
Engineering and Exploiting Immobilized Peptide Organocatalysts for Modern Synthesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
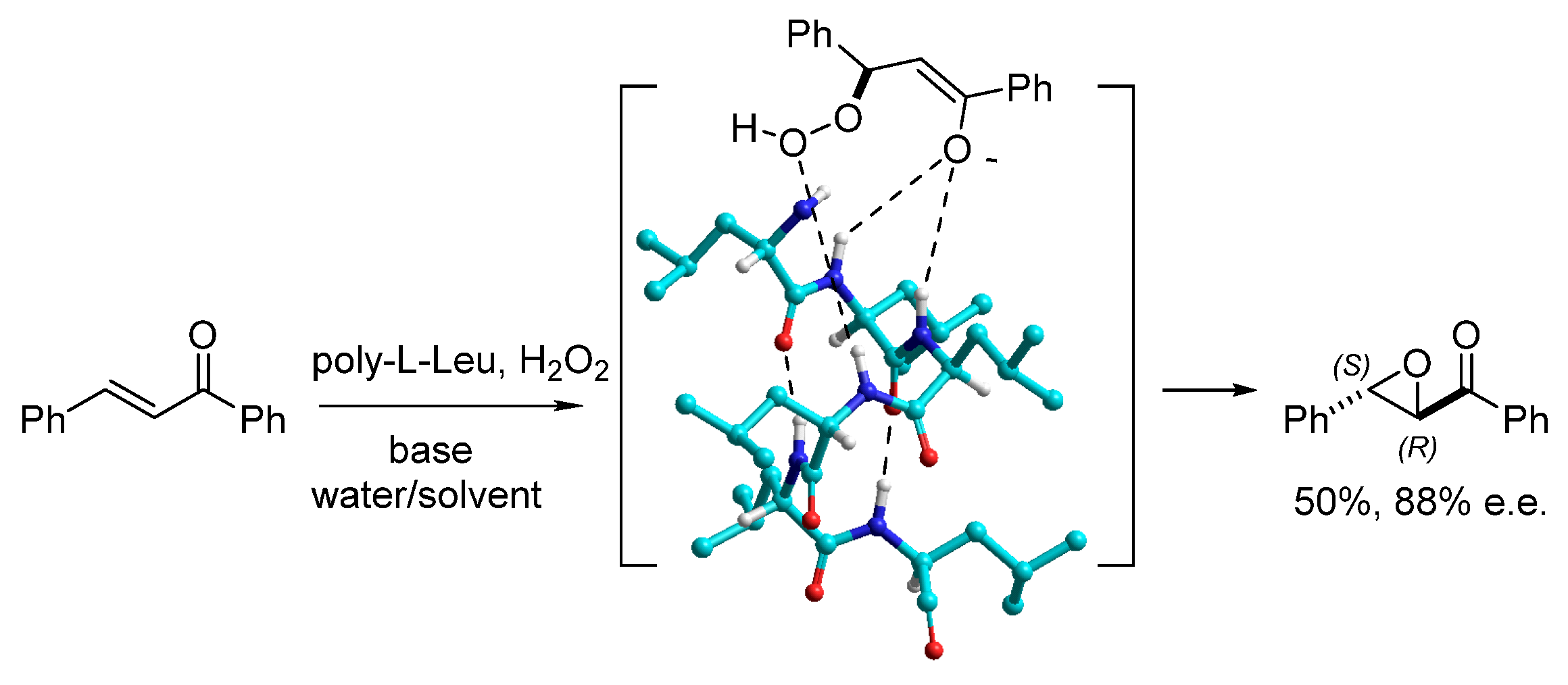
2. Heterogeneous Peptide Organocatalysis
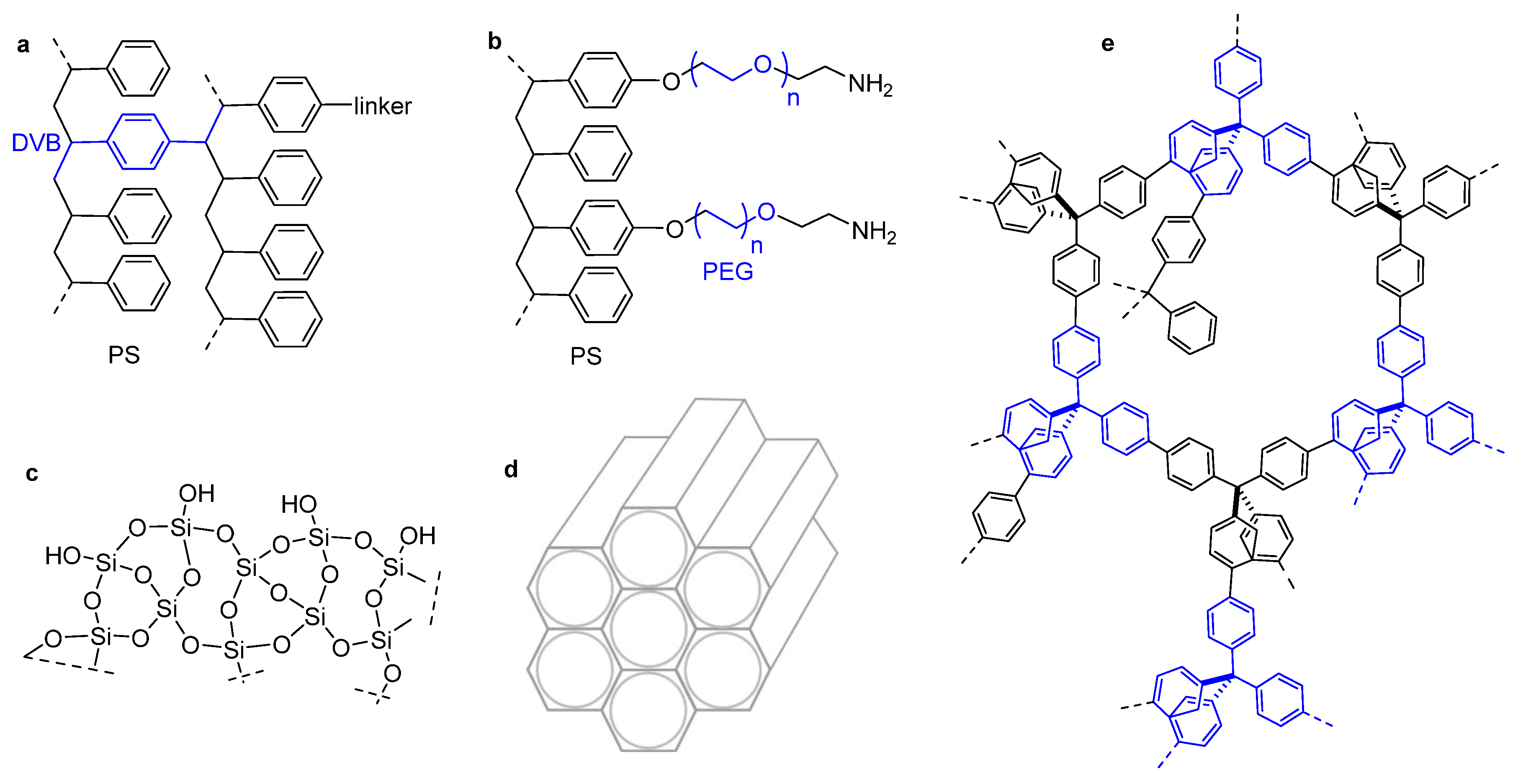
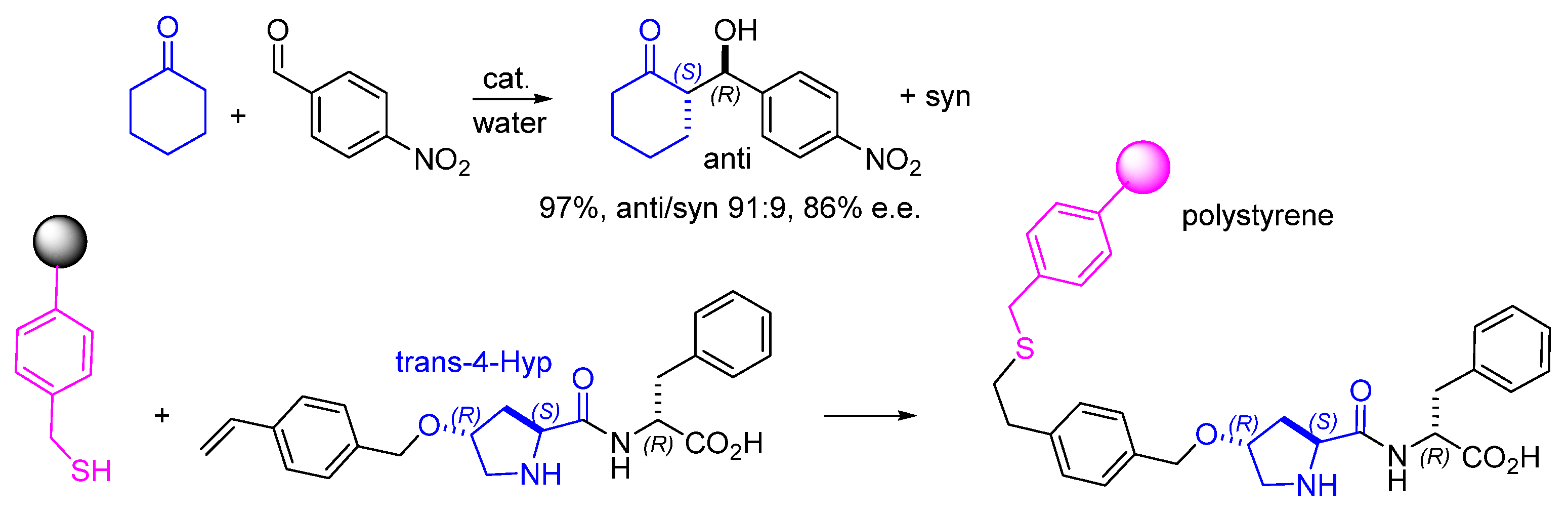
3. Immobilization of Peptide on Polystyrene Resin
4. Immobilization on Polyethylene Glycol-Polystyrene Resins
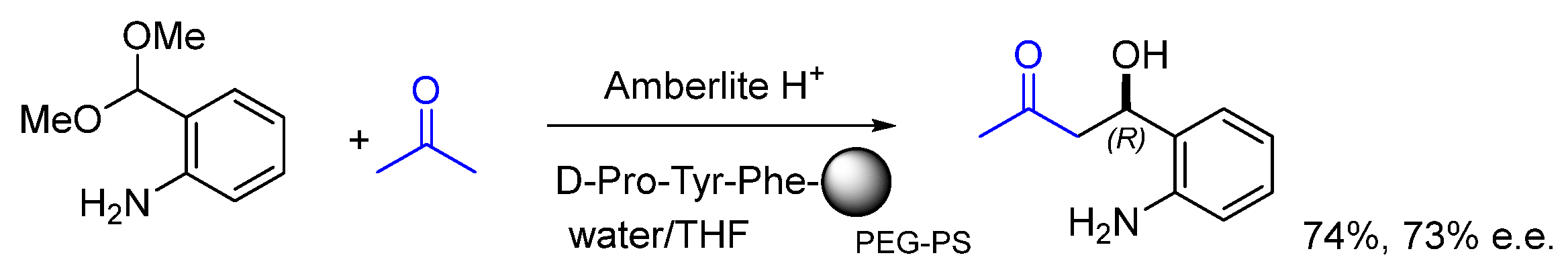
5. On-Bead Immobilization and Screening of Peptide Catalysts
6. Immobilization on Inorganic Supports
7. Porous Organic Frameworks (POFs)
8. Metal–Organic Frameworks (MOFs)
9. Adsorption or Inclusion of Peptides in Inorganic or Hybrid Materials
10. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| e.r. | enantiomeric ratio |
| e.e. | enantiomeric excess |
| d.r. | diastereomeric ratio |
| J-C | Juliá–Colonna |
| DKP | diketopiperazine |
| SPPS | solid-phase peptide synthesis |
| PEG | polyethylene glycol |
| PS | polystyrene |
| OMS | ordered mesoporous silica |
| CuAAC | Cu-catalyzed alkyne-azide cycloaddition |
| MBHA | methyl benzhydryl amine |
| MPNs | microporous polymer networks |
| COFs | covalent organic frameworks |
| POFs | porous organic frameworks |
| MOFs | metal–organic frameworks |
| Aib | 2-aminoisobutyric acid |
References
- Gentilucci, L.; Tolomelli, A.; Squassabia, F. Peptides and peptidomimetics in medicine, surgery and biotechnology. Curr. Med. Chem. 2006, 13, 2449–2466. [Google Scholar] [CrossRef] [PubMed]
- Kuster, T.H.R.; Schnitzer, T. Peptide catalysis: Trends and opportunities. Chem. Catalysis 2025, 5, 101339. [Google Scholar] [CrossRef]
- Juliá, S.; Masana, J.; Vega, J.C. “Synthetic enzymes”. Highly stereoselective epoxidation of chalcone in a triphasic toluene-water-poly[(S)-alanine] system. Angew. Chem. Int. Ed. 1980, 19, 929–931. [Google Scholar] [CrossRef]
- Bérubé, C.; Barbeau, X.; Lague, P.; Voyer, N. Revisiting the Juliá–Colonna enantioselective epoxidation: Supramolecular catalysis in water. Chem. Commun. 2017, 53, 5099–5102. [Google Scholar] [CrossRef]
- De Marco, R.; Tolomelli, A.; Greco, A.; Gentilucci, L. Controlled solid phase peptide bond formation using N-carboxyanhydrides and PEG resins in water. ACS Sustain. Chem. 2013, 1, 566–569. [Google Scholar] [CrossRef]
- Colonna, S.; Perdicchia, D.; Mauro, E.D. Enantioselective reactions catalyzed by synthetic enzymes. A model for chemical evolution. Tetrahedron Asymmetry 2009, 20, 1709–1714. [Google Scholar] [CrossRef]
- Wakeham, D.; Crivoi, D.G.; Medina, F.; Segarra, A.M.; Rutland, M.W. In-situ study of substrate–catalyst interactions in a Juliá–Colonna epoxidation using quartz crystal microbalance with dissipation. J. Colloid. Interface Sci. 2016, 469, 263–268. [Google Scholar] [CrossRef]
