
Structural Aspects of Arylpiperazines as Aminergic GPCR Ligands




Abstract

1. Introduction
2. Solid-State and Bioactive Conformations of Arylpiperazines
2.1. Aripiprazole
2.2. N-Arylpiperazines
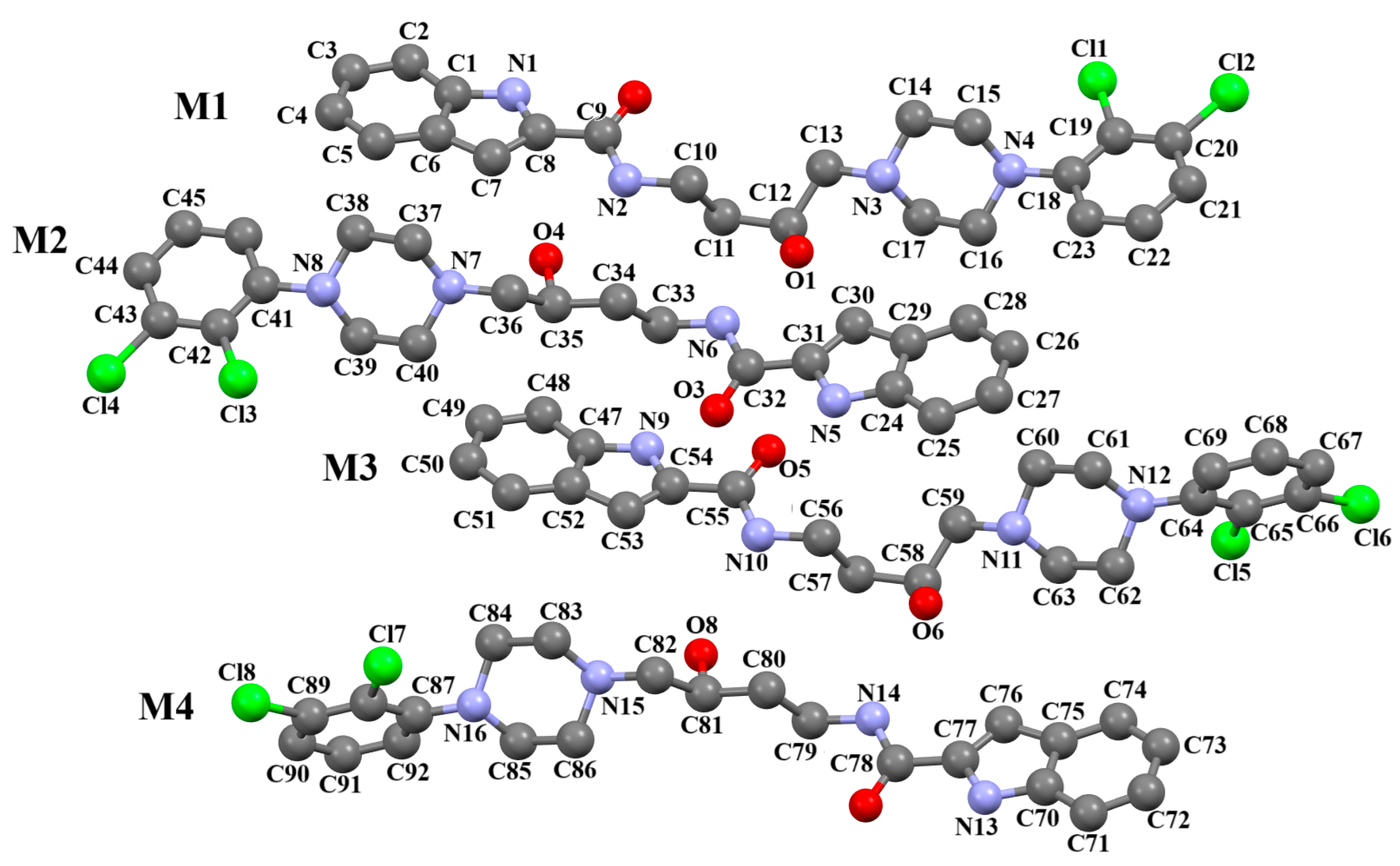
2.2.1. General Overview
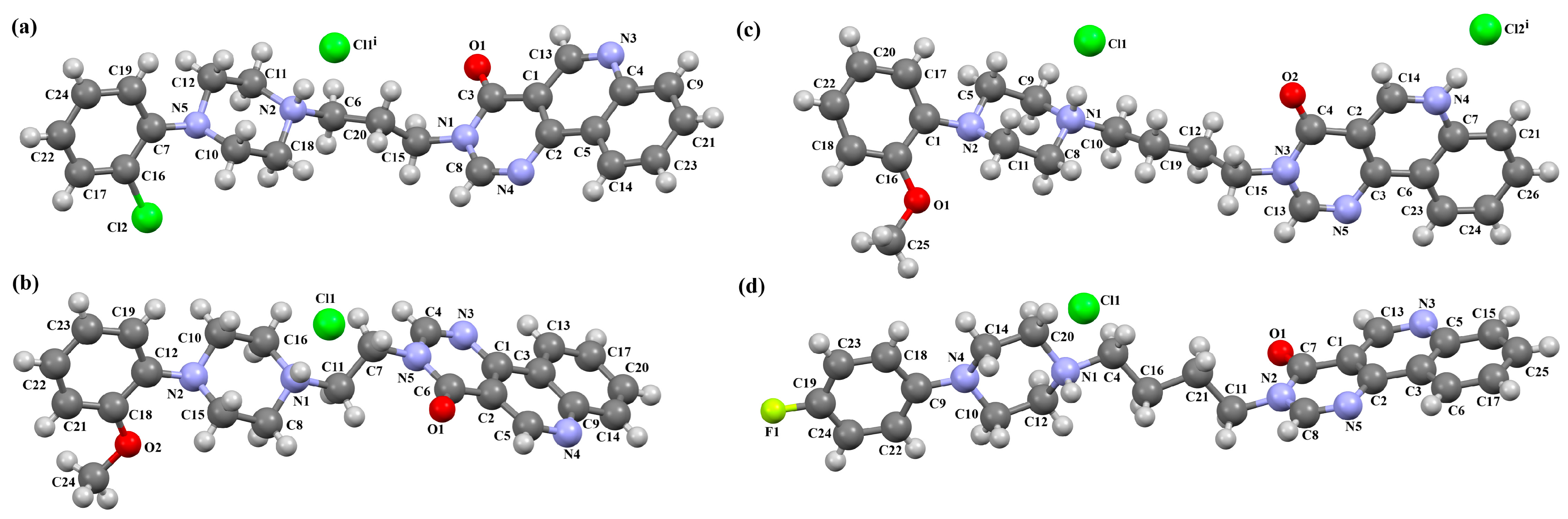
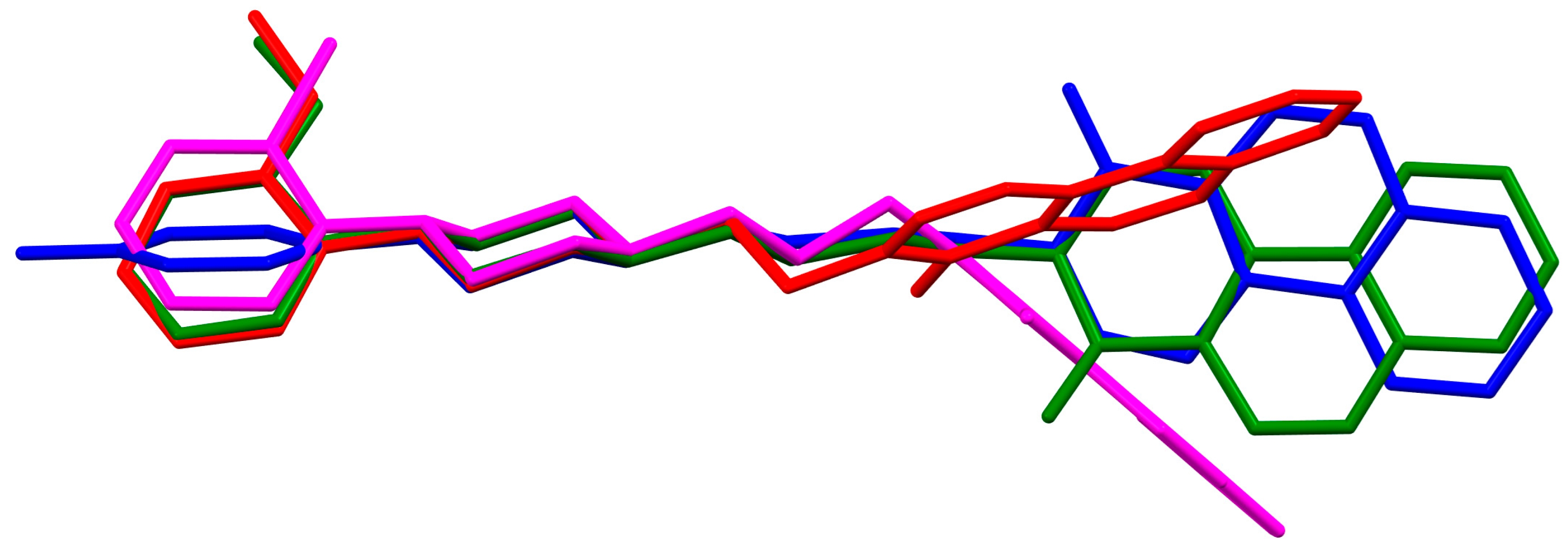
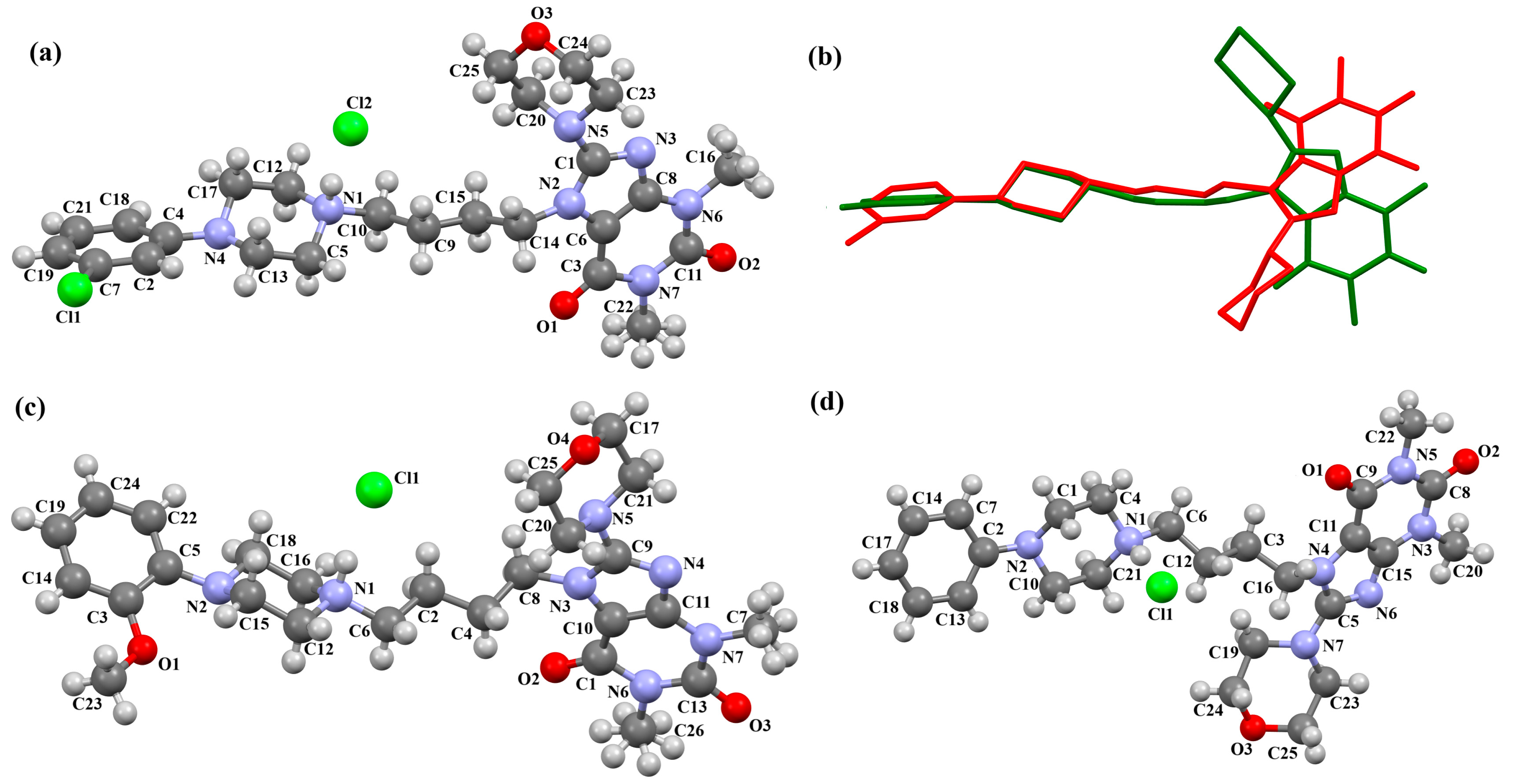
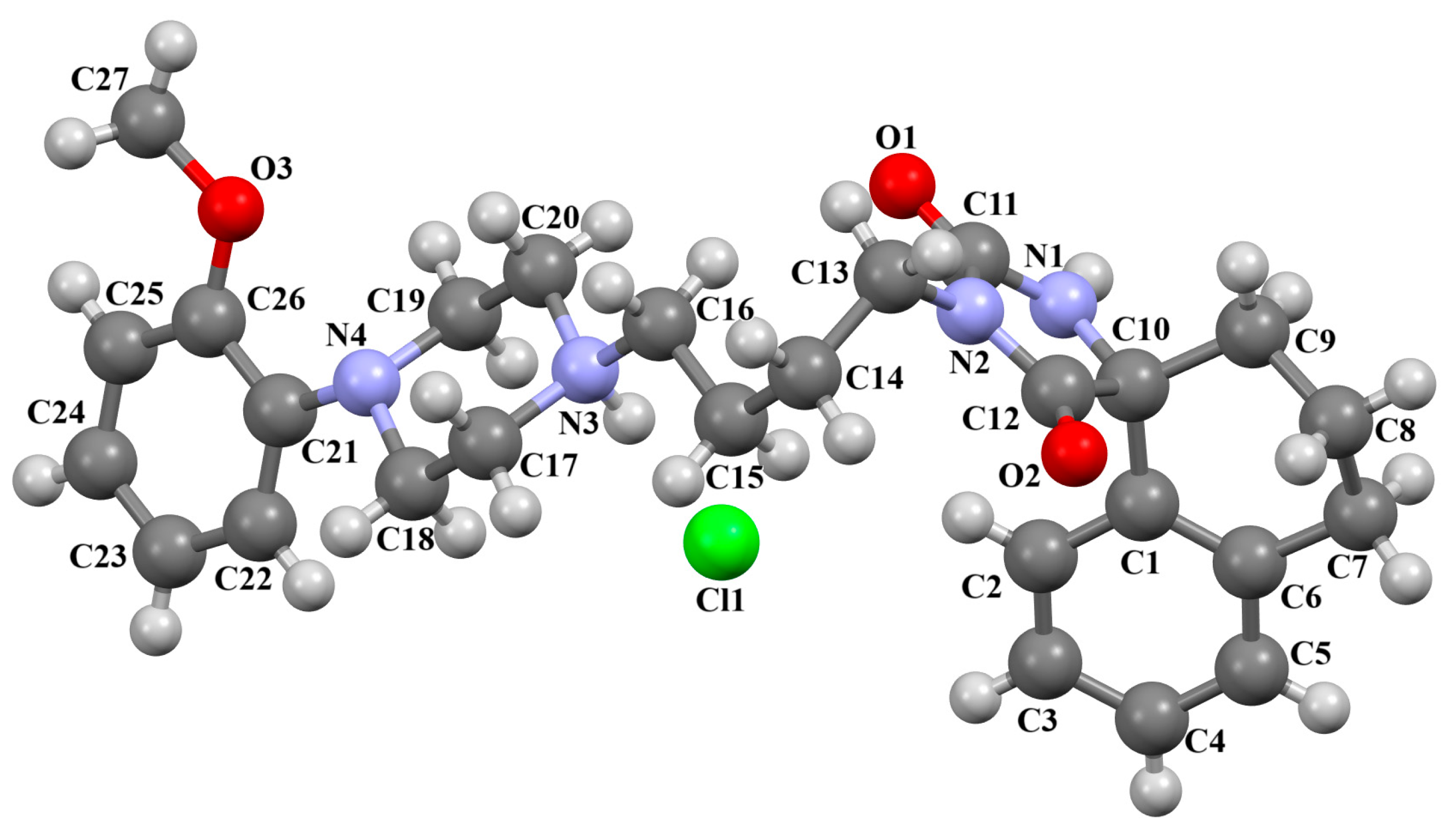
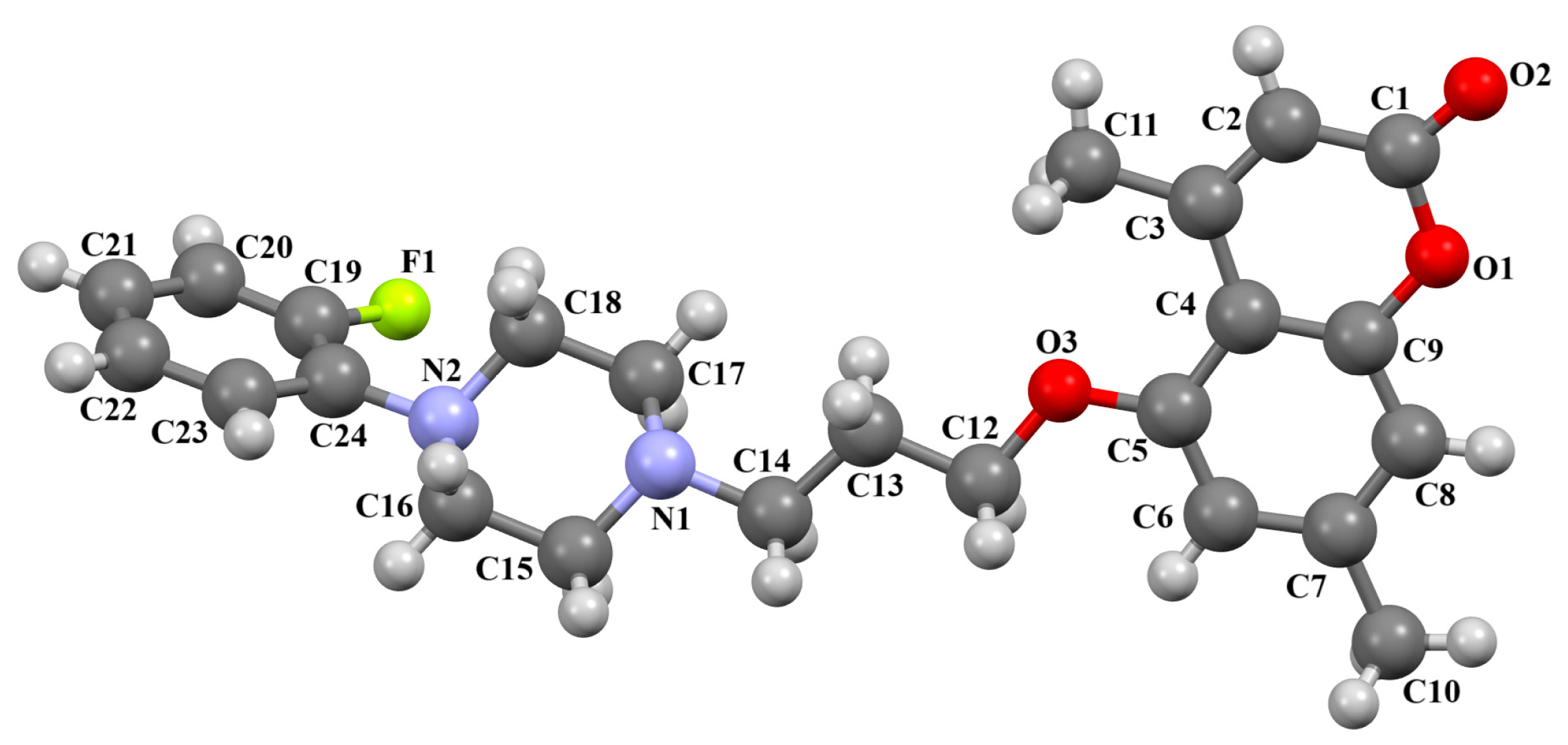
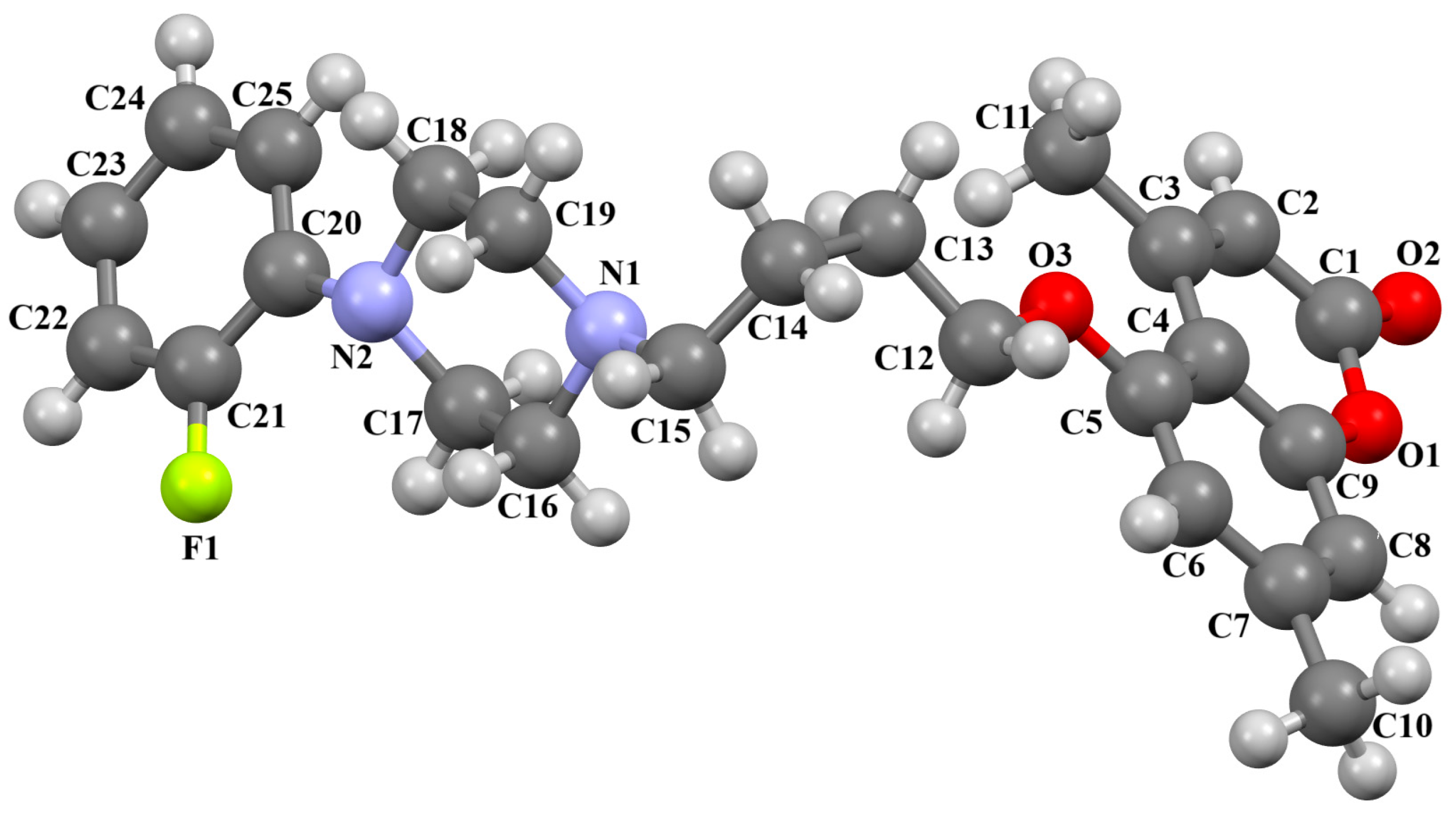
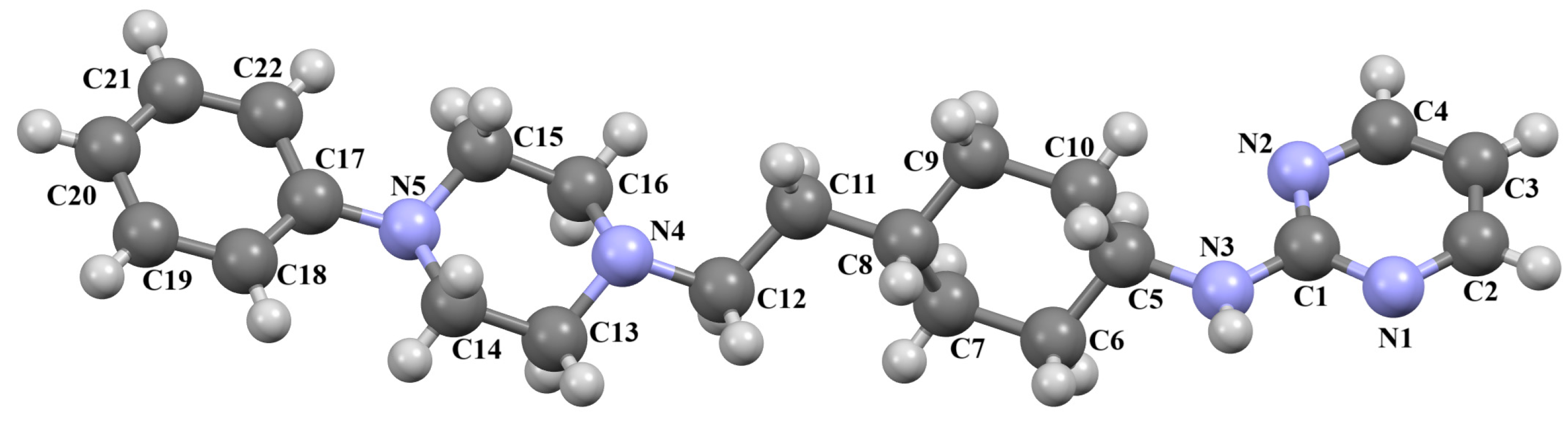
2.2.2. Arylpiperazine Derivatives Containing −(CH2)n− Spacer
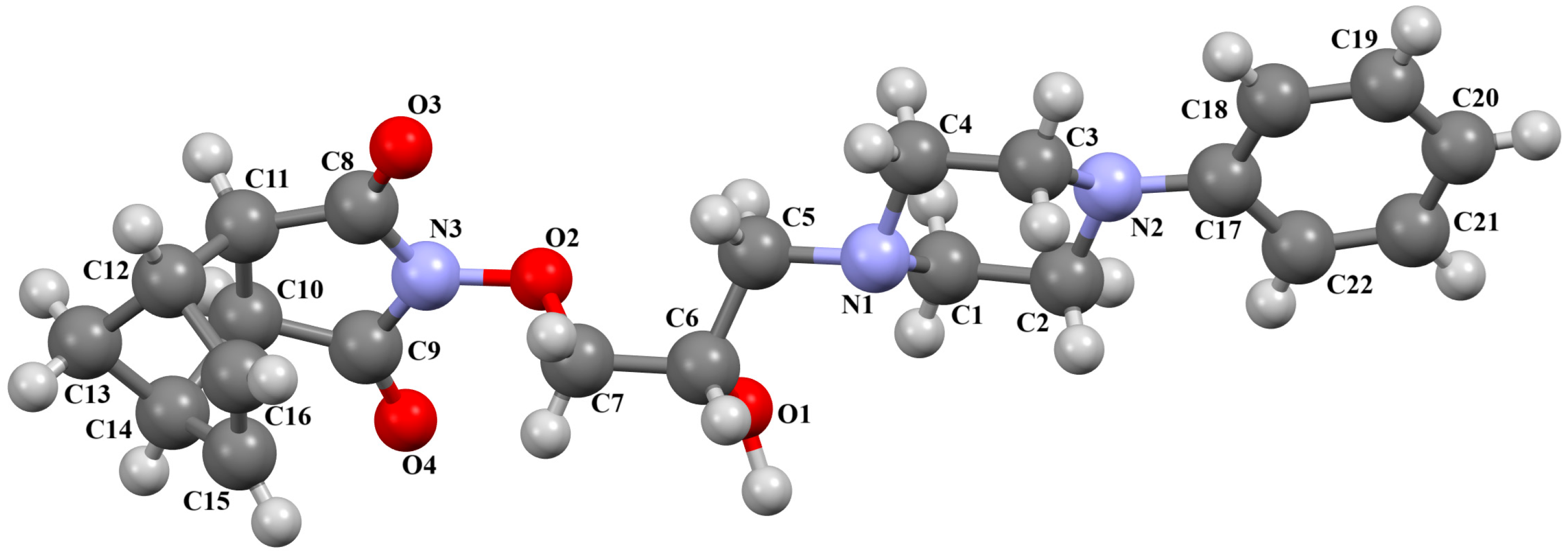
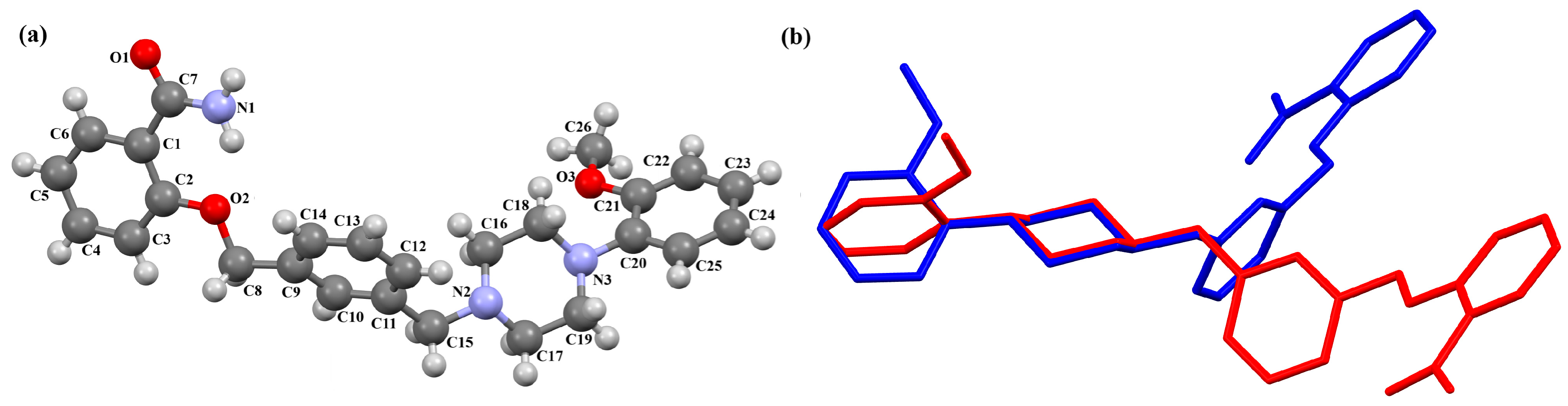
2.2.3. Arylpiperazine Derivatives Containing Flexible Aliphatic Chain with Heteroatom
2.2.4. Arylpiperazine Derivatives Containing Flexible Aliphatic Chain with an Electron-Donating Substituent
2.2.5. Other Arylpiperazine Derivatives
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CNS | Central nervous system |
| Cryo-EM | Cryogenic electron microscopy |
| ECL | Extracellular loop |
| ESR1 | Estrogen receptor 1 |
| GPCR | G protein-coupled receptors |
| MAPK3 | Mitogen-activated protein kinase 3 |
| LCAPs | Long chain arylpiperazines |
| PD | Parkinson’s disease |
| PPARG | Peroxisome proliferator-activated receptor gamma |
| TGA | Third-generation antipsychotics |
| TM | Transmembrane |
References
- Evans, B.E.; Rittle, K.E.; Bock, M.G.; DiPardo, R.M.; Freidinger, R.M.; Whitter, W.L.; Lundell, G.F.; Veber, D.F.; Anderson, P.S.; Chang, R.S. Methods for Drug Discovery: Development of Potent, Selective, Orally Effective Cholecystokinin Antagonists. J. Med. Chem. 1988, 31, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- Duarte, C.D.; Barreiro, E.J.; Fraga, C.A.M. Privileged Structures: A Useful Concept for the Rational Design of New Lead Drug Candidates. Mini Rev. Med. Chem. 2007, 7, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.J. Privileged Scaffolds in Medicinal Chemistry: An Introduction. Book Chapter 2015, 468. [Google Scholar] [CrossRef]
- Marson, C.M. New and Unusual Scaffolds in Medicinal Chemistry. Chem. Soc. Rev. 2011, 40, 5514–5533. [Google Scholar] [CrossRef]
- Kumar, B.; Kumar, N.; Thakur, A.; Kumar, V.; Kumar, R.; Kumar, V. A Review on the Arylpiperazine Derivatives as Potential Therapeutics for the Treatment of Various Neurological Disorders. Curr. Drug Targets 2022, 23, 729–751. [Google Scholar] [CrossRef]
- Andreozzi, G.; Corvino, A.; Severino, B.; Magli, E.; Perissutti, E.; Frecentese, F.; Santagada, V.; Caliendo, G.; Fiorino, F. Arylpiperazine Derivatives and Cancer: A New Challenge in Medicinal Chemistry. Pharmaceuticals 2024, 17, 1320. [Google Scholar] [CrossRef]
- Singh, D.; Singh, P.; Srivastava, P.; Kakkar, D.; Pathak, M.; Tiwari, A.K. Development and Challenges in the Discovery of 5-HT1A and 5-HT7 Receptor Ligands. Bioorg. Chem. 2023, 131, 106254. [Google Scholar] [CrossRef] [PubMed]
