
Nickel-Driven Electrochemical Upgrading of Kraft Lignin to Value-Added Aliphatic and Phenolic Products


 ,
, 
Abstract
1. Introduction
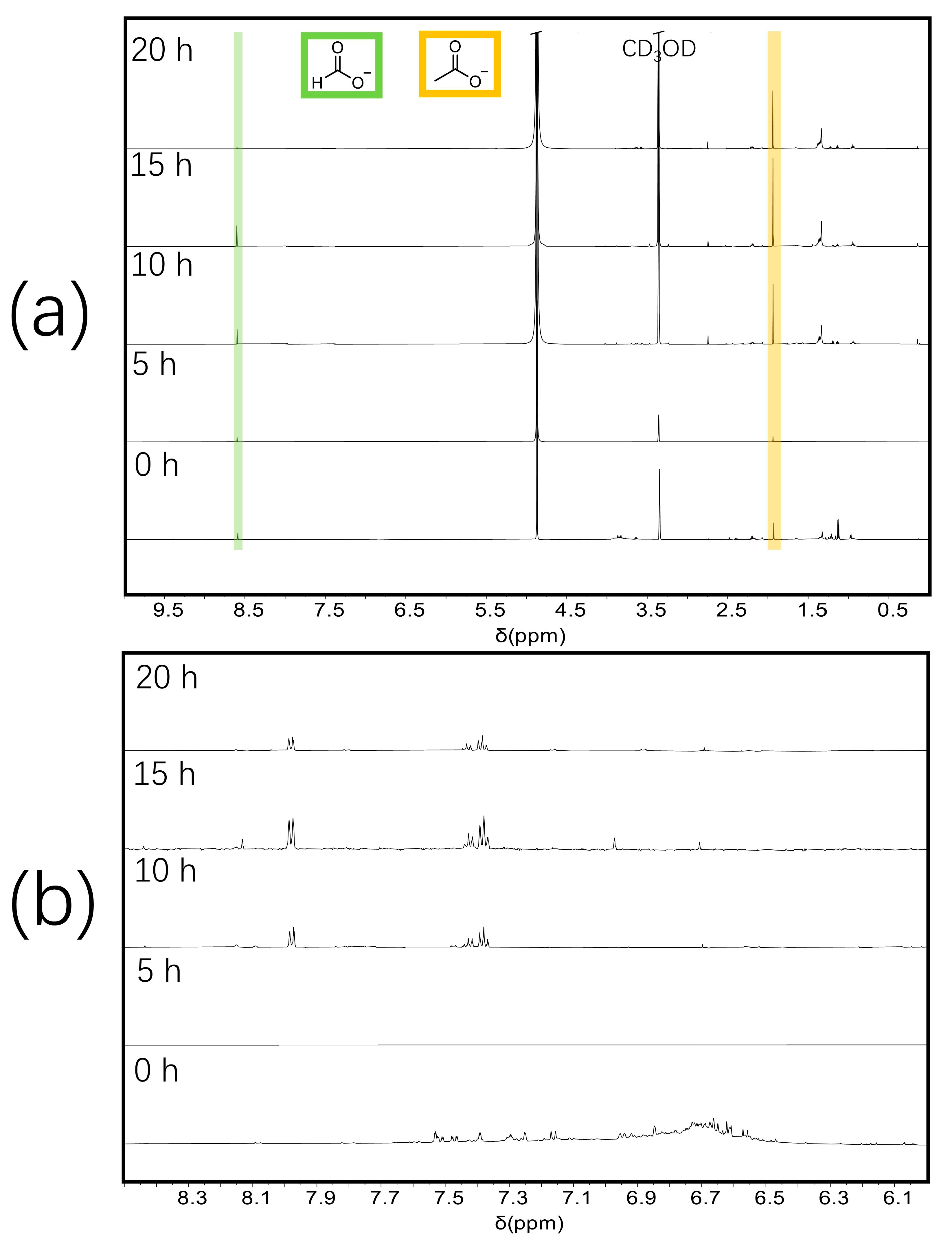
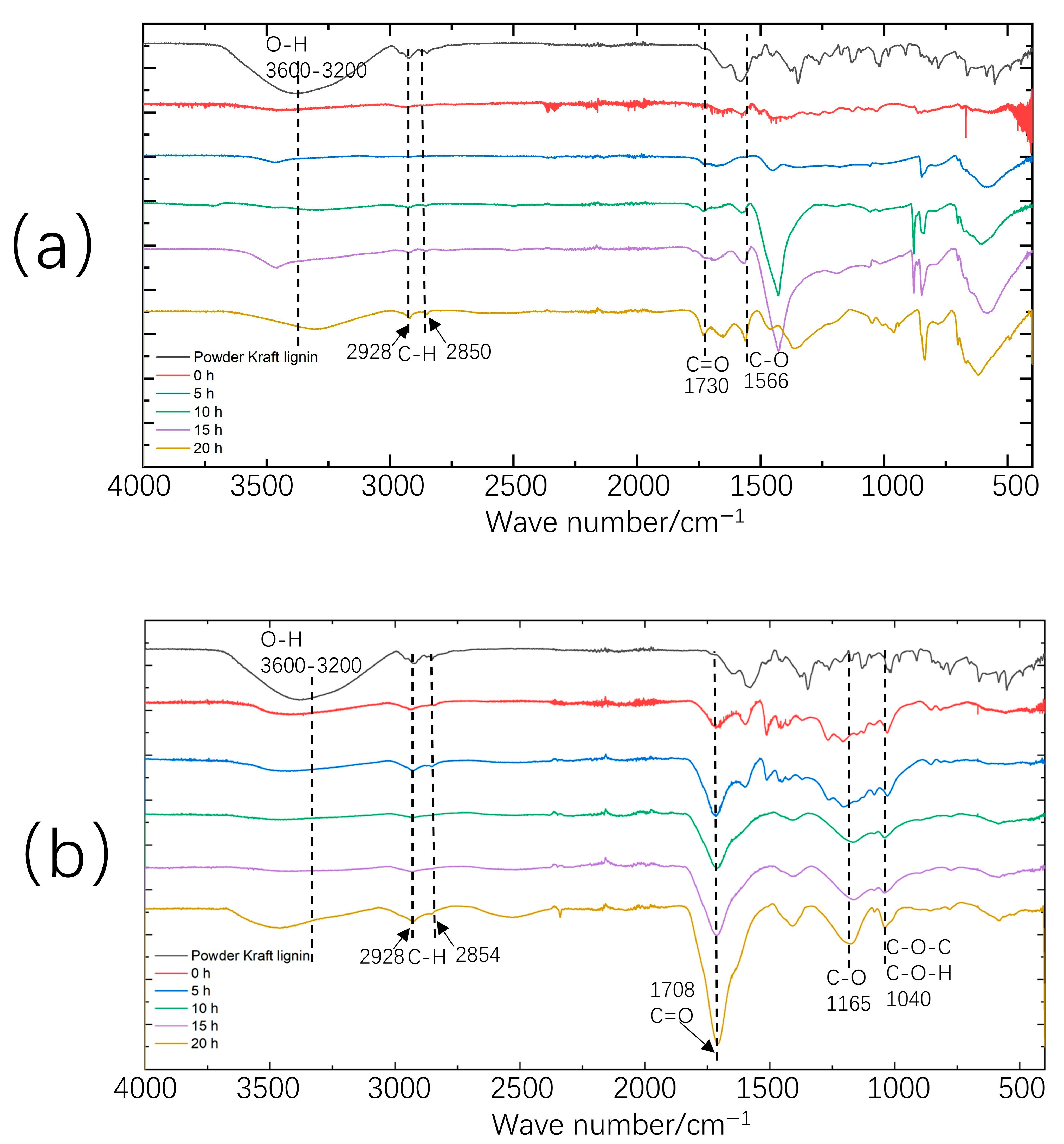
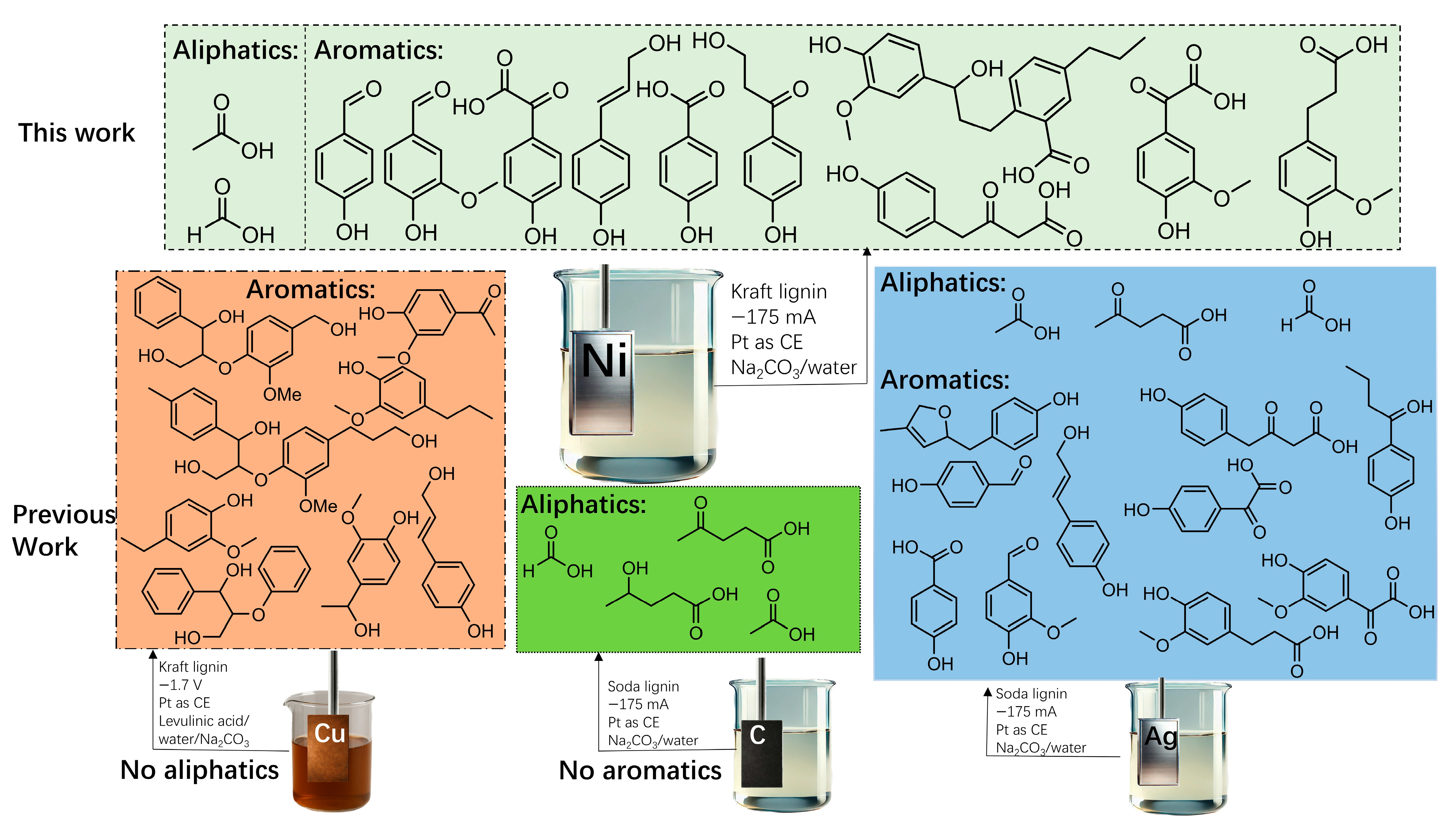
2. Results
3. Materials and Methods
3.1. Materials
3.2. Electrochemical Reactions
3.3. Analytical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boerjan, W.; Ralph, J.; Baucher, M. Lignin Biosynthesis. Annu. Rev. Plant Biol. 2003, 54, 519–546. [Google Scholar] [CrossRef] [PubMed]
- Crestini, C.; Lange, H.; Sette, M.; Argyropoulos, D.S. On the Structure of Softwood Kraft Lignin. Green Chem. 2017, 19, 4104–4121. [Google Scholar] [CrossRef]
- Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin Biosynthesis and Structure. Plant Physiol. 2010, 153, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.K.; Gu, S.; Luo, K.H.; Wang, S.R.; Fang, M.X. The Pyrolytic Degradation of Wood-Derived Lignin from Pulping Process. Bioresour. Technol. 2010, 101, 6136–6146. [Google Scholar] [CrossRef]
- Argyropoulos, D.D.S.; Crestini, C.; Dahlstrand, C.; Furusjö, E.; Gioia, C.; Jedvert, K.; Henriksson, G.; Hulteberg, C.; Lawoko, M.; Pierrou, C.; et al. Kraft Lignin: A Valuable, Sustainable Resource, Opportunities and Challenges. ChemSusChem 2023, 16, e202300492. [Google Scholar] [CrossRef]
- Gavrilescu, D.; Puitel, A.C.; Dutuc, G.; Craciun, G. ENVIRONMENTAL IMPACT OF PULP AND PAPER MILLS. Environ. Eng. Manag. J. 2012, 11, 81–85. [Google Scholar] [CrossRef]
- Rawal, T.B.; Zahran, M.; Dhital, B.; Akbilgic, O.; Petridis, L. The Relation between Lignin Sequence and Its 3D Structure. Biochim. Biophys. Acta (BBA) Gen. Subj. 2020, 1864, 129547. [Google Scholar] [CrossRef]
