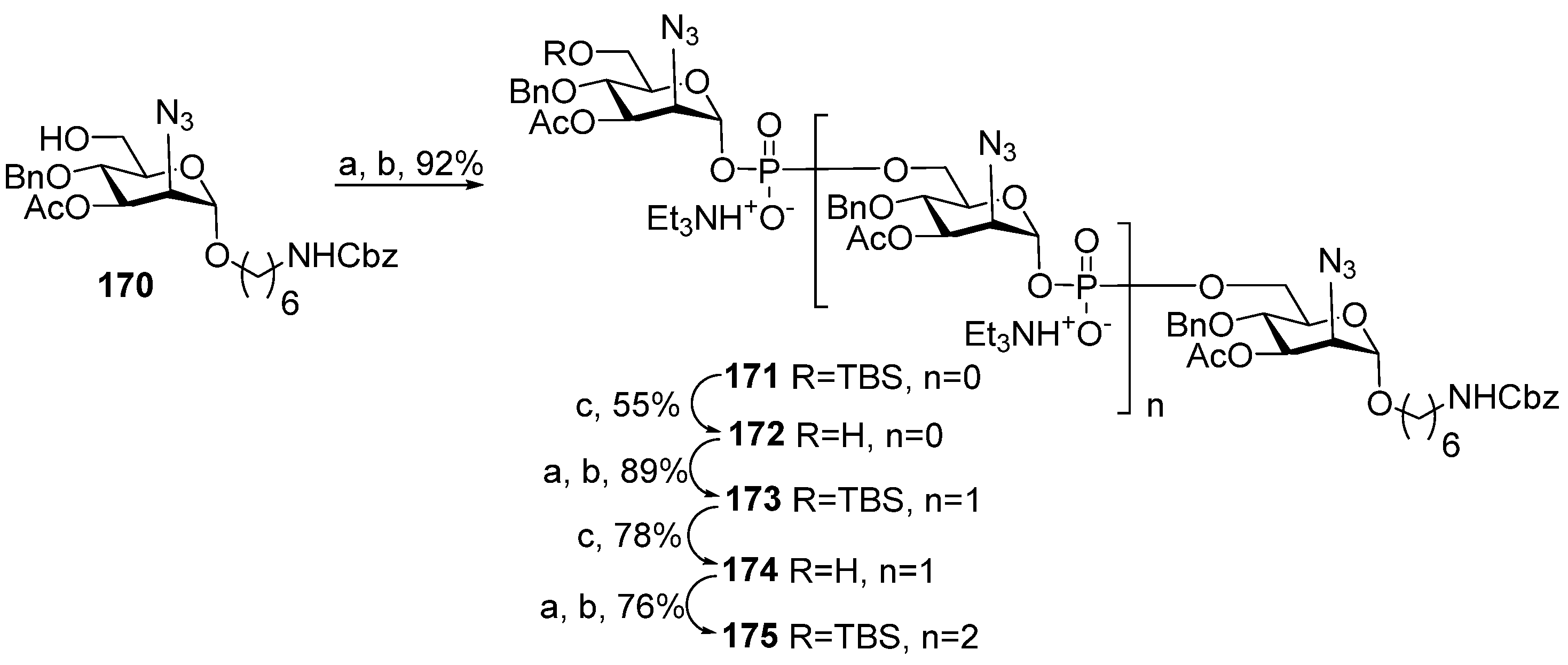
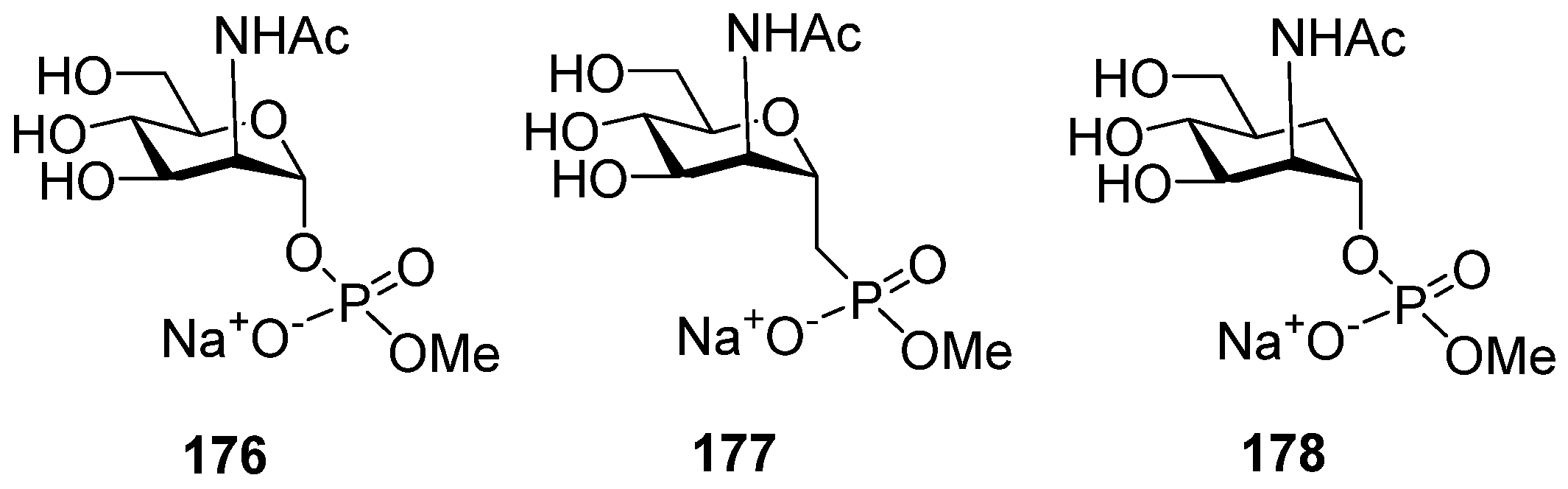