- Rague, A.; Tidgewell, K. Pharmacophore Comparison and Development of Recently Discovered Long Chain Arylpiperazine and Sulfonamide Based 5-HT7 Ligands. Mini Rev. Med. Chem. 2018, 18, 552–560. [Google Scholar] [CrossRef]
- Soskic, V.; Sukalovic, V.; Kostic-Rajacic, S. Exploration of N-Arylpiperazine Binding Sites of D2 Dopaminergic Receptor. Mini Rev. Med. Chem. 2015, 15, 988–1001. [Google Scholar] [CrossRef]
- Manetti, F.; Corelli, F.; Strappaghetti, G.; Botta, M. Arylpiperazines with Affinity toward Alpha(1)-Adrenergic Receptors. Curr. Med. Chem. 2002, 9, 1303–1321. [Google Scholar] [CrossRef]
- Giorgioni, G.; Bonifazi, A.; Botticelli, L.; Cifani, C.; Matteucci, F.; Bonaventura, E.M.D.; Bonaventura, M.V.M.D.; Giannella, M.; Piergentili, A.; Piergentili, A.; et al. Advances in Drug Design and Therapeutic Potential of Selective or Multitarget 5-HT1A Receptor Ligands. Med. Res. Rev. 2024, 44, 2640–2706. [Google Scholar] [CrossRef] [PubMed]
- Mastromarino, M.; Niso, M.; Abate, C.; Proschak, E.; Dubiel, M.; Stark, H.; Castro, M.; Lacivita, E.; Leopoldo, M. Design and Synthesis of Arylpiperazine Serotonergic/Dopaminergic Ligands with Neuroprotective Properties. Molecules 2022, 27, 1297. [Google Scholar] [CrossRef] [PubMed]
- Bielenica, A.; Kozioł, A.E.; Struga, M. Binding Modes of Chain Arylpiperazines to 5-HT1a, 5-HT2a and 5-HT7 Receptors. Mini Rev. Med. Chem. 2013, 13, 1516–1539. [Google Scholar] [CrossRef]
- Thepchatri, P.; Eliseo, T.; Cicero, D.O.; Myles, D.; Snyder, J.P. Relationship among Ligand Conformations in Solution, in the Solid State, and at the Hsp90 Binding Site: Geldanamycin and Radicicol. J. Am. Chem. Soc. 2007, 129, 3127–3134. [Google Scholar] [CrossRef]
- Saini, M.; Mehra, N.; Kumar, G.; Paul, R.; Kovács, B. Molecular and Structure-Based Drug Design: From Theory to Practice. Adv. Pharmacol. 2025, 103, 121–138. [Google Scholar] [CrossRef]
- García-Nafría, J.; Tate, C.G. Structure Determination of GPCRs: Cryo-EM Compared with X-Ray Crystallography. Biochem. Soc. Trans. 2021, 49, 2345–2355. [Google Scholar] [CrossRef]
- García-Nafría, J.; Tate, C.G. Cryo-Electron Microscopy: Moving Beyond X-Ray Crystal Structures for Drug Receptors and Drug Development. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 51–71. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Tessler, L.; Goldberg, I. Crystal Structures of Aripiprazole, a New Anti-Psychotic Drug, and of Its Inclusion Compounds with Methanol, Ethanol and Water. J. Incl. Phenom. Macrocycl. Chem. 2006, 55, 255–261. [Google Scholar] [CrossRef]
- Braun, D.E.; Gelbrich, T.; Kahlenberg, V.; Tessadri, R.; Wieser, J.; Griesser, U.J. Conformational Polymorphism in Aripiprazole: Preparation, Stability and Structure of Five Modifications. J. Pharm. Sci. 2009, 98, 2010–2026. [Google Scholar] [CrossRef] [PubMed]
- Nanubolu, J.B.; Sridhar, B.; Babu, V.S.P.; Jagadeesh, B.; Ravikumar, K. Sixth Polymorph of Aripiprazole—An Antipsychotic Drug. Cryst. Eng. Comm. 2012, 14, 4677–4685. [Google Scholar] [CrossRef]