- da Cruz, M.G.A.; Gueret, R.; Chen, J.; Piątek, J.; Beele, B.; Sipponen, M.H.; Frauscher, M.; Budnyk, S.; Rodrigues, B.V.M.; Slabon, A. Electrochemical Depolymerization of Lignin in a Biomass-Based Solvent. ChemSusChem 2022, 15, e202200718. [Google Scholar] [CrossRef]
- Lindenbeck, L.M.; Barra, V.C.; Dahlhaus, S.; Brand, S.; Wende, L.M.; Beele, B.B.; Schebb, N.H.; Rodrigues, B.V.M.; Slabon, A. Organic Chemicals from Wood: Selective Depolymerization and Dearomatization of Lignin via Aqueous Electrocatalysis. ChemSusChem 2024, 17, e202301617. [Google Scholar] [CrossRef]
- Lindenbeck, L.; Brand, S.; Stallmann, F.; Barra, V.; Frauscher, M.; Beele, B.B.; Slabon, A.; Rodrigues, B.V.M. Silver-Catalyzed Aqueous Electrochemical Valorization of Soda Lignin into Aliphatics and Phenolics. Polymers 2024, 16, 3325. [Google Scholar] [CrossRef]
- Borella, M.; Casazza, A.A.; Garbarino, G.; Riani, P.; Busca, G. A Study of the Pyrolysis Products of Kraft Lignin. Energies 2022, 15, 991. [Google Scholar] [CrossRef]
- Wang, S.; Li, X.; Ma, R.; Song, G. Catalytic Hydrogenolysis of Lignin into Serviceable Products. Acc. Chem. Res. 2025, 58, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, F. Catalytic Lignin Depolymerization to Aromatic Chemicals. Acc. Chem. Res. 2020, 53, 470–484. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Y.; Duan, H. Recent Progress in Electrocatalytic Conversion of Lignin: From Monomers, Dimers, to Raw Lignin. Precis. Chem. 2024, 2, 428–446. [Google Scholar] [CrossRef]
- Kärkäs, M.D.; Matsuura, B.S.; Monos, T.M.; Magallanes, G.; Stephenson, C.R.J. Transition-Metal Catalyzed Valorization of Lignin: The Key to a Sustainable Carbon-Neutral Future. Org. Biomol. Chem. 2016, 14, 1853–1914. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, Z.; Luo, Y.; Wei, H.; Chen, J.; Liu, D.; Zhang, W. Nickel-Catalyzed Asymmetric Hydrogenation for the Preparation of α-Substituted Propionic Acids. Nat. Commun. 2024, 15, 5482. [Google Scholar] [CrossRef]