- Weyer, A.; Díaz, D.; Nierth, A.; Schlorer, N.E.; Berkessel, A. The L-Leu hexamer, a short and highly enantioselective peptide catalyst for the Juliá–Colonna epoxidation: Stabilization of a helical conformation in DMSO. ChemCatChem 2012, 4, 337–340. [Google Scholar] [CrossRef]
- Oku, J.; Inoue, S. Asymmetric cyanohydrin synthesis catalyzed by a synthetic cyclic dipeptide. J. Chem. Soc. Chem. Commun. 1981, 5, 229–230. [Google Scholar] [CrossRef]
- List, B.; Lerner, R.A.; Barbas III, C.F. Proline-catalyzed direct asymmetric aldol reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Jarvo, E.R.; Miller, S.J. Amino acids and peptides as asymmetric organocatalysts. Tetrahedron 2002, 58, 2481–2495. [Google Scholar] [CrossRef]
- Wennemers, H. Asymmetric catalysis with peptides. Chem. Commun. 2011, 47, 12036–12041. [Google Scholar] [CrossRef]
- Lewandowski, B.; Wennemers, H. Asymmetric catalysis with short-chain peptides. Curr. Opin. Chem. Biol. 2014, 22, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Akagawa, K.; Kudo, K. Development of selective peptide catalysts with secondary structural frameworks. Acc. Chem. Res. 2017, 50, 2429–2439. [Google Scholar] [CrossRef] [PubMed]
- Davie, E.A.C.; Mennen, S.M.; Xu, Y.; Miller, S.J. Asymmetric catalysis mediated by synthetic peptides. Chem. Rev. 2007, 107, 5759–5812. [Google Scholar] [CrossRef] [PubMed]
- Metrano, A.J.; Chinn, A.J.; Shugrue, C.R.; Stone, E.A.; Kim, B.; Miller, S.J. Asymmetric catalysis mediated by synthetic peptides, version 2.0: Expansion of scope and mechanisms. Chem. Rev. 2020, 120, 11479–11615. [Google Scholar] [CrossRef]
- Lewis, J.C. Artificial metalloenzymes and metallopeptide catalysts for organic synthesis. ACS Catal. 2013, 3, 2954–2975. [Google Scholar] [CrossRef]
- Hamley, I.W. Biocatalysts based on peptide and peptide conjugate nanostructures. Biomacromolecules 2021, 22, 1835–1855. [Google Scholar] [CrossRef]
- Sinibaldi, A.; Della Penna, F.; Ponzetti, M.; Fini, F.; Marchesan, S.; Baschieri, A.; Pesciaioli, F.; Carlone, A. Asymmetric organocatalysis accelerated via self assembled minimal structures. Eur. J. Org. Chem. 2021, 39, 5403–5406. [Google Scholar] [CrossRef]
- Qing Liu, Q.; Kuzuya, A.; Wang, Z.-G. Supramolecular enzyme-mimicking catalysts self-assembled from peptides. iScience 2023, 26, 05831. [Google Scholar]
- Grünenfelder, C.E.; Kisunzu, J.K.; Wennemers, H. Peptide-catalyzed stereoselective conjugate addition reactions of aldehydes to maleimide. Angew. Chem. Int. Ed. 2016, 55, 8571–8574. [Google Scholar] [CrossRef] [PubMed]
- Möhler, J.S.; Beiersdörfer, L.K.; Masina, B.; Wechsler, P.; Wennemers, H. Tripeptide organocatalysts for stereoselective conjugate addition reactions with N-heterocyclic substituents. Adv. Synth. Catal. 2022, 364, 3354–3359. [Google Scholar] [CrossRef]