- Delaney, S.P.; Pan, D.; Yin, S.X.; Smith, T.M.; Korter, T.M. Evaluating the Roles of Conformational Strain and Cohesive Binding in Crystalline Polymorphs of Aripiprazole. Cryst. Growth Des. 2013, 13, 2943–2952. [Google Scholar] [CrossRef]
- Delaney, S.P.; Smith, T.M.; Pan, D.; Yin, S.X.; Korter, T.M. Low-Temperature Phase Transition in Crystalline Aripiprazole Leads to an Eighth Polymorph. Cryst. Growth Des. 2014, 14, 5004–5010. [Google Scholar] [CrossRef]
- Zeidan, T.A.; Trotta, J.T.; Tilak, P.A.; Oliveira, M.A.; Chiarella, R.A.; Foxman, B.M.; Almarsson, Ö.; Hickey, M.B. An Unprecedented Case of Dodecamorphism: The Twelfth Polymorph of Aripiprazole Formed by Seeding with Its Active Metabolite. Cryst. Eng. Comm. 2016, 18, 1486–1488. [Google Scholar] [CrossRef]
- Tyler, A.R.; Ragbirsingh, R.; McMonagle, C.J.; Waddell, P.G.; Heaps, S.E.; Steed, J.W.; Thaw, P.; Hall, M.J.; Probert, M.R. Encapsulated Nanodroplet Crystallization of Organic-Soluble Small Molecules. Chem 2020, 6, 1755–1765. [Google Scholar] [CrossRef]
- Shapiro, D.A.; Renock, S.; Arrington, E.; Chiodo, L.A.; Liu, L.-X.; Sibley, D.R.; Roth, B.L.; Mailman, R. Aripiprazole, A Novel Atypical Antipsychotic Drug with a Unique and Robust Pharmacology. Neuropsychopharmacol. 2003, 28, 1400–1411. [Google Scholar] [CrossRef]
- Stępnicki, P.; Kondej, M.; Kaczor, A.A. Current Concepts and Treatments of Schizophrenia. Molecules 2018, 23, 2087. [Google Scholar] [CrossRef]
- Kondej, M.; Stępnicki, P.; Kaczor, A.A. Multi-Target Approach for Drug Discovery against Schizophrenia. Int. J. Mol. Sci. 2018, 19, 3105. [Google Scholar] [CrossRef]
- Stępnicki, P.; Kondej, M.; Koszła, O.; Żuk, J.; Kaczor, A.A. Multi-Targeted Drug Design Strategies for the Treatment of Schizophrenia. Expert Opin. Drug Discov. 2021, 16, 101–114. [Google Scholar] [CrossRef]
- Ekhteiari Salmas, R.; Serhat Is, Y.; Durdagi, S.; Stein, M.; Yurtsever, M. A QM Protein-Ligand Investigation of Antipsychotic Drugs with the Dopamine D2 Receptor (D2R). J. Biomol. Struct. Dyn. 2018, 36, 2668–2677. [Google Scholar] [CrossRef] [PubMed]
- Amani, P.; Habibpour, R.; Karami, L.; Hofmann, A. Docking Screens of Noncovalent Interaction Motifs of the Human Subtype-D2 Receptor-75 Schizophrenia Antipsychotic Complexes with Physicochemical Appraisal of Antipsychotics. ACS Chem. Neurosci. 2021, 12, 2218–2232. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Yang, L.; Zhang, Q.; Li, C.; Tian, H.; Zhuo, C. Aripiprazole Alleviates the High Prolactin Levels Induced by Amisulpride via Distinct Molecular Mechanisms: A Network Pharmacology and Molecular Docking Study. BMC Psychiatry 2025, 25, 373. [Google Scholar] [CrossRef]
- Xu, P.; Huang, S.; Zhang, H.; Mao, C.; Zhou, X.E.; Cheng, X.; Simon, I.A.; Shen, D.-D.; Yen, H.-Y.; Robinson, C.V.; et al. Structural Insights into the Lipid and Ligand Regulation of Serotonin Receptors. Nature 2021, 592, 469–473. [Google Scholar] [CrossRef]