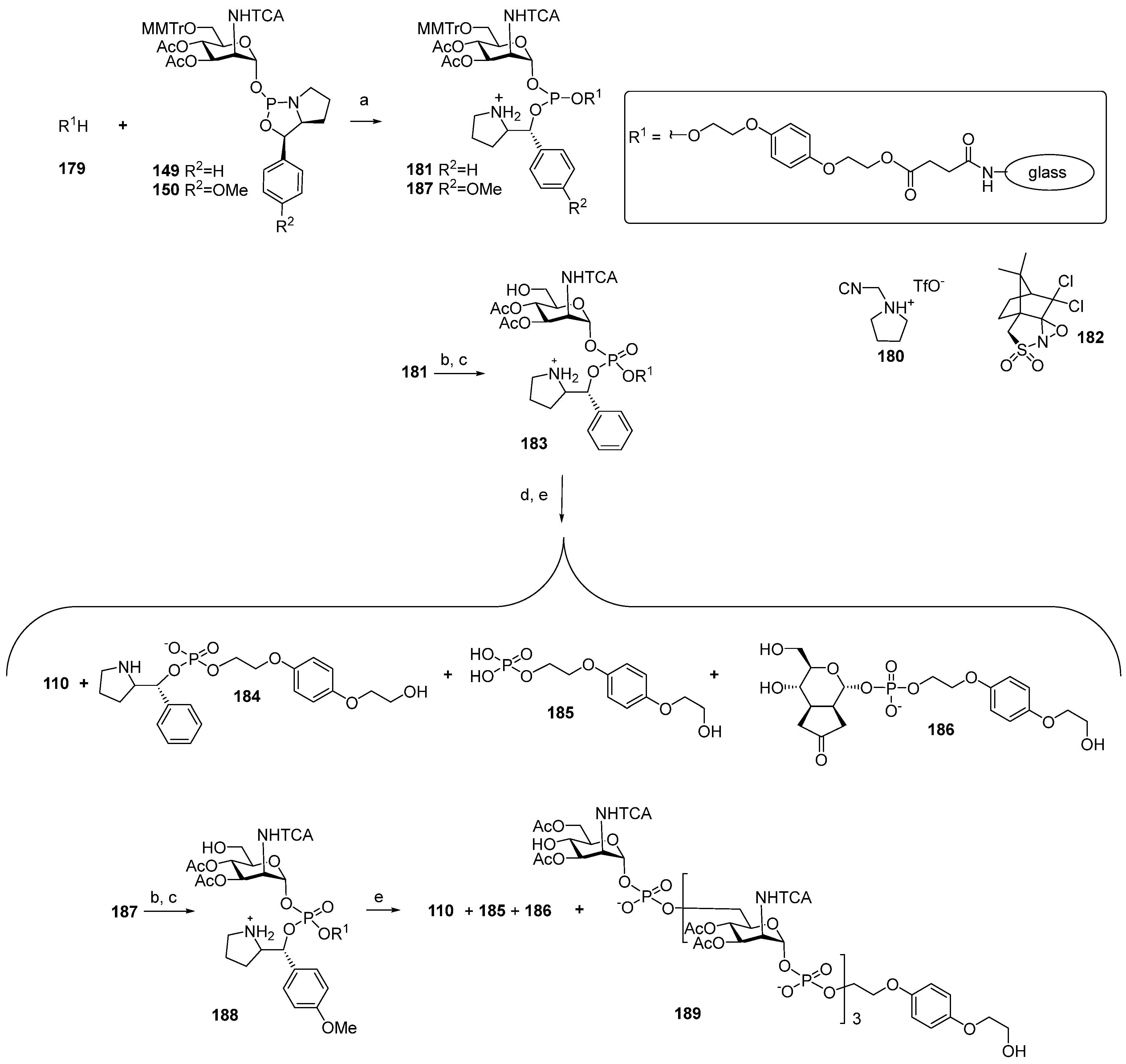


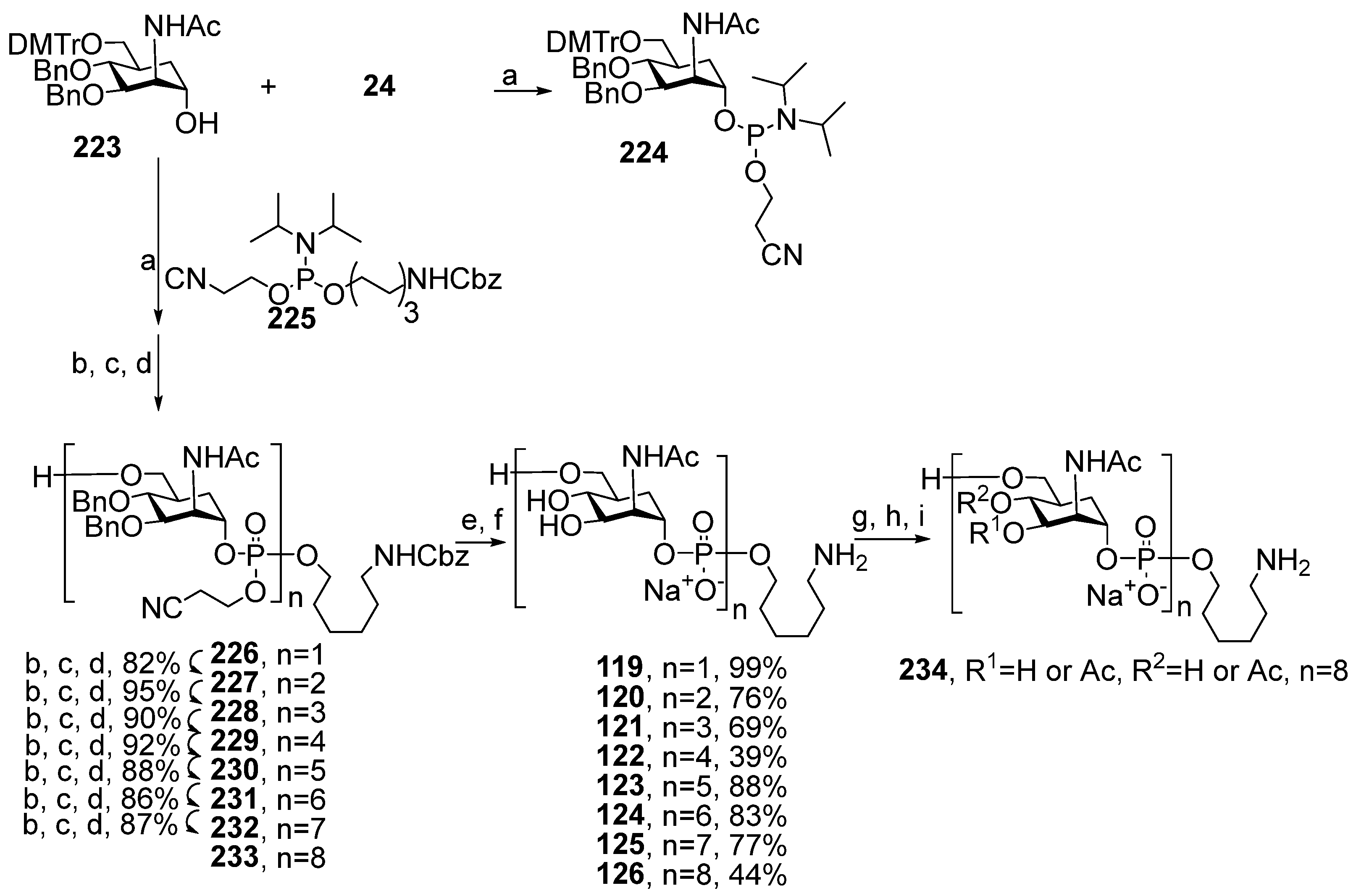