- Rigling, C.; Kisunzu, J.K.; Duschmalé, J.; Haussinger, D.; Wiesner, M.; Ebert, M.-O.; Wennemers, H. Conformational properties of a peptidic catalyst: Insights from NMR spectroscopic studies. J. Am. Chem. Soc. 2018, 140, 10829–10838. [Google Scholar] [CrossRef]
- Metrano, A.J.; Miller, S.J. Peptide-based catalysts reach the outer sphere through remote desymmetrization and atroposelectivity. Acc. Chem. Res. 2019, 52, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Kamanna, K. Amino acids and peptides organocatalysts: A brief overview on its evolution and applications in organic asymmetric synthesis. Curr. Organocat. 2021, 8, 126–146. [Google Scholar]
- Antenucci, A.; Dughera, S.; Renzi, P. Green Chemistry meets asymmetric organocatalysis: A critical overview on catalysts synthesis. ChemSusChem 2021, 14, 2785–2853. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, R.; Adamala, K.; Gasperi, T.; Polticelli, F.; Stano, P. Small and random peptides: An unexplored reservoir of potentially functional primitive organocatalysts. The case of Seryl-Histidine. Life 2017, 7, 19. [Google Scholar]
- Varnava, K.G.; Sarojini, V. Making solid-phase peptide synthesis greener: A review of the literature. Chem. Asian J. 2019, 14, 1088–1097. [Google Scholar] [CrossRef]
- Ferrazzano, L.; Catani, M.; Cavazzini, A.; Martelli, G.; Corbisiero, D.; Cantelmi, P.; Fantoni, T.; Mattellone, A.; De Luca, C.; Felletti, S.; et al. Sustainability in peptide chemistry: Current synthesis and purification technologies and future challenges. Green. Chem. 2022, 24, 975–1020. [Google Scholar] [CrossRef]
- Sánchez-Antonio, O.; Romero-Sedglach, K.A.; Vázquez-Orta, E.C.; Juaristi, E. New mesoporous silica-supported organocatalysts based on (2S)-(1,2,4-triazol-3-yl)-proline: Efficient, reusable, and heterogeneous catalysts for the asymmetric aldol reaction. Molecules 2020, 25, 4532. [Google Scholar] [CrossRef]
- Pasuparthy, S.D.; Maiti, B. Enantioselective organocatalytic Michael addition reactions catalyzed by proline/prolinol/supported proline based organocatalysts: An overview. ChemistrySelect 2022, 7, e202104261. [Google Scholar] [CrossRef]
- Franconetti, A.; de Gonzalo, G. Recent developments on supported hydrogen-bond organocatalysts. ChemCatChem 2018, 10, 5554–5572. [Google Scholar] [CrossRef]
- Bartók, M. Advances in immobilized organocatalysts for the heterogeneous asymmetric direct aldol reactions. Cat. Rev. 2015, 57, 192–255. [Google Scholar] [CrossRef]
- Lu, J.; Toy, P.H. Organic polymer supports for synthesis and for reagent and catalyst immobilization. Chem. Rev. 2009, 109, 815–838. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.S.; Bao, X.Y.; Guo, W.; Lee, F.Y. Immobilizing catalysts on porous materials. Mater. Today 2006, 9, 32–39. [Google Scholar] [CrossRef]