- Chen, Z.; Fan, L.; Wang, H.; Yu, J.; Lu, D.; Qi, J.; Nie, F.; Luo, Z.; Liu, Z.; Cheng, J.; et al. Structure-Based Design of a Novel Third-Generation Antipsychotic Drug Lead with Potential Antidepressant Properties. Nat. Neurosci 2022, 25, 39–49. [Google Scholar] [CrossRef]
- Karolak-Wojciechowska, J.; Fruziński, A.; Czylkowski, R.; Paluchowska, M.H.; Mokrosz, M.J. Spacer Conformation in Biologically Active Molecules. Part 2. Structure and Conformation of 4-[2-(Diphenylmethylamino)Ethyl]-1-(2-Methoxyphenyl) Piperazine and Its Diphenylmethoxy Analog—Potential 5-HT1A Receptor Ligands. J. Mol. Struct. 2003, 657, 7–17. [Google Scholar] [CrossRef]
- Caliendo, G.; Santagada, V.; Perissutti, E.; Fiorino, F. Derivatives as 5HT1A Receptor Ligands--Past and Present. Curr. Med. Chem. 2005, 12, 1721–1753. [Google Scholar] [CrossRef]
- Bojarski, A.J.; Duszyńska, B.; Kołaczkowski, M.; Kowalski, P.; Kowalska, T. The Impact of Spacer Structure on 5-HT7 and 5-HT1A Receptor Affinity in the Group of Long-Chain Arylpiperazine Ligands. Bioorg. Med. Chem. Lett. 2004, 14, 5863–5866. [Google Scholar] [CrossRef]
- Michino, M.; Boateng, C.A.; Donthamsetti, P.; Yano, H.; Bakare, O.M.; Bonifazi, A.; Ellenberger, M.P.; Keck, T.M.; Kumar, V.; Zhu, C.; et al. Toward Understanding the Structural Basis of Partial Agonism at the Dopamine D3 Receptor. J. Med. Chem. 2017, 60, 580–593. [Google Scholar] [CrossRef]
- Cappelli, A.; Manini, M.; Valenti, S.; Castriconi, F.; Giuliani, G.; Anzini, M.; Brogi, S.; Butini, S.; Gemma, S.; Campiani, G.; et al. Synthesis and Structure-Activity Relationship Studies in Serotonin 5-HT(1A) Receptor Agonists Based on Fused Pyrrolidone Scaffolds. Eur. J. Med. Chem. 2013, 63, 85–94. [Google Scholar] [CrossRef]
- Chłoń-Rzepa, G.; Bucki, A.; Kołaczkowski, M.; Partyka, A.; Jastrzębska-Więsek, M.; Satała, G.; Bojarski, A.J.; Kalinowska-Tłuścik, J.; Kazek, G.; Mordyl, B.; et al. Arylpiperazinylalkyl Derivatives of 8-Amino-1,3-Dimethylpurine-2,6-Dione as Novel Multitarget 5-HT/D Receptor Agents with Potential Antipsychotic Activity. J. Enzyme Inhib. Med. Chem. 2016, 31, 1048–1062. [Google Scholar] [CrossRef] [PubMed]
- Kucwaj-Brysz, K.; Kurczab, R.; Jastrzębska-Więsek, M.; Żesławska, E.; Satała, G.; Nitek, W.; Partyka, A.; Siwek, A.; Jankowska, A.; Wesołowska, A.; et al. Computer-Aided Insights into Receptor-Ligand Interaction for Novel 5-Arylhydantoin Derivatives as Serotonin 5-HT7 Receptor Agents with Antidepressant Activity. Eur. J. Med. Chem. 2018, 147, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Lewgowd, W.; Bojarski, A.J.; Szczesio, M.; Olczak, A.; Glowka, M.L.; Mordalski, S.; Stanczak, A. Synthesis and Structural Investigation of Some Pyrimido[5,4-c]Quinolin-4(3H)-One Derivatives with a Long-Chain Arylpiperazine Moiety as Potent 5-HT(1A/2A) and 5-HT(7) Receptor Ligands. Eur. J. Med. Chem. 2011, 46, 3348–3361. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.H.; Grundt, P.; Cyriac, G.; Deschamps, J.R.; Taylor, M.; Kumar, R.; Ho, D.; Luedtke, R.R. N-(4-(4-(2,3-Dichloro- or 2-Methoxyphenyl)Piperazin-1-Yl)Butyl)Heterobiarylcarboxamides with Functionalized Linking Chains as High Affinity and Enantioselective D3 Receptor Antagonists. J. Med. Chem. 2009, 52, 2559–2570. [Google Scholar] [CrossRef]