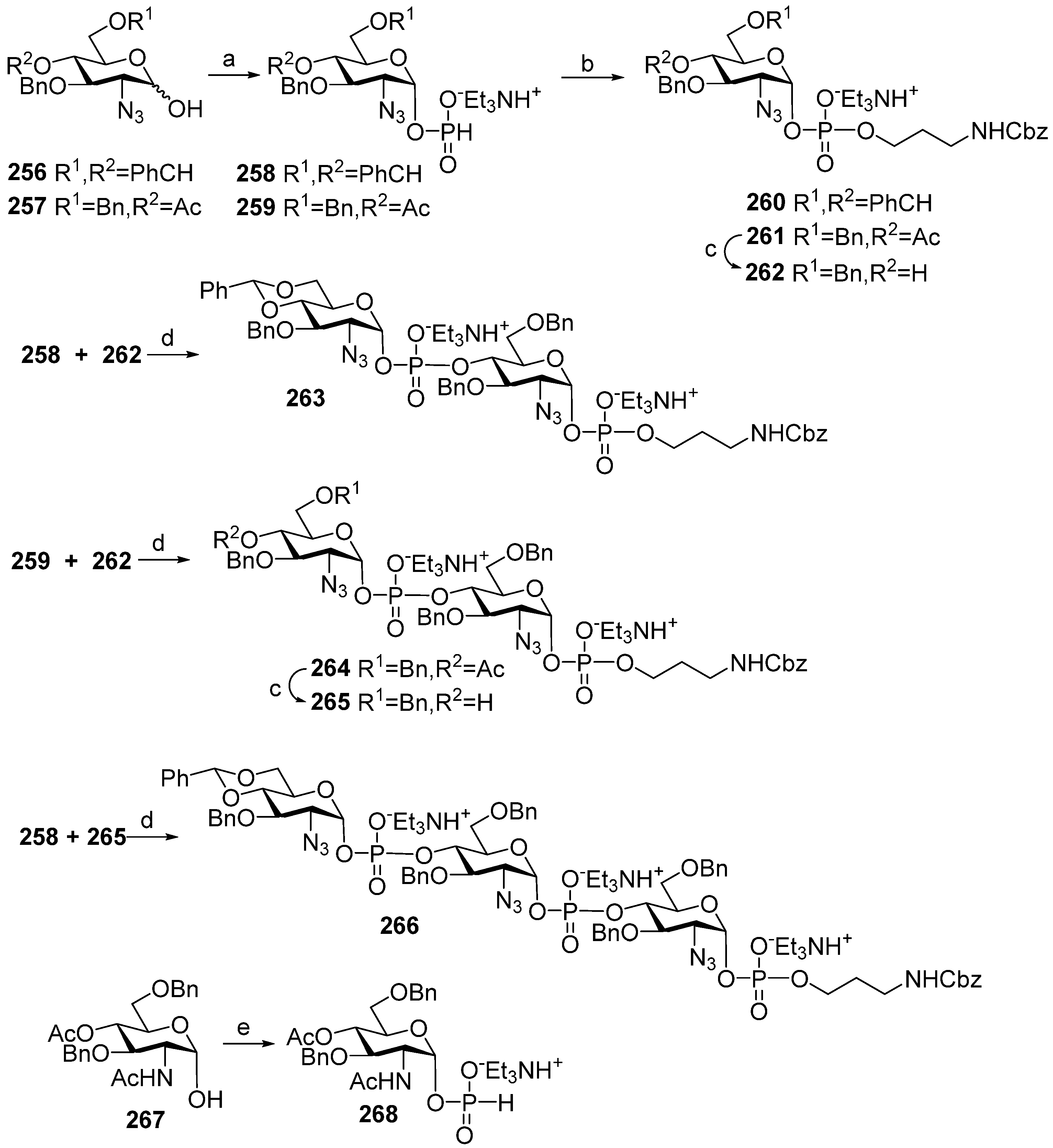
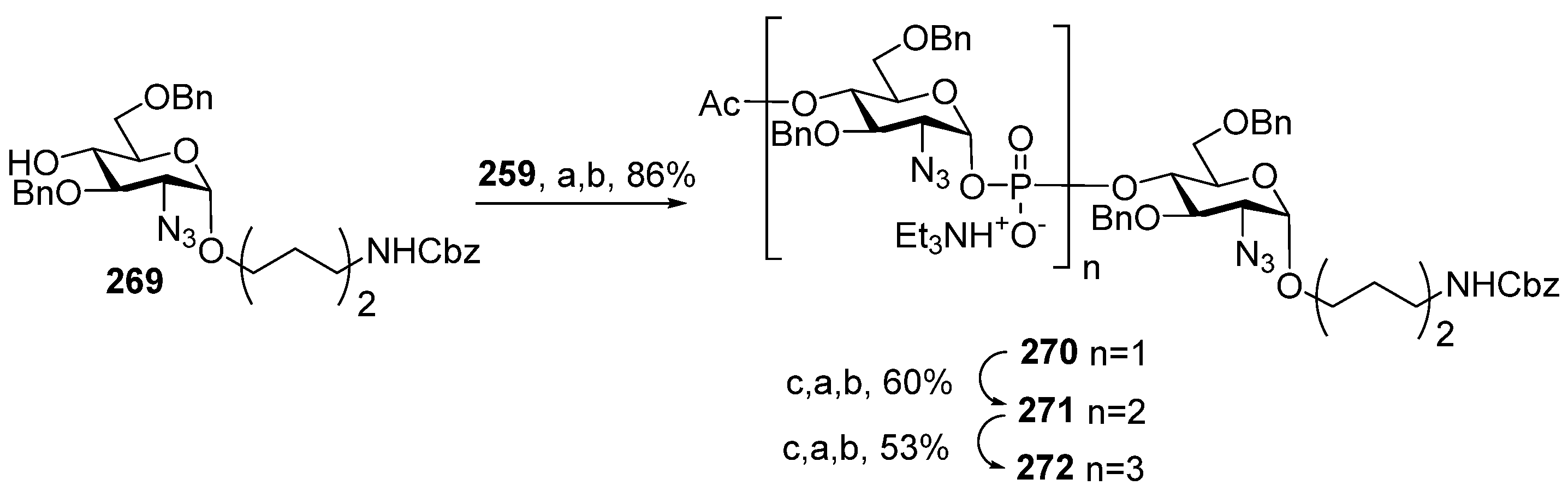
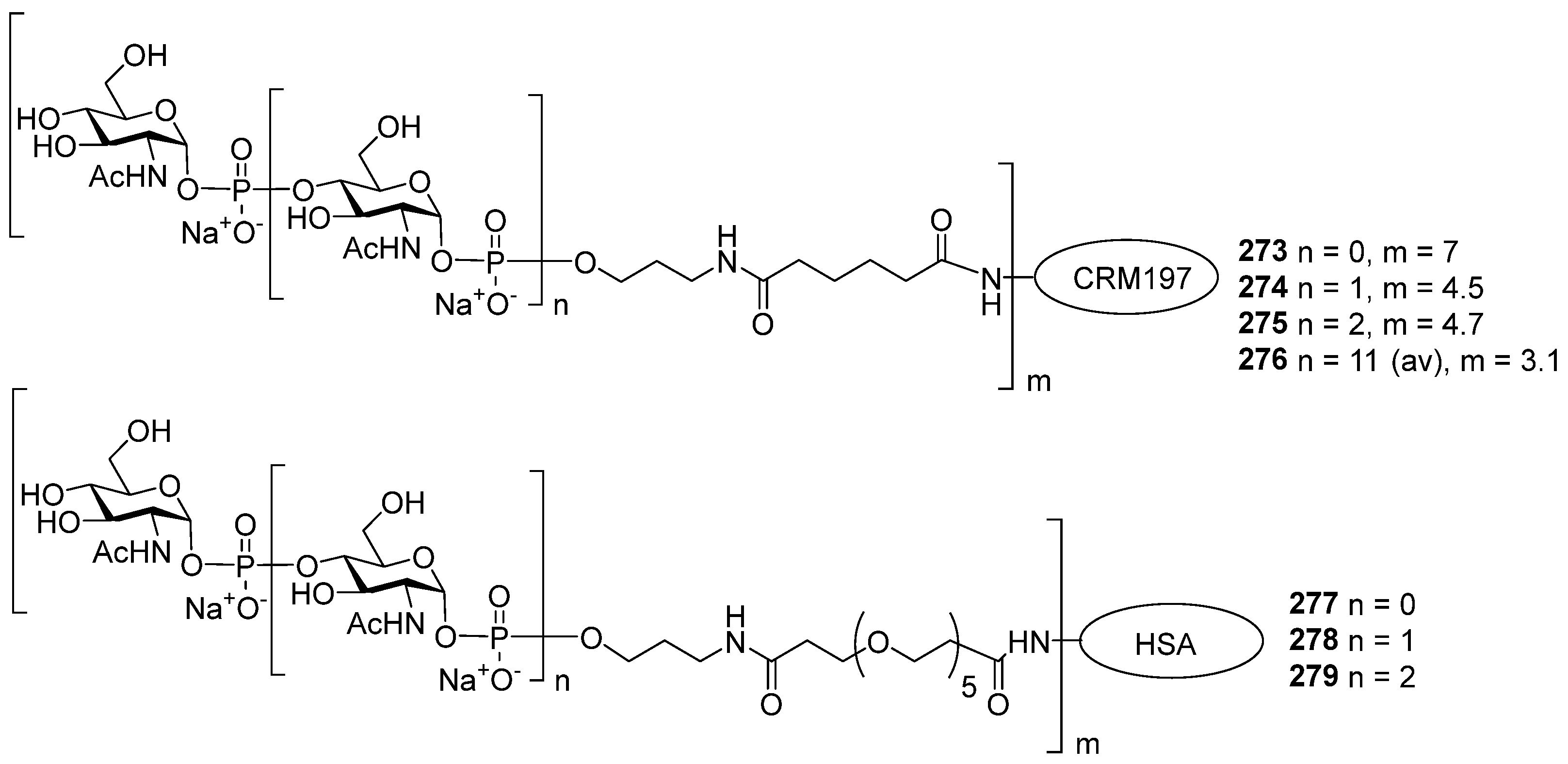