- Chaoui, N.; Trunk, M.; Dawson, R.; Schmidt, J.; Thomas, A. Trends and challenges for microporous polymers. Chem. Soc. Rev. 2017, 46, 3302. [Google Scholar] [CrossRef]
- Geng, K.; He, T.; Liu, R.; Dalapati, S.; Tan, K.T.; Li, Z.; Tao, S.; Gong, Y.; Jiang, Q.; Jiang, D. Covalent organic frameworks: Design, synthesis, and functions. Chem. Rev. 2020, 120, 8814. [Google Scholar] [CrossRef]
- Alegre-Requena, J.V.; Marqués-López, E.; Herrera, R.P.; Díaz Díaz, D. Metal–organic frameworks (MOFs) bring new life to hydrogen-bonding organocatalysts in confined spaces. Cryst. Eng. Comm. 2016, 18, 3985–3995. [Google Scholar] [CrossRef]
- Gang, L.; Lam, K.S. The One-bead one-compound combinatorial library method. Chem. Rev. 1997, 97, 411–448. [Google Scholar]
- Amenós, L.; Alza, E.; Pericàs, M.A. Organocatalysis in continuous flow for drug discovery. In Flow Chemistry in Drug Discovery; Alcazar, J., de la Hoz, A., Díaz-Ortiz, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2021. [Google Scholar]
- De Risi, C.; Bortolini, O.; Brandolese, A.; Di Carmine, G.; Ragno, D.; Massi, A. Recent advances in continuous-flow organocatalysis for process intensification. React. Chem. Eng. 2020, 5, 1017–1052. [Google Scholar] [CrossRef]
- Qiu, W.; He, L.; Chen, Q.; Luo, W.; Yu, Z.; Yang, F.; Tang, J. Imidazolium-modified poly(l-leucine) catalyst: An efficient and recoverable catalyst for Juliá–Colonna reactions. Tetrahedron Lett. 2009, 50, 5225–5227. [Google Scholar] [CrossRef]
- Cozzi, F. Immobilization of organic catalysts: When, why, and how. Adv. Synth. Cat. 2006, 348, 1367–1390. [Google Scholar] [CrossRef]
- Hernández, J.G.; Juaristi, E. Recent efforts directed to the development of more sustainable asymmetric organocatalysis. Chem. Commun. 2012, 48, 5396–5409. [Google Scholar] [CrossRef]
- Benaglia, M.; Puglisi, A. Catalyst Immobilization: Methods and Applications; Wiley–VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2020. [Google Scholar]
- Guillena, G.; Alonso, D.; Baeza, A.; Chinchilla, R.; Flores-Ferrándiz, J.; Gómez-Martínez, M.; Trillo, P. Pursuing chemical efficiency by using supported organocatalysts for asymmetric reactions under aqueous conditions. Curr. Organocat. 2015, 2, 102–123. [Google Scholar] [CrossRef]
- Banfi, S.; Colonna, S.; Molinari, H.; Juliá, S.; Guixer, J. Asymmetric epoxidation of electron-poor olefins-V: Influence on stereoselectivity of the structure of poly-α-aminoacids used as catalysts. Tetrahedron 1984, 40, 5207–5211. [Google Scholar] [CrossRef]
- Itsuno, S.; Sakakura, M.; Koichi, I. Polymer-supported poly(amino acids) as new asymmetric epoxidation catalyst of α,β-unsaturated ketones. J. Org. Chem. 1990, 55, 6047–6049. [Google Scholar] [CrossRef]
- Gruttadauria, M.; Salvo, A.M.P.; Giacalone, F.; Agrigento, P.; Noto, R. Enhanced activity and stereoselectivity of polystyrene-supported proline-based organic catalysts for direct asymmetric aldol reaction in water. Eur. J. Org. Chem. 2009, 31, 5437–5444. [Google Scholar] [CrossRef]