- Ostrowska, K.; Grzeszczuk, D.; Głuch-Lutwin, M.; Gryboś, A.; Siwek, A.; Dobrzycki, Ł.; Trzaskowski, B. Development of Selective Agents Targeting Serotonin 5HT1A Receptors with Subnanomolar Activities Based on a Coumarin Core. Med. Chem. Comm 2017, 8, 1690–1696. [Google Scholar] [CrossRef]
- Herold, F.; Kleps, J.; Wolska, I.; Nowak, G. Synthesis of New Hexahydro- and Octahydropyrido[1,2-c]Pyrimidine Derivatives with an Arylpiperazine Moiety as Ligands for 5-HT1A and 5-HT2A Receptors. Farmaco 2002, 57, 959–971. [Google Scholar] [CrossRef]
- Enguehard-Gueiffier, C.; Hübner, H.; El Hakmaoui, A.; Allouchi, H.; Gmeiner, P.; Argiolas, A.; Melis, M.R.; Gueiffier, A. 2-[(4-Phenylpiperazin-1-Yl)Methyl]Imidazo(Di)Azines as Selective D4-Ligands. Induction of Penile Erection by 2-[4-(2-Methoxyphenyl)Piperazin-1-Ylmethyl]Imidazo[1,2-a]Pyridine (PIP3EA), a Potent and Selective D4 Partial Agonist. J. Med. Chem. 2006, 49, 3938–3947. [Google Scholar] [CrossRef]
- Kossakowski, J.; Bielenica, A.; Struga, M.; Koziol, A.E. Synthesis and Pharmacological Evaluation of 4-[2-Hydroxy-3-(4-Phenyl-Piperazin-1-Yl)-Propoxy]-4-Azatricyclo[5.2.1.02,6]Dec-8-Ene-3,5-Dione. Med. Chem. Res. 2008, 17, 507–514. [Google Scholar] [CrossRef]
- Pańczyk, K.; Pytka, K.; Jakubczyk, M.; Rapacz, A.; Siwek, A.; Głuch-Lutwin, M.; Gryboś, A.; Słoczyńska, K.; Koczurkiewicz, P.; Ryszawy, D.; et al. Synthesis of N-(Phenoxyalkyl)-, N-2-[2-(Phenoxy)Ethoxy]Ethyl- or N-(Phenoxyacetyl)Piperazine Derivatives and Their Activity Within the Central Nervous System. ChemistrySelect 2019, 4, 9381–9391. [Google Scholar] [CrossRef]
- Wustrow, D.; Belliotti, T.; Glase, S.; Kesten, S.R.; Johnson, D.; Colbry, N.; Rubin, R.; Blackburn, A.; Akunne, H.; Corbin, A.; et al. Aminopyrimidines with High Affinity for Both Serotonin and Dopamine Receptors. J. Med. Chem. 1998, 41, 760–771. [Google Scholar] [CrossRef]
- Pindelska, E.; Marczewska-Rak, A.; Jaśkowska, J.; Madura, I.D. Solvates of New Arylpiperazine Salicylamide Derivative-a Multi-Technique Approach to the Description of 5 HTR Ligand Structure and Interactions. Int. J. Mol. Sci. 2021, 22, 4992. [Google Scholar] [CrossRef] [PubMed]
- Kucwaj-Brysz, K.; Kurczab, R.; Żesławska, E.; Lubelska, A.; Marć, M.A.; Latacz, G.; Satała, G.; Nitek, W.; Kieć-Kononowicz, K.; Handzlik, J. The Role of Aryl-Topology in Balancing between Selective and Dual 5-HT7R/5-HT1A Actions of 3,5-Substituted Hydantoins. Med. Chem. Comm. 2018, 9, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Kucwaj-Brysz, K.; Latacz, G.; Podlewska, S.; Żesławska, E.; Handzlik, J.; Lubelska, A.; Satała, G.; Nitek, W.; Handzlik, J. The Relationship between Stereochemical and Both, Pharmacological and ADME-Tox, Properties of the Potent Hydantoin 5-HT7R Antagonist MF-8. Bioorg. Chem. 2021, 106, 104466. [Google Scholar] [CrossRef]