- He, J.; Zhao, C.; Lercher, J.A. Ni-Catalyzed Cleavage of Aryl Ethers in the Aqueous Phase. J. Am. Chem. Soc. 2012, 134, 20768–20775. [Google Scholar] [CrossRef]
- Ferreira, E.B.; Jerkiewicz, G. On the Electrochemical Reduction of β-Ni(OH)2 to Metallic Nickel. Electrocatalysis 2021, 12, 199–209. [Google Scholar] [CrossRef]
- Mott, N.F.; Wills, H.H. The Electrical Conductivity of Transition Metals. Proc. R. Soc. Lond. A 1936, 153, 699–717. [Google Scholar]
- Wang, S.; Shen, Z.; Osatiashtiani, A.; Nabavi, S.A.; Clough, P.T. Ni-Based Bimetallic Catalysts for Hydrogen Production via (Sorption-Enhanced) Steam Methane Reforming. Chem. Eng. J. 2024, 486, 150170. [Google Scholar] [CrossRef]
- Schweitzer, P.A. Metallic Materials: Physical, Mechanical, and Corrosion Properties (Corrosion Technology); CRC Press: Boca Raton, FL, USA, 2003; ISBN 978-0-8247-0878-8. [Google Scholar]
- Garedew, M.; Lin, F.; Song, B.; DeWinter, T.M.; Jackson, J.E.; Saffron, C.M.; Lam, C.H.; Anastas, P.T. Greener Routes to Biomass Waste Valorization: Lignin Transformation Through Electrocatalysis for Renewable Chemicals and Fuels Production. ChemSusChem 2020, 13, 4214–4237. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Waldvogel, S.R. Selective Electrochemical Degradation of Lignosulfonate to Bio-Based Aldehydes. ChemSusChem 2023, 16, e202202300. [Google Scholar] [CrossRef]
- Lin, F.; Tse, H.-Y.; Erythropel, H.C.; Petrović, P.V.; Garedew, M.; Chen, J.; Lam, J.C.-H.; Anastas, P.T. Development of a Ni-Promoted, Selective Electrochemical Reductive Cleavage of the C–O Bond in Lignin Model Compound Benzyl Phenyl Ether. Green Chem. 2022, 24, 6295–6305. [Google Scholar] [CrossRef]