- Wiesner, M.; Revell, J.D.; Wennemers, H. Tripeptides as efficient asymmetric catalysts for 1,4-addition reactions of aldehydes to nitroolefins—A rational approach. Angew. Chem. Int. Ed. 2008, 47, 1871–1874. [Google Scholar] [CrossRef]
- Arakawa, Y.; Wennemers, H. Enamine catalysis in flow with an immobilized peptidic catalyst. ChemSusChem 2013, 6, 242–245. [Google Scholar] [CrossRef]
- Tong, M.L.; Huber, F.; Taghuo Kaptouom, E.S.; Cellnik, T.; Kirsch, S.F. Enhanced site-selectivity in acylation reactions with substrate-optimized catalysts on solid supports. Chem. Commun. 2017, 53, 3086–3089. [Google Scholar] [CrossRef]
- Szőllősi, G.; Csampai, A.; Somlai, C.; Fekete, M.; Bartok, M. Unusual enantioselectivities in heterogeneous organocatalyzed reactions: Reversal of direction using proline di- versus tri-peptides in the aldol addition. J. Mol. Catal. A Chem. 2014, 382, 86–92. [Google Scholar] [CrossRef]
- Machuca, E.; Granados, G.; Hinojosa, B.; Juaristi, E. Synthesis and evaluation of (S)-proline-containing dipeptidic organocatalysts bound to MBHA resin in asymmetric aldol reactions. Tetrahedron Lett. 2015, 56, 6047–6051. [Google Scholar] [CrossRef]
- García-Monzón, I.; Borges-González, J.; Martín, T. Solid-supported tetrahydropyran-based hybrid dipeptide catalysts for Michael addition of aldehydes to nitrostyrenes. Adv. Synth. Catal. 2022, 364, 2822–2829. [Google Scholar] [CrossRef]
- Arakawa, Y.; Yamanomoto, K.; Kita, H.; Minagawa, K.; Tanaka, M.; Haraguchi, N.; Itsuno, S.; Imada, Y. Design of peptide-containing N5-unmodified neutral flavins that catalyze aerobic oxygenations. Chem. Sci. 2017, 8, 5468–5475. [Google Scholar] [CrossRef]
- Seitz, A.; Wende, R.C.; Schreiner, P.R. Site–selective acylation of pyranosides with immobilized oligopeptide catalysts in flow. Chem. Eur. J. 2023, 29, e202203002. [Google Scholar] [CrossRef] [PubMed]
- Cappi, W.; Chen, W.P.; Flood, R.W.; Liao, Y.W.; Roberts, S.M.; Skidmore, J.; Smith, J.A.; Williamson, N.M. New procedures for the Julia–Colonna asymmetric epoxidation: Synthesis of (+)-clausenamide. Chem. Commun. 1998, 2, 1159–1160. [Google Scholar] [CrossRef]
- Berkessel, A.; Gasch, N.; Glaubitz, K.; Koch, C. Highly enantioselective enone epoxidation catalyzed by short solid phase-bound peptides: Dominant role of peptide helicity. Org. Lett. 2001, 3, 3839–3842. [Google Scholar] [CrossRef]
- Akagawa, K.; Sakamoto, S.; Kudo, K. Direct asymmetric aldol reaction in aqueous media using polymer-supported peptide. Tetrahedron Lett. 2005, 46, 8185–8187. [Google Scholar] [CrossRef]
- Akagawa, K.; Sakamoto, S.; Kudo, K. Resin-supported acid and base-catalyzed one-pot sequential reaction including an enantioselective step. Tetrahedron Lett. 2007, 48, 985–987. [Google Scholar] [CrossRef]