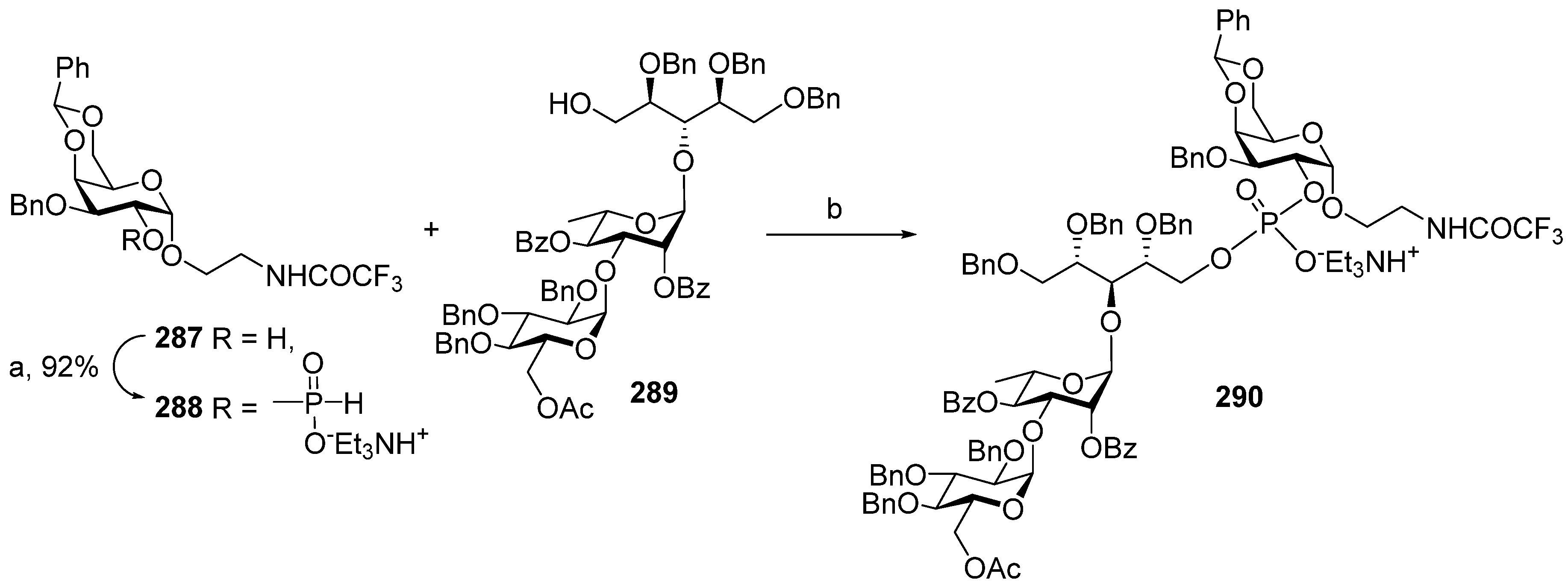

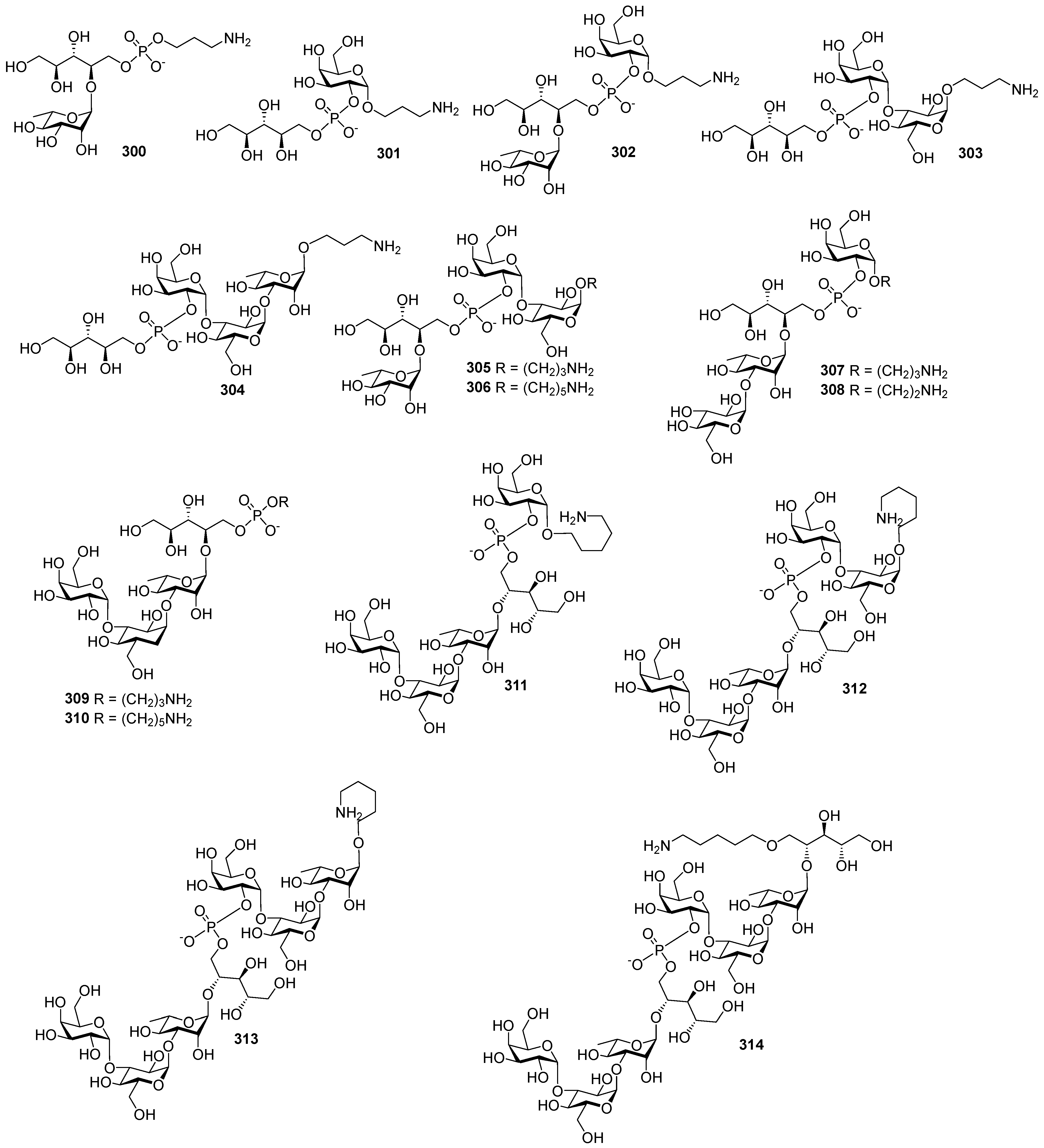