- Lin, F.; Petrović, P.V.; Tse, H.Y.; Erythropel, H.C.; Lam, J.C.; Anastas, P.T. Mechanistic Investigation of a Ni-Catalyzed Electrochemical Reductive Cleavage of the α-O-4 Bond in the Lignin Model Compound Benzyl Phenyl Ether. Green Chem. 2023, 25, 9720–9732. [Google Scholar] [CrossRef]
- Tofani, G.; Cornet, I.; Tavernier, S. Multiple Linear Regression to Predict the Brightness of Waste Fibres Mixtures before Bleaching. Chem. Pap. 2022, 76, 4351–4365. [Google Scholar] [CrossRef]
- Wijaya, Y.P.; Smith, K.J.; Kim, C.S.; Gyenge, E.L. Electrocatalytic Hydrogenation and Depolymerization Pathways for Lignin Valorization: Toward Mild Synthesis of Chemicals and Fuels from Biomass. Green Chem. 2020, 22, 7233–7264. [Google Scholar] [CrossRef]
- Amit, T.A.; Roy, R.; Raynie, D.E. Thermal and Structural Characterization of Two Commercially Available Technical Lignins for Potential Depolymerization via Hydrothermal Liquefaction. Curr. Res. Green Sustain. Chem. 2021, 4, 100106. [Google Scholar] [CrossRef]
- Deepa, A.K.; Dhepe, P.L. Lignin Depolymerization into Aromatic Monomers over Solid Acid Catalysts. ACS Catal. 2015, 5, 365–379. [Google Scholar] [CrossRef]
- Lindenbeck, L.M.; Beele, B.B.; Lützenkirchen-Hecht, D.F.; Rodrigues, B.V.M.; Slabon, A. Electrochemical Biomass Depolymerization: Will Complex Catalysts Trigger High Product Selectivity? Chem. Mater. 2024, 36, 9173–9188. [Google Scholar] [CrossRef]
- Da Cruz, M.G.A.; Onwumere, J.N.; Chen, J.; Beele, B.; Yarema, M.; Budnyk, S.; Slabon, A.; Rodrigues, B.V.M. Solvent-Free Synthesis of Photoluminescent Carbon Nanoparticles from Lignin-Derived Monomers as Feedstock. Green Chem. Lett. Rev. 2023, 16, 2196031. [Google Scholar] [CrossRef]