- Akagawa, K.; Akabane, H.; Sakamoto, S.; Kudo, K. Organocatalytic asymmetric transfer hydrogenation in aqueous media using resin-supported peptide having a polyleucine tether. Org. Lett. 2008, 10, 2035–2037. [Google Scholar] [CrossRef]
- Gentilucci, L.; De Marco, R.; Cerisoli, L. Chemical modifications designed to improve peptide stability: Incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203. [Google Scholar] [CrossRef] [PubMed]
- Akagawa, K.; Fujiwara, T.; Sakamoto, S.; Kudo, K. Efficient asymmetric α-oxyamination of aldehydes by resin-supported peptide catalyst in aqueous media. Org. Lett. 2010, 12, 1804–1807. [Google Scholar] [CrossRef]
- Akagawa, K.; Takigawa, S.; Mano, E.; Kudo, K. Sequential oxidation/asymmetric aldol reaction of primary alcohols by resin-supported catalysts. Tetrahedron Lett. 2011, 52, 770–773. [Google Scholar] [CrossRef]
- Akagawa, K.; Hirata, T.; Kudo, K. Asymmetric epoxidation of enones by peptide-based catalyst: A strategy inverting Juliá–Colonna stereoselectivity. Synlett 2016, 27, 1217–1222. [Google Scholar] [CrossRef]
- Akagawa, K.; Kudo, K. Asymmetric epoxidation of α,β-unsaturated aldehydes in aqueous media catalyzed by resin-supported peptide-containing unnatural amino acids. Adv. Synth. Catal. 2011, 353, 843–847. [Google Scholar] [CrossRef]
- Arakawa, Y.; Wiesner, M.; Wennemers, H. Efficient recover and reuse of an immobilized peptidic organocatalyst. Adv. Synth. Catal. 2011, 353, 1201–1206. [Google Scholar] [CrossRef]
- Ötvös, S.B.; Mándity, I.M.; Fülöp, F. Asymmetric aldol reaction in a continuous-flow reactor catalyzed by a highly reusable heterogeneous peptide. J. Catal. 2012, 295, 179–185. [Google Scholar] [CrossRef]
- Akagawa, K.; Kudo, K. Asymmetric induction by helical poly (amino acid)s in cyanosilylation of aldehydes. Tetrahedron Lett. 2012, 53, 5981–5983. [Google Scholar] [CrossRef]
- Akagawa, K.; Yamashita, T.; Sakamoto, S.; Kudo, K. Friedel–Crafts-type alkylation in aqueous media using resin-supported peptide catalyst having polyleucine. Tetrahedron Lett. 2009, 50, 5602–5604. [Google Scholar] [CrossRef]
- Akagawa, K.; Suzuki, R.; Kudo, K. Effect of the helical tether of a resin-supported peptide catalyst for Friedel–Crafts-type alkylation in water. Adv. Synth. Catal. 2012, 354, 1280–1286. [Google Scholar] [CrossRef]
- Akagawa, K.; Nishi, N.; Sen, J.; Kudo, K. Peptide-catalyzed consecutive 1,6- and 1,4-additions of thiols to α,β,γ,δ-unsaturated aldehydes. Org. Biomol. Chem. 2014, 12, 3581–3585. [Google Scholar] [CrossRef] [PubMed]
- Akagawa, K.; Kudo, K. Construction of an all-carbon quaternary stereocenter by the peptide-catalyzed asymmetric michael addition of nitromethane to β-disubstituted α,β-unsaturated aldehydes. Angew. Chem. Int. Ed. 2012, 51, 12786–12789. [Google Scholar] [CrossRef] [PubMed]