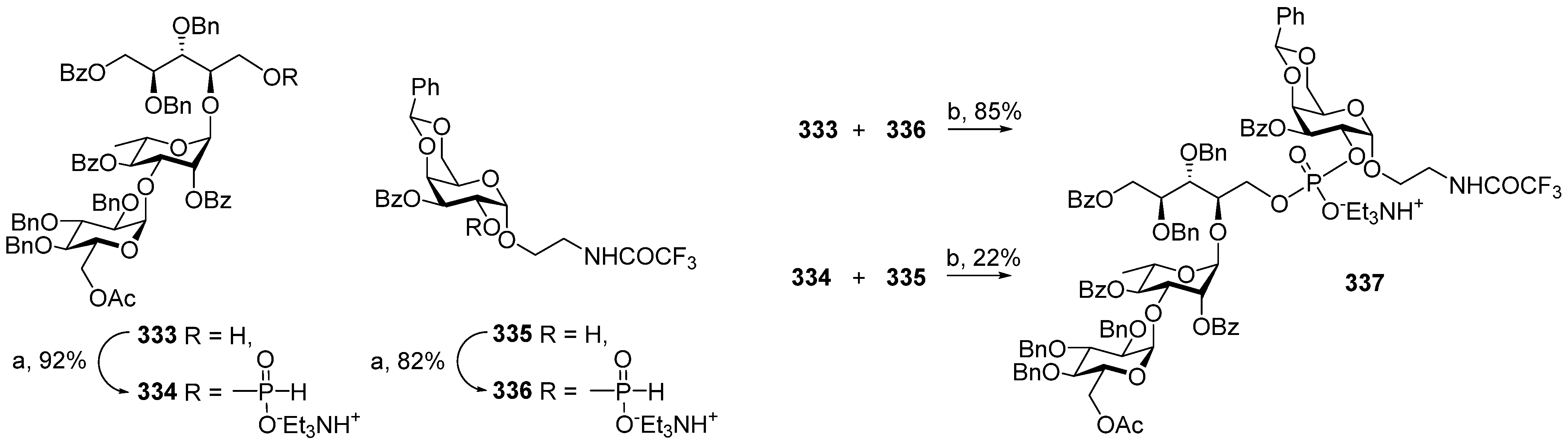


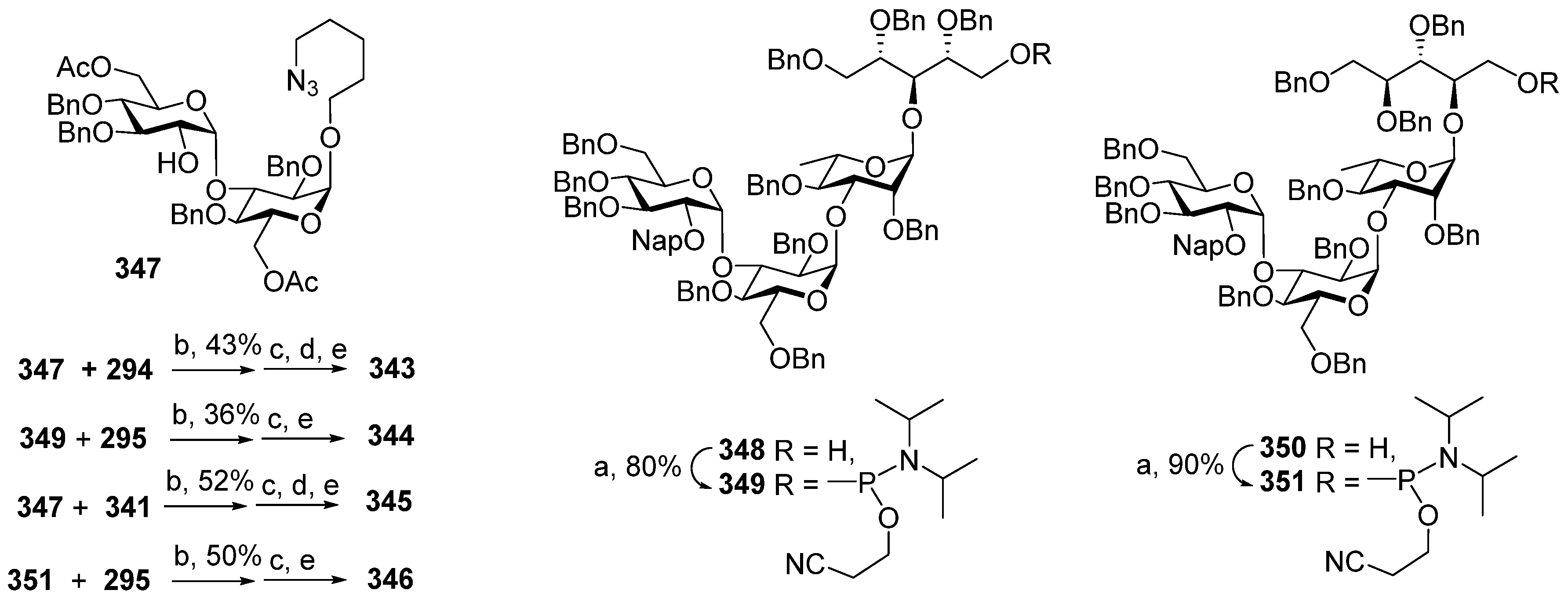