- Kumar, V.; Banala, A.K.; Garcia, E.G.; Cao, J.; Keck, T.M.; Bonifazi, A.; Deschamps, J.R.; Newman, A.H. Chiral Resolution and Serendipitous Fluorination Reaction for the Selective Dopamine D3 Receptor Antagonist BAK2-66. ACS Med. Chem. Lett. 2014, 5, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Chłoń-Rzepa, G.; Żmudzki, P.; Pawłowski, M.; Wesołowska, A.; Satała, G.; Bojarski, A.J.; Jabłoński, M.; Kalinowska-Tłuścik, J. New 7-Arylpiperazinylalkyl-8-Morpholin-4-Yl-Purine-2,6-Dione Derivatives withAnxiolytic Activity—Synthesis, Crystal Structure and Structure–Activity Study. J. Mol. Struct. 2014, 1067, 243–251. [Google Scholar] [CrossRef]
- Stępnicki, P.; Wronikowska-Denysiuk, O.; Zięba, A.; Targowska-Duda, K.M.; Bartyzel, A.; Wróbel, M.Z.; Wróbel, T.M.; Szałaj, K.; Chodkowski, A.; Mirecka, K.; et al. Novel Multi-Target Ligands of Dopamine and Serotonin Receptors for the Treatment of Schizophrenia Based on Indazole and Piperazine Scaffolds-Synthesis, Biological Activity, and Structural Evaluation. J Enzyme Inhib. Med. Chem. 2023, 38, 2209828. [Google Scholar] [CrossRef]
- Keck, T.M.; Banala, A.K.; Slack, R.D.; Burzynski, C.; Bonifazi, A.; Okunola-Bakare, O.M.; Moore, M.; Deschamps, J.R.; Rais, R.; Slusher, B.S.; et al. Using Click Chemistry toward Novel 1,2,3-Triazole-Linked Dopamine D3 Receptor Ligands. Bioorg. Med. Chem. 2015, 23, 4000–4012. [Google Scholar] [CrossRef]
- Czopek, A.; Kołaczkowski, M.; Bucki, A.; Byrtus, H.; Pawłowski, M.; Kazek, G.; Bojarski, A.J.; Piaskowska, A.; Kalinowska-Tłuścik, J.; Partyka, A.; et al. Novel Spirohydantoin Derivative as a Potent Multireceptor-Active Antipsychotic and Antidepressant Agent. Bioorg. Med. Chem. 2015, 23, 3436–3447. [Google Scholar] [CrossRef]
- Stępnicki, P.; Targowska-Duda, K.M.; Martínez, A.L.; Zięba, A.; Wronikowska-Denysiuk, O.; Wróbel, M.Z.; Bartyzel, A.; Trzpil, A.; Wróbel, T.M.; Chodkowski, A.; et al. Discovery of Novel Arylpiperazine-Based DA/5-HT Modulators as Potential Antipsychotic Agents—Design, Synthesis, Structural Studies and Pharmacological Profiling. Eur. J. Med. Chem. 2023, 252, 115285. [Google Scholar] [CrossRef]
- Ostrowska, K.; Grzeszczuk, D.; Maciejewska, D.; Młynarczuk-Biały, I.; Czajkowska, A.; Sztokfisz, A.; Dobrzycki, Ł.; Kruszewska, H. Synthesis and Biological Screening of a New Series of 5-[4-(4-Aryl-1-Piperazinyl)Butoxy]Coumarins. Monatsh. Chem. 2016, 147, 1615–1627. [Google Scholar] [CrossRef]




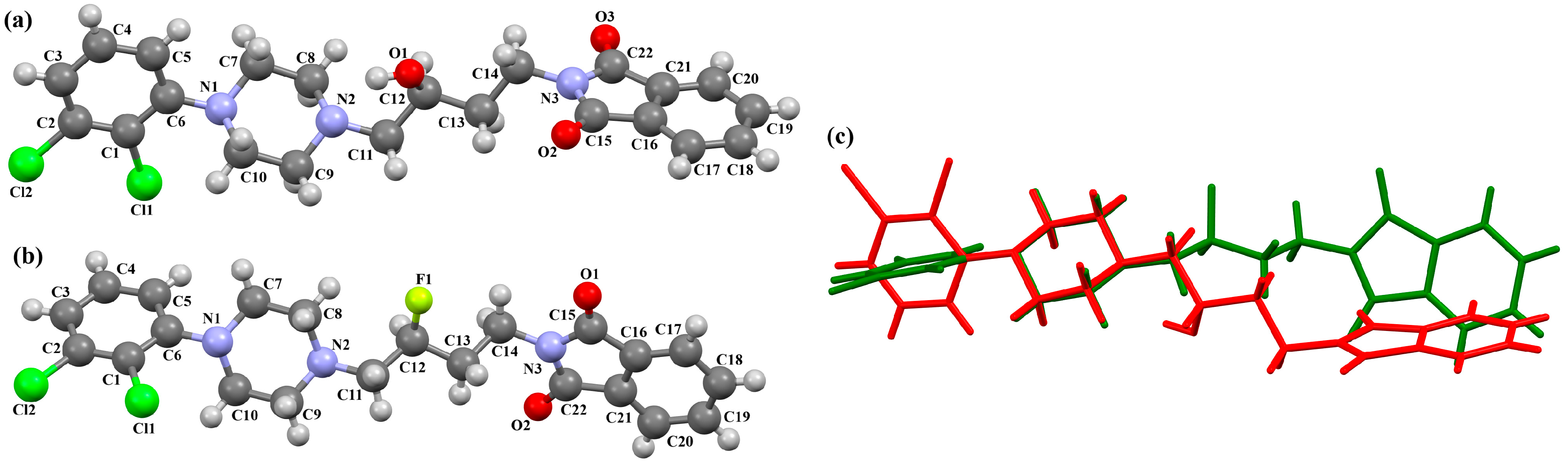
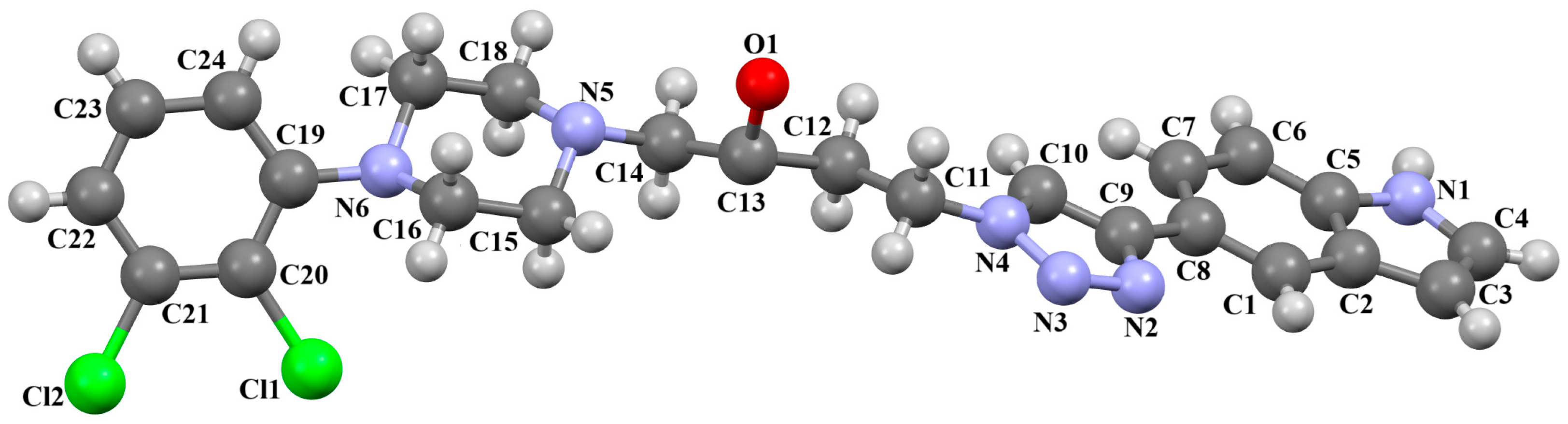
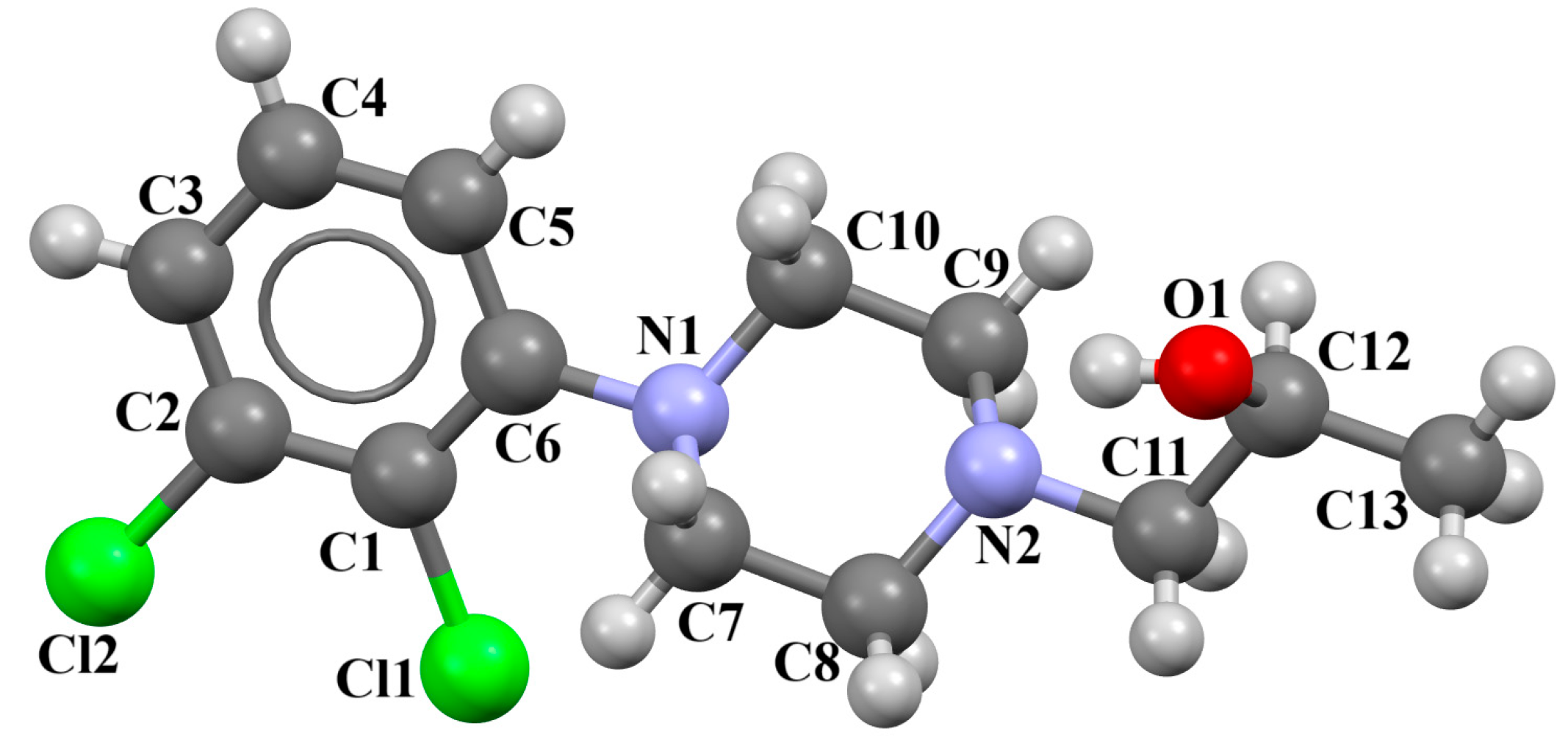
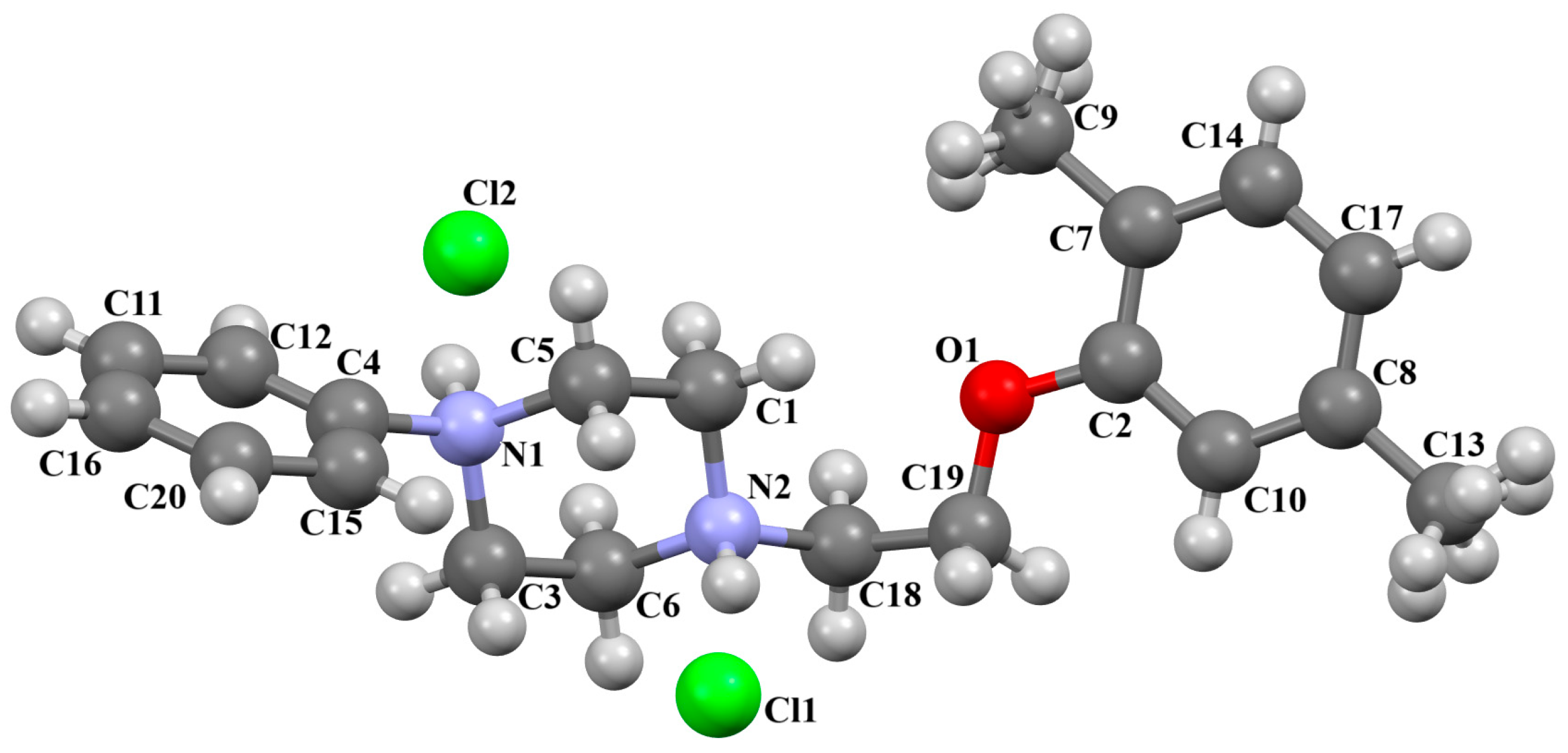

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
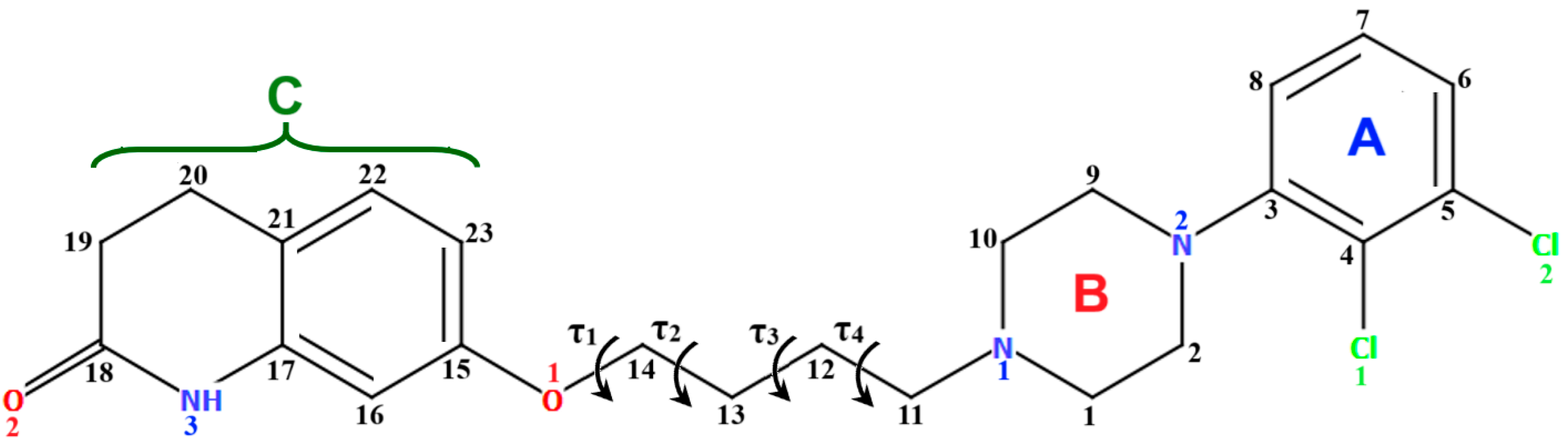
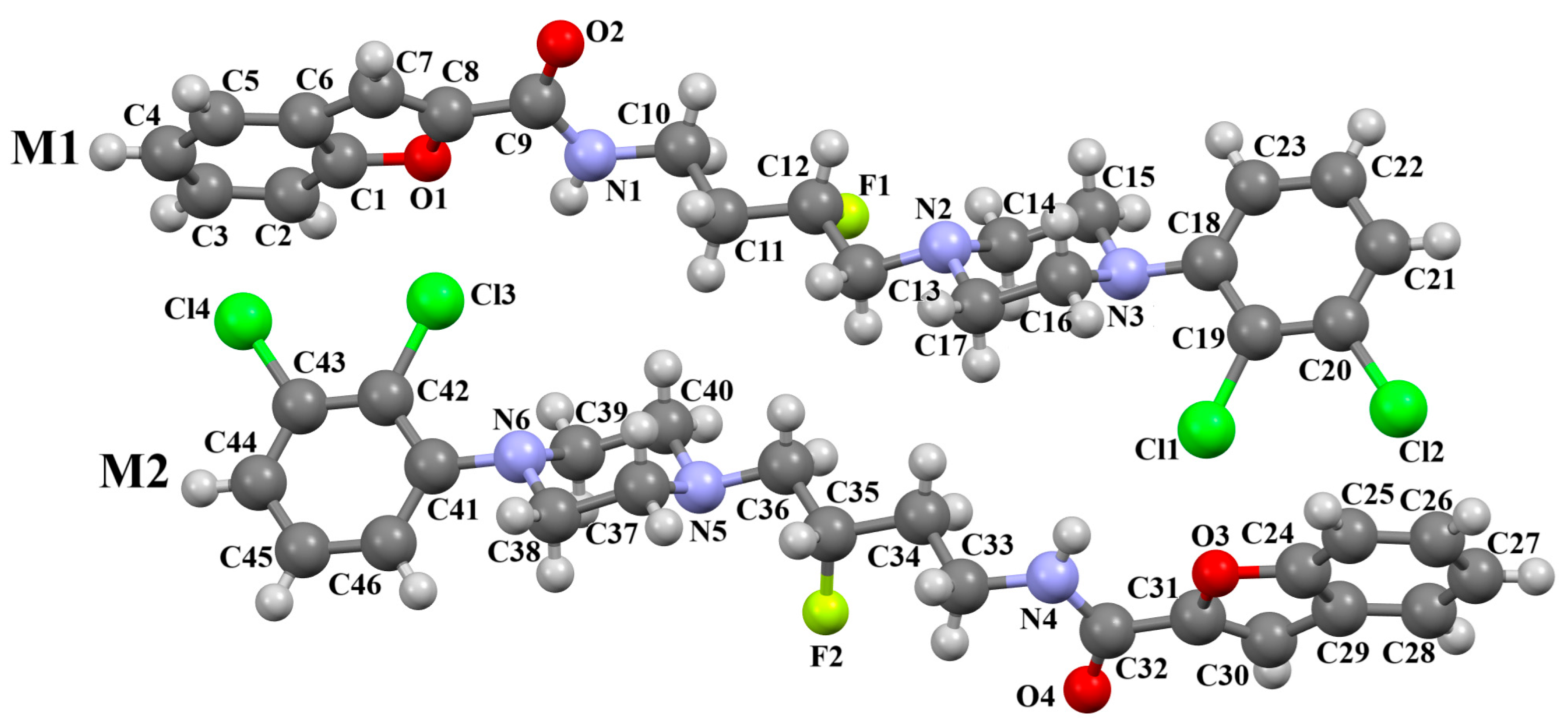
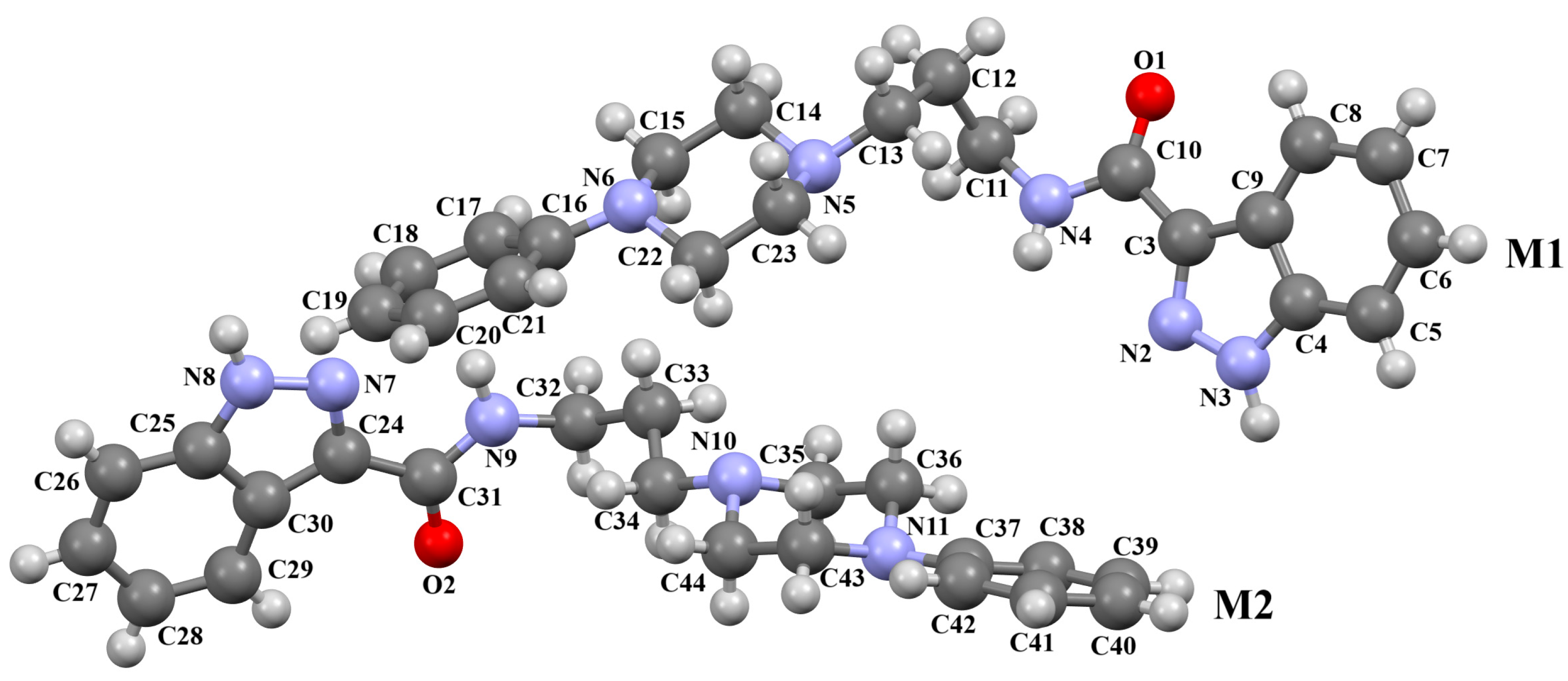
| CSD Refcode | Space Group | ∠A/B (°) | ∠C/B (°) | τ1 (°) | τ2 (°) | τ3 (°) | τ4 (°) | Ref. |
|---|---|---|---|---|---|---|---|---|