- Slabon, A.; Rodrigues, B.V.M. To Break, or Not to Break: Is Selective Depolymerization of Lignin a Riemann Hypothesis Rather than a Solution? Green Chem. 2024, 27, 2178–2183. [Google Scholar] [CrossRef]
- Zilbermann, I.; Maimon, E.; Cohen, H.; Meyerstein, D. Redox Chemistry of Nickel Complexes in Aqueous Solutions. Chem. Rev. 2005, 105, 2609–2626. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Guo, H.; Li, C.; Zhou, P.; Zhang, Z. Selective Cleavage of Lignin and Lignin Model Compounds without External Hydrogen, Catalyzed by Heterogeneous Nickel Catalysts. Chem. Sci. 2019, 10, 4458–4468. [Google Scholar] [CrossRef] [PubMed]
- Ryabchuk, P.; Agostini, G.; Pohl, M.-M.; Lund, H.; Agapova, A.; Junge, H.; Junge, K.; Beller, M. Intermetallic Nickel Silicide Nanocatalyst—A Non-Noble Metal–Based General Hydrogenation Catalyst. Sci. Adv. 2018, 4, eaat0761. [Google Scholar] [CrossRef]
- Song, Q.; Wang, F.; Xu, J. Hydrogenolysis of Lignosulfonate into Phenols over Heterogeneous Nickel Catalysts. Chem. Commun. 2012, 48, 7019. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Time | Kraft Lignin | 0 h | 5 h | 10 h | 15 h | 20 h |
| Yield of Work-up A [wt%] | 0.9 | 1.5 | 1.8 | 3.1 | 4.9 | 5.7 |
| Yield of Work-up B [wt%] | 5.9 | 6.6 | 13.7 | 20.1 | 33.2 | 42.3 |
| m/z | 121.0295 | 137.0246 | 149.0610 | 151.0402 |
|---|---|---|---|---|
| Chemical structure |  | 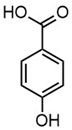 |  | 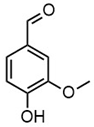 |
| Name | 4-hydroxybenzaldehyde | 4-hydroxybenzoic acid | (E)-4-(3-hydroxyprop-1-en-1-yl)phenol; Cumarylalcohol | 4-hydroxy-3-methoxybenzaldehyde; Vanillin |
| m/z | 165.0195 | 165.0559 | 193.0507 | 195.0300 |