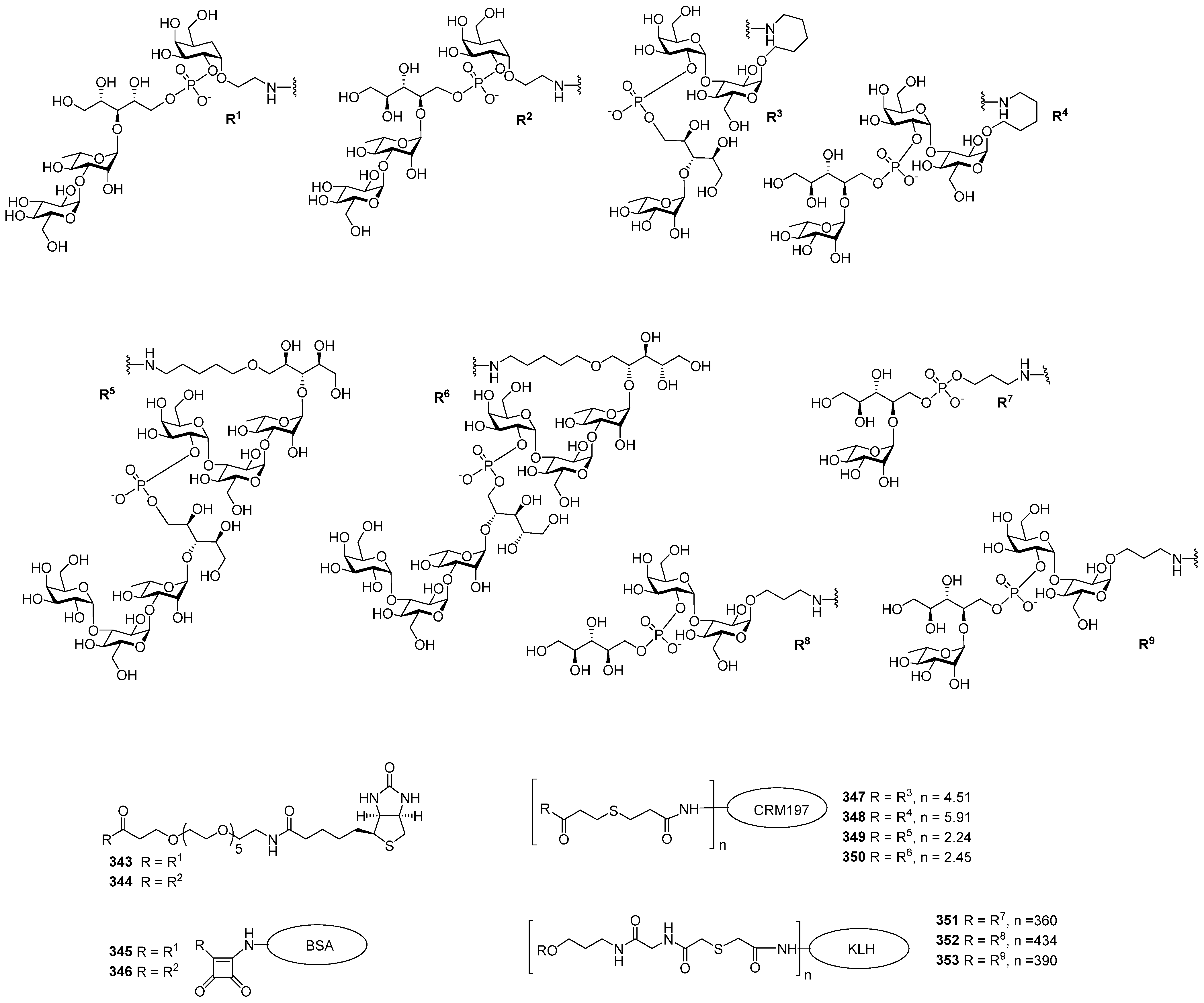
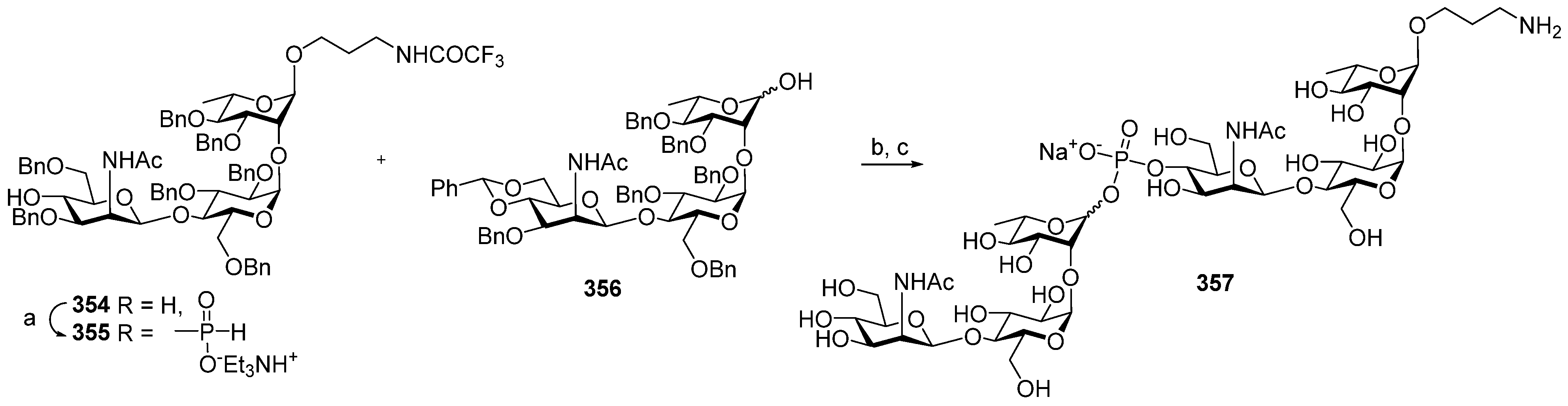