- Akagawa, K.; Takigawa, S.; Nagamine, I.S.; Umezawa, R.; Kudo, K. Peptide-catalyzed diastereo- and enantioselective cyclopropanation of aromatic α,β-unsaturated aldehydes. Org. Lett. 2013, 15, 4964–4967. [Google Scholar] [CrossRef]
- Akiyama, M.; Akagawa, K.; Seino, H.; Kudo, K. Peptide-catalyzed kinetic resolution of planar-chiral metallocenes. Chem. Commun. 2014, 50, 7893–7896. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Akagawa, K.; Mitsuda, M.; Kudo, K. Asymmetric α-amination of aldehydes by a recyclable resin-supported peptide catalyst. Adv. Synth. Catal. 2013, 355, 294–296. [Google Scholar] [CrossRef]
- Ötvös, S.B.; Szloszár, A.; Mándity, I.M.; Fülöp, F. Heterogeneous dipeptide-catalyzed α-amination of aldehydes in a continuous-flow reactor: Effect of residence time on enantioselectivity. Adv. Synth. Catal. 2015, 357, 3671–3680. [Google Scholar] [CrossRef]
- Akagawa, K.; Higuchi, J.; Yoshikawa, I.; Kudo, K. Kinetic resolution of ansa cyclophanes by peptide-catalyzed aldol/retro-aldol reactions. Eur. J. Org. Chem. 2018, 2018, 5278–5281. [Google Scholar] [CrossRef]
- Akagawa, K.; Suzuki, R.; Kudo, K. Development of a peptide-based primary aminocatalyst with a helical structure. Asian J. Org. Chem. 2014, 3, 514–522. [Google Scholar] [CrossRef]
- Lichtor, P.A.; Miller, S.J. Combinatorial evolution of site and enantioselective catalysts for polyene epoxidation. Nat. Chem. 2012, 4, 990–995. [Google Scholar] [CrossRef]
- Lichtor, P.A.; Miller, S.J. Experimental lineage and functional analysis of a remotely directed peptide epoxidation catalyst. J. Am. Chem. Soc. 2014, 136, 5301–5308. [Google Scholar] [CrossRef]
- Sambasivan, R.; Ball, Z.T. Screening rhodium metallopeptide libraries “on bead”: Asymmetric cyclopropanation and a solution to the enantiomer problem. Angew. Chem. Int. Ed. 2012, 51, 8568–8572. [Google Scholar] [CrossRef] [PubMed]
- Sambasivan, R.; Zheng, W.; Burya, S.J.; Popp, B.V.; Turro, C.; Clementi, C.; Ball, Z.T. A tripodal peptide ligand for asymmetric Rh(II) catalysis highlights unique features of on-bead catalyst development. Chem. Sci. 2014, 5, 1401–1407. [Google Scholar] [CrossRef]
- Akagawa, K.; Sakai, N.; Kudo, K. Histidine-containing peptide catalysts developed by a facile library screening method. Angew. Chem. Int. Ed. 2015, 54, 1822–1826. [Google Scholar] [CrossRef]
- Akagawa, K.; Satou, J.; Kudo, K. Exploration of structural frameworks for reactive and enantioselective peptide catalysts by library screenings. J. Org. Chem. 2016, 81, 9396–9401. [Google Scholar] [CrossRef]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 1992, 359, 710–712. [Google Scholar] [CrossRef]