| MELFIT | P-1 | M1: 45.47 M2: 48.18 | M1: 52.22 M2:59.70 | M1: −163.7(2) M2: 178.2(2) | M1: −178.9(2) M2: −175.5(2) | M1: −177.5(2) M2: 161.9(2) | M1: 172.4(2) M2: −174.7(2) | [20] |
| MELFIT01 | P21 | 44.90 | 52.61 | 173.7(6) | 63.0(9) | −174.7(6) | −60.2(7) | [21] |
| MELFIT02 | Pna21 | 44.80 | 86.20 | −176.2(9) | 178.9(9) | 173.3(9) | −178.0(9) | [21] |
| MELFIT03 | P-1 | M1: 46.81 M2: 49.86 | M1: 52.80 M2: 59.71 | M1: 166.1(7) M2: 179.7(8) | M1: 178.9(6) M2: −177.5(8) | M1: 175.9(7) M2: 161.8(9) | M1: −173.0(6) M2: −174.2(8) | [21] |
| MELFIT04 | P-1 | M1: 43.83 M2: 42.14 | M1: 46.88 M2: 41.28 | M1: 176.7(2) M2: −175.3(3) | M1: 179.6(3) M2: 175.2(3) | M1: 176.5(3) M2: 178.3(3) | M1: −169.4(3) M2: −179.6(3) | [21] |
| MELFIT05 | P21 | 41.03 | 41.96 | 178.8(3) | 174.4(3) | 169.6(3) | 170.6(3) | [21] |
| MELFIT06 | P-1 | M1: 35.23 M2: 41.85 | M1: 33.18 M2: 41.32 | M1: 178.5(2) M2: −170.6(2) | M1: 178.6(2) M2: −178.5(2) | M1: −175.9(2) M2: −178.2(2) | M1: 168.9(2) M2: −171.0(2) | [22] |
| MELFIT07 | P-1 | 45.48 | 81.35 | −175.8(1) | −176.4(1) | −58.3(2) | −53.8(2) | [23] |
| MELFIT08 | Pna21 | 44.15 | 86.06 | 176.3(2) | −178.4(3) | −171.6(3) | 179.0(2) | [24] |
| MELFIT09 | Pna21 | 47.86 | 87.61 | 169.2(1) | 68.5(2) | 179.8(2) | −56.0(2) | [24] |
| MELFIT10 | Pna21 | 47.78 | 87.64 | −169.5(3) | −68.2(4) | −180.0(3) | 56.5(4) | [24] |
| MELFIT11 | Pna21 | 47.80 | 87.68 | −169.7(2) | −68.2(3) | −179.8(2) | 55.9(3) | [24] |
| MELFIT12 | Pna21 | 47.87 | 87.70 | 169.9(3) | 68.0(4) | 179.8(3) | −56.2(4) | [24] |
| MELFIT13 | Pna21 | 47.99 | 87.87 | 170.2(2) | 67.9(3) | 179.6(3) | −56.4(3) | [24] |
| MELFIT14 | Pna21 | 47.69 | 87.64 | −170.0(3) | −67.6(3) | −179.5(2) | 56.8(3) | [24] |
| MELFIT15 | Pna21 | 44.24 | 86.16 | 176.0(2) | −178.3(2) | −171.2(2) | 178.9(2) | [24] |
| MELFIT16 | Pna21 | 44.14 | 86.15 | 175.9(2) | −178.4(2) | −171.1(2) | 179.1(2) | [24] |
| MELFIT17 | Pna21 | 44.30 | 86.16 | 176.0(2) | −178.8(2) | −171.5(2) | 179.1(2) | [24] |
| MELFIT18 | Pna21 | 47.71 | 87.63 | −168.9(2) | −68.1(3) | 179.6(2) | 53.8(3) | [24] |
| MELFIT19 | P21/n | 53.11 | 82.61 | 179.4(1) | −177.5(1) | 177.9(2) | 55.3(2) | [25] |
| MELFIT20 | P-1 | M1: 40.16 M2: 42.78 | M1: 40.46 M2: 45.41 | M1: 175.1(2) M2: 177.8(2) | M1: −175.6(2) M2: −179.6(2) | M1: −177.9(2) M2: 176.9(2) | M1: −179.6(2) M2: −168.5(2) | [26] |
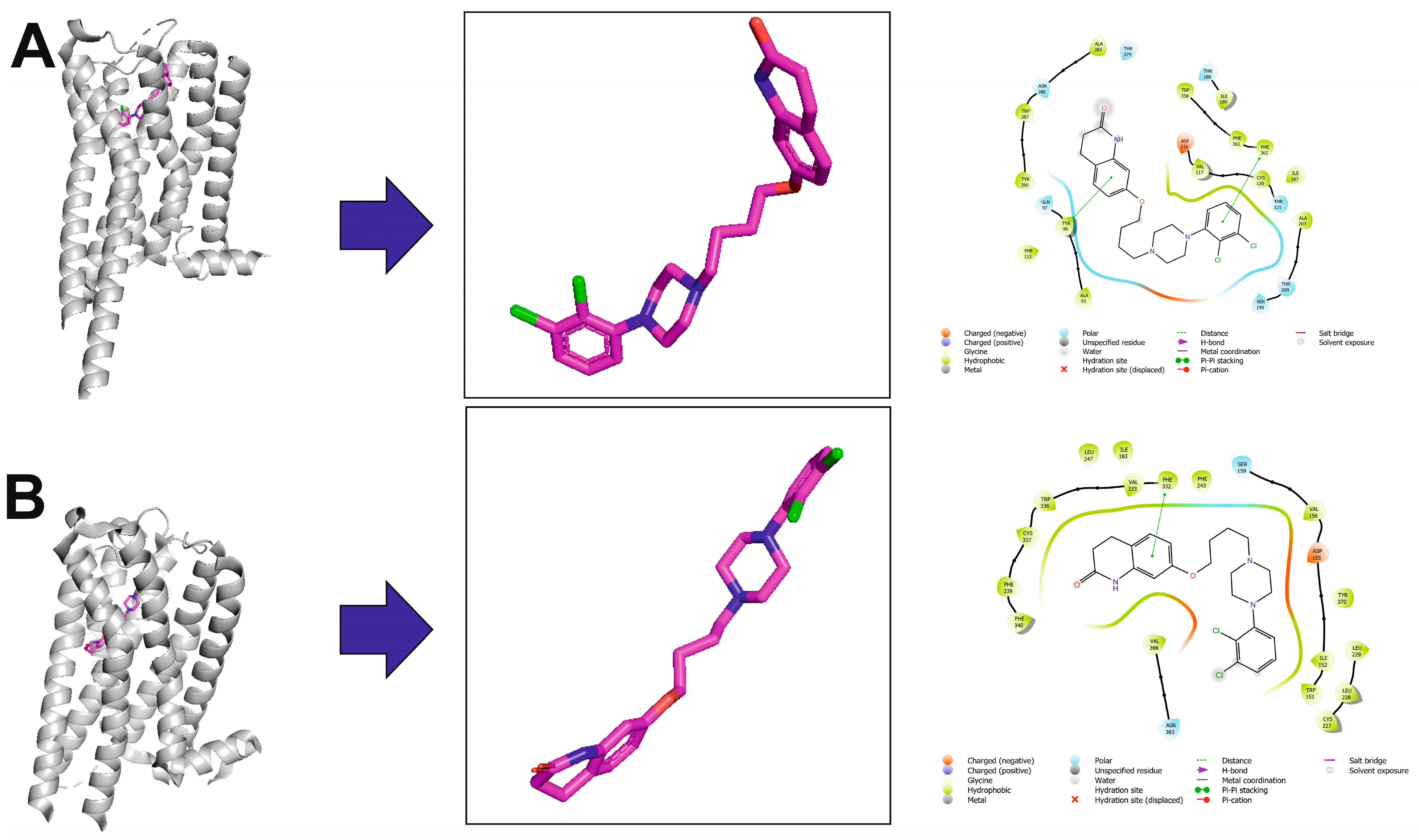
| Receptor | Ki [nM] |
|---|---|
| D1 | 1960 ± 670 |
| D2L | 0.74 ± 0.09 |
| D2 | 3.3 ± 1.1 |
| D3 | 9.7 ± 5.4 |
| D3 | 1.0 ± 0.40 |
| rD4 | 510 ± 93 |
| D5 | 2590 ± 1350 |
| 5-HT1A | 5.6 ± 0.8 |
| 5-HT1B | 830 ± 260 |
| 5-HT1D | 68 ± 11 |
| 5-HT1E | 8000 ± 5000 |
| 5-HT2A | 8.7 ± 2.0 |
| 5-HT2A | 35 ± 4 |
| 5-HT2B | 0.36 ± 0.11 |
| 5-HT2C | 76 ± 8 |
| 5-HT5A | 1240 ± 280 |
| 5-HT6 | 570 ± 95 |
| 5-HT7 | 10.3 ± 3.7 |
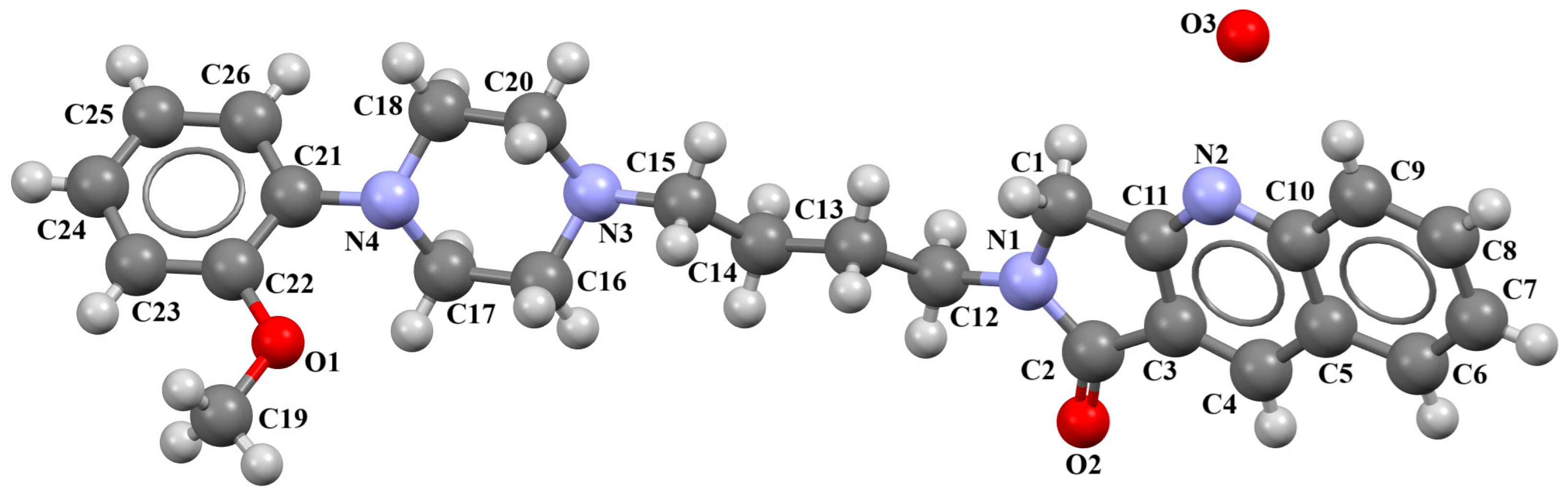
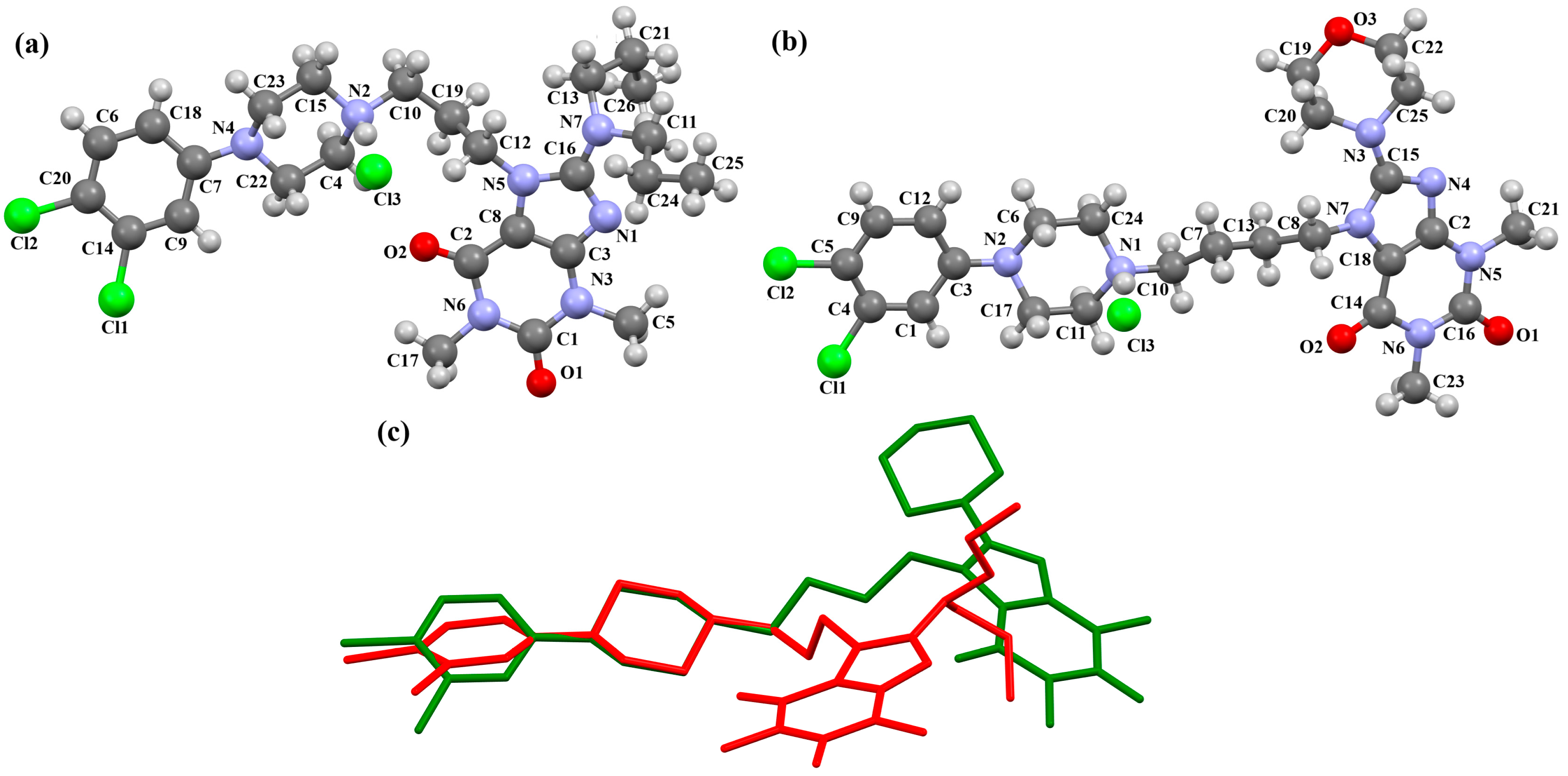
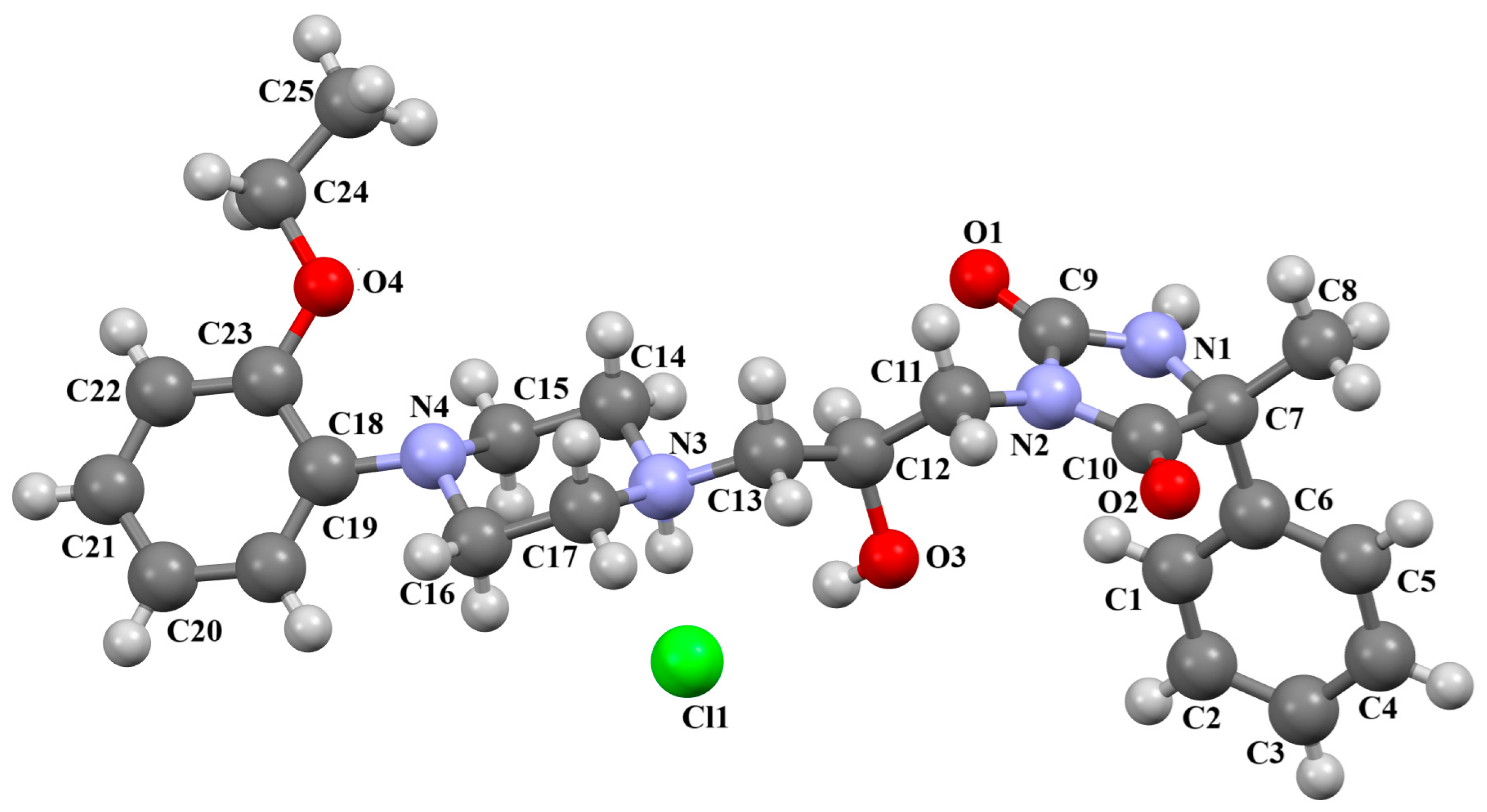
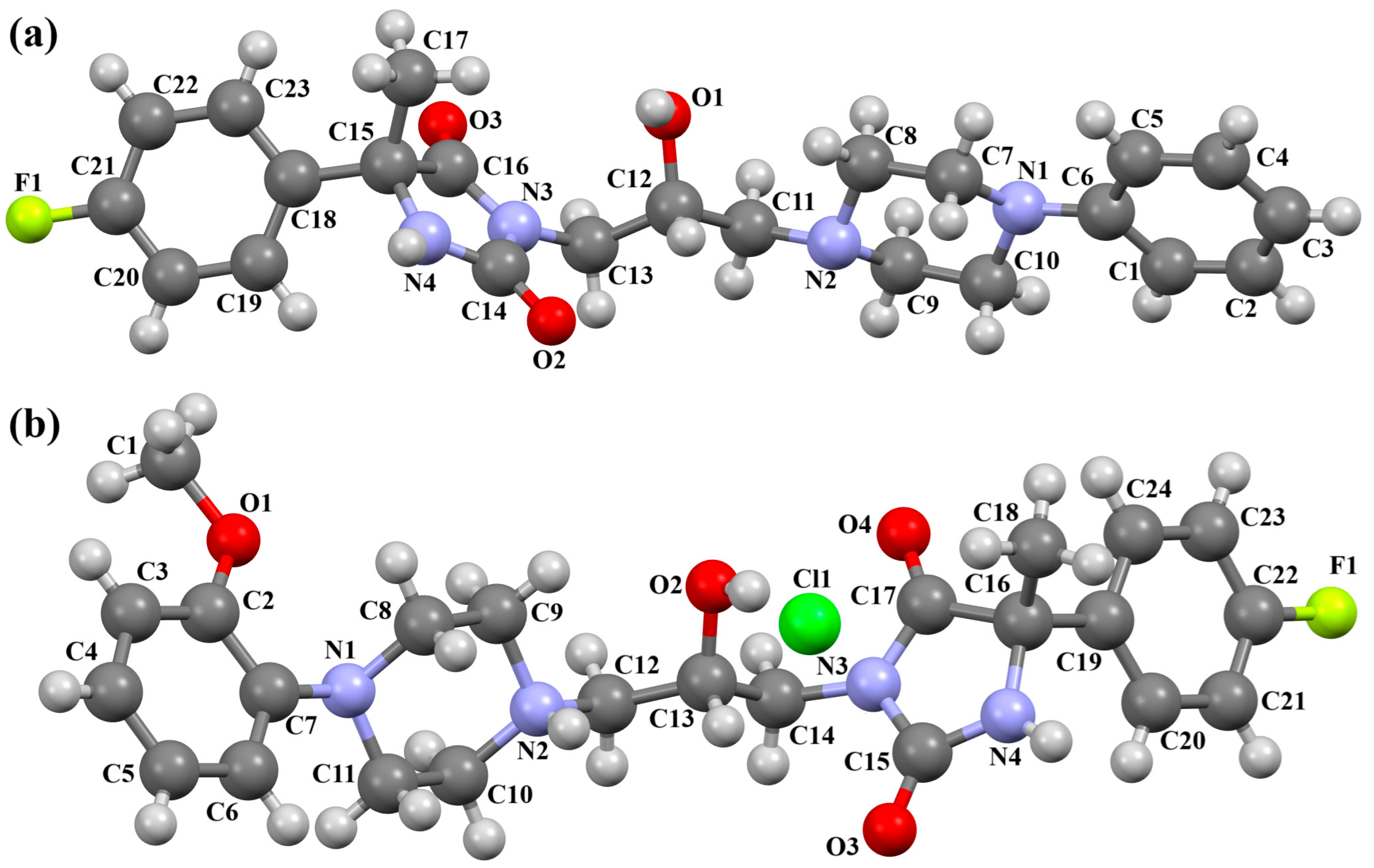