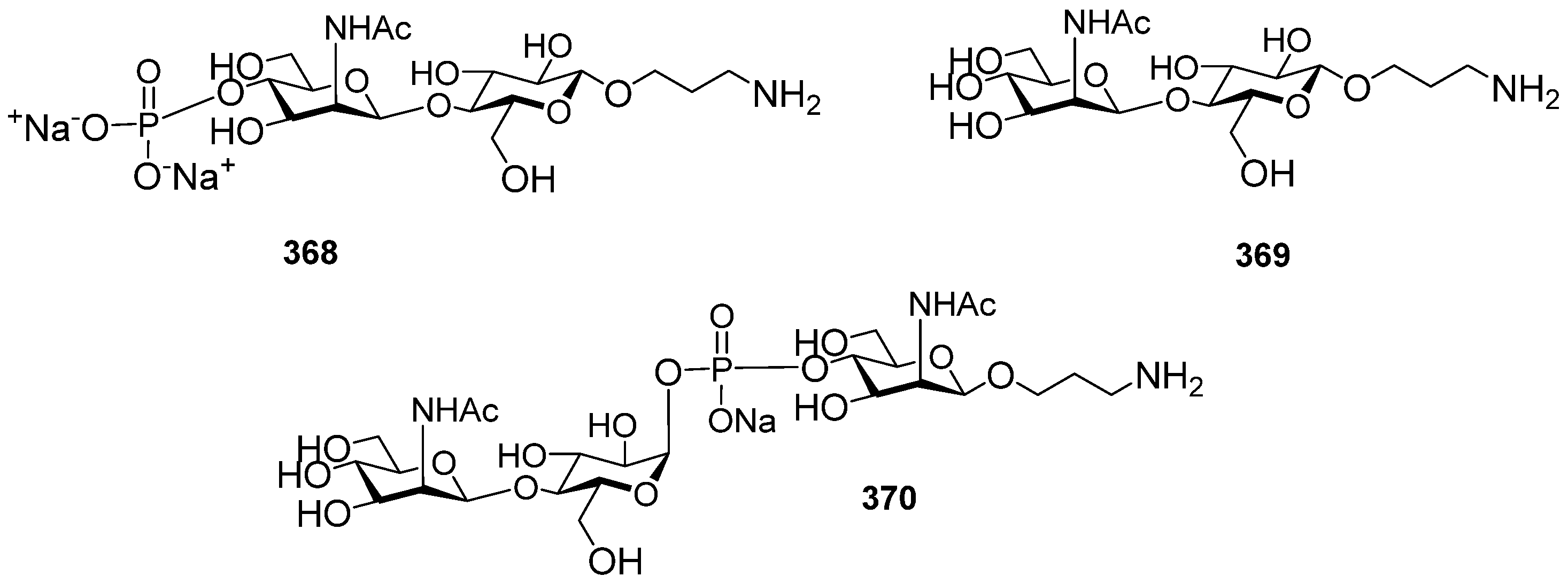
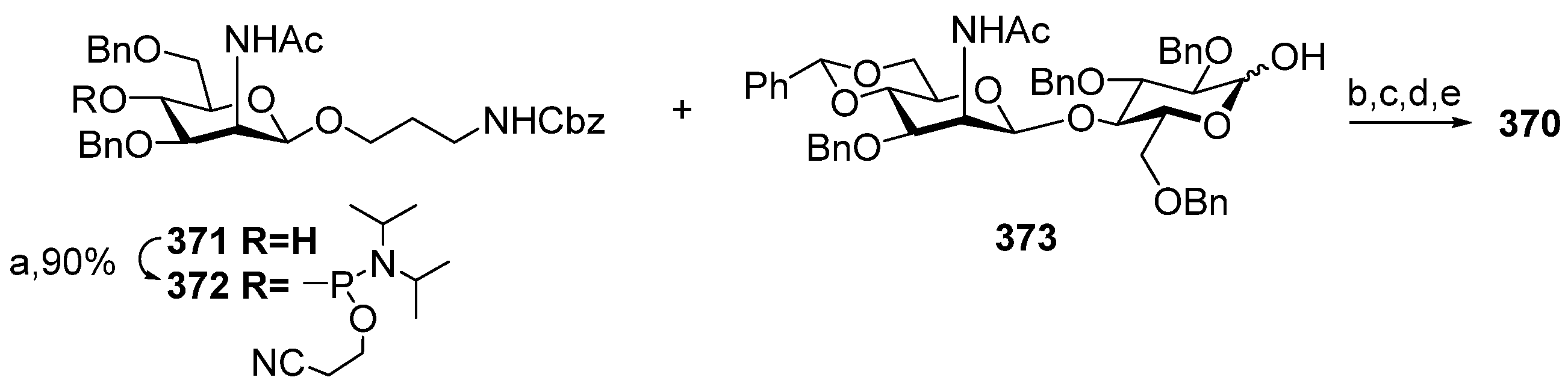