| Chemical structure |  | 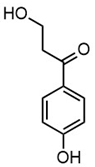 | 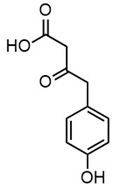 | 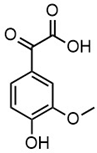 |
| Name | 2-(4-hydroxyphenyl)-2-oxoacetic acid | 3-hydroxy-1-(4-hydroxyphenyl)-1-propanone | 4-(4-hydroxyphenyl)-3-oxobutanoic acid | 2-(4-hydroxy-3-methoxyphenyl)-2-oxoacetic acid |
| m/z | 195.0664 | 343.1558 | ||
| Chemical structure | 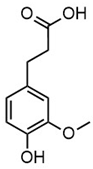 | 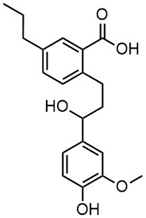 | ||
| Name | 3-(4-hydroxy-3-methoxyphenyl)propanoic acid | 2-(3-hydroxy-3-(4-hydroxy-3-methoxyphenyl)propyl)-5-propylbenzoic acid |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Lindenbeck, L.M.; Frauscher, M.; Beele, B.B.; Rodrigues, B.V.M.; Slabon, A. Nickel-Driven Electrochemical Upgrading of Kraft Lignin to Value-Added Aliphatic and Phenolic Products. Molecules 2025, 30, 2544. https://doi.org/10.3390/molecules30122544
Liu Y, Lindenbeck LM, Frauscher M, Beele BB, Rodrigues BVM, Slabon A. Nickel-Driven Electrochemical Upgrading of Kraft Lignin to Value-Added Aliphatic and Phenolic Products. Molecules. 2025; 30(12):2544. https://doi.org/10.3390/molecules30122544
Chicago/Turabian StyleLiu, Yanbing, Lucie M. Lindenbeck, Marcella Frauscher, Björn B. Beele, Bruno V. Manzolli Rodrigues, and Adam Slabon. 2025. "Nickel-Driven Electrochemical Upgrading of Kraft Lignin to Value-Added Aliphatic and Phenolic Products" Molecules 30, no. 12: 2544. https://doi.org/10.3390/molecules30122544
APA StyleLiu, Y., Lindenbeck, L. M., Frauscher, M., Beele, B. B., Rodrigues, B. V. M., & Slabon, A. (2025). Nickel-Driven Electrochemical Upgrading of Kraft Lignin to Value-Added Aliphatic and Phenolic Products. Molecules, 30(12), 2544. https://doi.org/10.3390/molecules30122544