| Scheme of the Compound | CSD Refcode | ∠A/B (°) | Ref. |
|---|---|---|---|
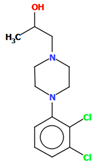 | BAHRIO | 43.40 | [39] |
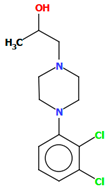 | BAHROU | 43.48 | [39] |
 | BIJBON | 47.39 | [40] |
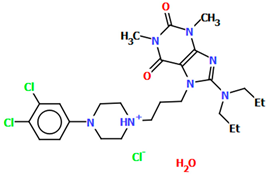 | CAHLAB | 27.26 | [41] |
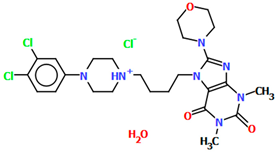 | CAHLEF | 5.39 | [41] |
 | DEMSAS | 43.76 | [42] |
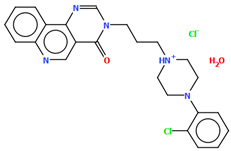 | EZEYUE | 53.69 | [43] |
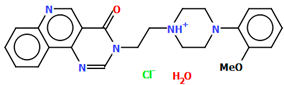 | EZEZAL | 49.03 | [43] |
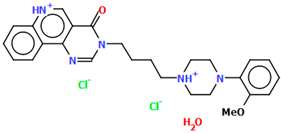 | EZEZEP | 45.75 | [43] |
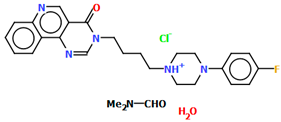 | EZEZIT | 3.77 | [43] |
 | KADXOE | M1: 61.13 M2: 48.68 M3: 45.86 M4: 47.87 | [44] |
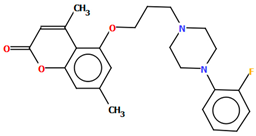 | KAXQEI | 41.73 | [45] |
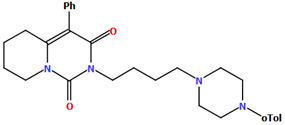 | LUYQOL | 65.07 | [46] |
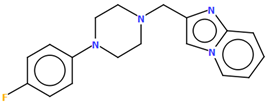 | MIDVOL | 34.56 | [47] |
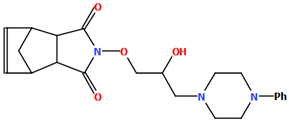 | POPDUU | 13.00 | [48] |
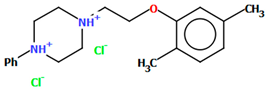 | POVSAX | 75.82 | [49] |
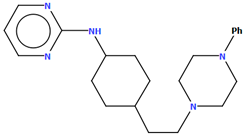 | PUDGOK | 5.68 | [50] |
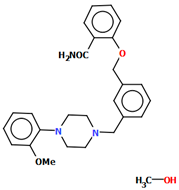 | QENRUA QENRUA01 QENRUA02 QENRUA03 | 47.74 47.72 46.62 46.61 | [51] |
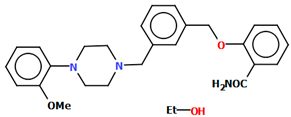 | QENSOV QENSOV01 | 48.74 48.73 | [51] |
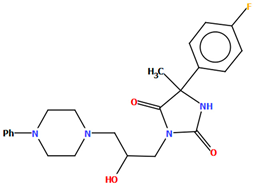 | QEYBAA QEYBAA01 | 4.75 - | [52] [53] |
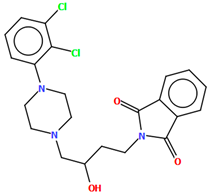 | QOQXOL | 55.32 | [54] |
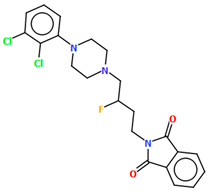 | QOQXUR | 49.93 | [54] |
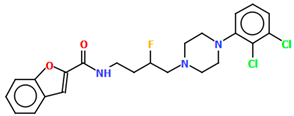 | QOQYAY | M1: 48.47 M2: 48.87 | [54] |
 | SOBVOW | 27.98 | [55] |
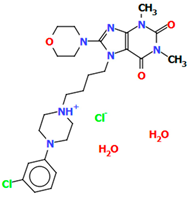 | SOBVUC | 8.33 | [55] |
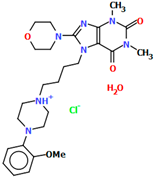 | SOBWAJ | 39.29 | [55] |
 | SOBWEN | 17.57 | [55] |
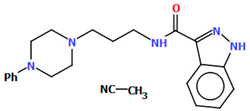 | TOYTIO | M1: 24.51 M2: 19.35 | [56] |
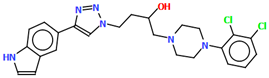 | VOTDUF | 44.33 | [57] |
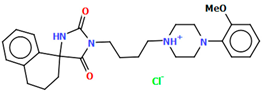 | VUFTIB | 41.84 | [58] |
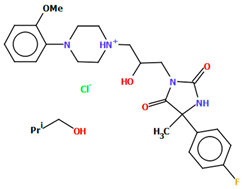 | WAGMOK | 45.28 | [52] |
 | ZIGWOF | M1: 25.13 M2: 10.68 | [59] |
 | ZOQPOM | 37.59 | [60] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartyzel, A.; Cristóvão, B.; Kaczor, A.A. Structural Aspects of Arylpiperazines as Aminergic GPCR Ligands. Molecules 2025, 30, 2545. https://doi.org/10.3390/molecules30122545
Bartyzel A, Cristóvão B, Kaczor AA. Structural Aspects of Arylpiperazines as Aminergic GPCR Ligands. Molecules. 2025; 30(12):2545. https://doi.org/10.3390/molecules30122545
Chicago/Turabian StyleBartyzel, Agata, Beata Cristóvão, and Agnieszka A. Kaczor. 2025. "Structural Aspects of Arylpiperazines as Aminergic GPCR Ligands" Molecules 30, no. 12: 2545. https://doi.org/10.3390/molecules30122545
APA StyleBartyzel, A., Cristóvão, B., & Kaczor, A. A. (2025). Structural Aspects of Arylpiperazines as Aminergic GPCR Ligands. Molecules, 30(12), 2545. https://doi.org/10.3390/molecules30122545