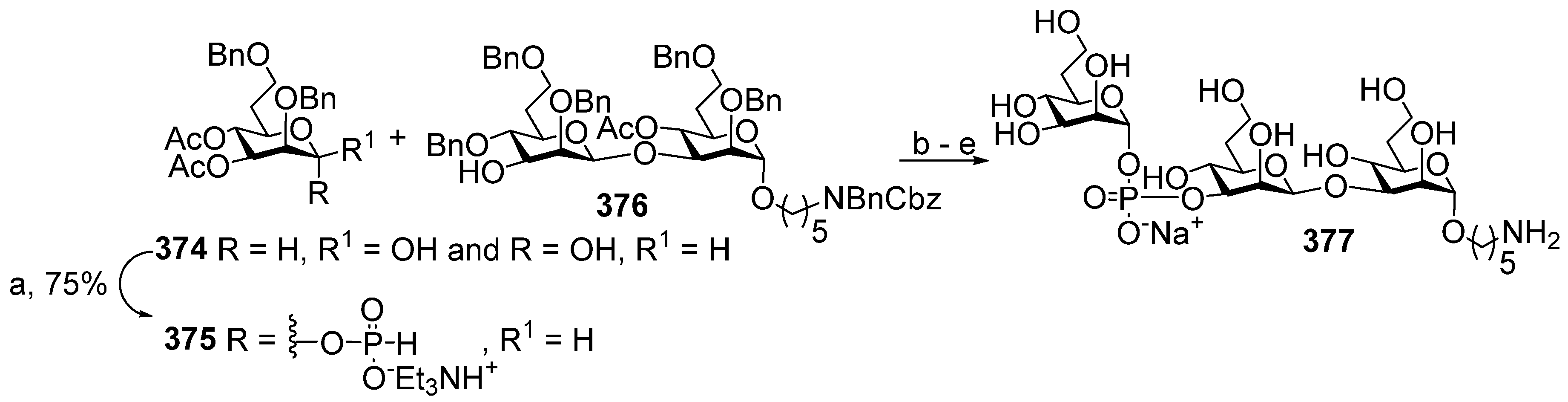

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}