- Zhao, D.; Huo, Q.; Feng, J.; Chmelka, B.F.; Stucky, G.D. Nonionic triblock and star diblock copolymer and oligomeric surfactant syntheses of highly ordered, hydrothermally stable, mesoporous silica structures. J. Am. Chem. Soc. 1998, 120, 6024–6036. [Google Scholar] [CrossRef]
- Yan, J.; Wang, L. Asymmetric aldol reactions catalyzed by efficient and recyclable silica-supported proline-based peptides. Chirality 2009, 21, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Jebors, S.; Enjalbal, C.; Amblard, M.; Mehdi, A.; Subra, G.; Martinez, J. Bioorganic hybrid oms by straightforward grafting of trialkoxysilyl peptides. J. Mater. Chem. B 2013, 1, 2921–2925. [Google Scholar] [CrossRef]
- Scatena, G.S.; de la Torre, A.F.; Cass, Q.B.; Rivera, D.G.; Paixão, M.W. Multicomponent approach to silica-grafted peptide catalysts: A 3D continuous-flow organocatalytic system with on-line monitoring of conversion and stereoselectivity. ChemCatChem 2014, 6, 3208–3214. [Google Scholar] [CrossRef]
- Brand, R.D.; Busche, S.A.; Börner, H.G.; Smarsly, B.M. Peptide-based organocatalyst on stage: Functionalizing mesoporous silica by tetrazine-norbornene ligation. ChemCatChem 2023, 15, e202300778. [Google Scholar] [CrossRef]
- Lin, Z.-J.; Lü, J.; Li, L.; Li, H.-F.; Cao, R. Defect porous organic frameworks (dPOFs) as a platform for chiral organocatalysis. J. Catal. 2017, 355, 131–138. [Google Scholar] [CrossRef]
- Busche, S.A.; Traxler, M.; Thomas, A.; Börner, H.G. Ligating catalytically active peptides onto microporous polymers: A general route toward specifically-functional high surface area platforms. ChemSusChem 2024, 17, e202301045. [Google Scholar] [CrossRef] [PubMed]
- Mantion, A.; Massüger, L.; Rabu, P.; Palivan, C.; McCusker, L.B.; Taubert, A. Metal–peptide frameworks (MPFs): “Bioinspired” metal organic. J. Am. Chem. Soc. 2008, 130, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy, J.; Legrand, A.; Quadrelli, E.A.; Canivet, J.; Farrusseng, D. Enantiopure peptide-functionalized metal-organic frameworks. J. Am. Chem. Soc. 2015, 137, 9409–9416. [Google Scholar] [CrossRef]
- Fracaroli, A.M.; Siman, P.; Nagib, D.A.; Suzuki, M.; Furukawa, H.; Dean-Toste, F.; Yaghi, O.M. Seven post-synthetic covalent reactions in tandem leading to enzyme-like complexity within metal–organic framework crystals. J. Am. Chem. Soc. 2016, 138, 8352–8355. [Google Scholar] [CrossRef]
- Cirujano, F.G.; Martín, N.; Almora-Barrios, N.; Martí-Gastaldo, C. Catalytic activity of a CuGHK peptide-basedb porous material. Catal. Sci. Technol. 2021, 11, 6053–6057. [Google Scholar] [CrossRef]
- Aprile, C.; Giacalone, F.; Gruttadauria, M.; Marculescu, A.M.; Noto, R.; Revell, J.D.; Wennemers, H. New ionic liquid-modified silica gels as recyclable materials for l-proline- or H-Pro-Pro-Asp-NH2-catalyzed aldol reaction. Green. Chem. 2007, 9, 1328–1334. [Google Scholar] [CrossRef]
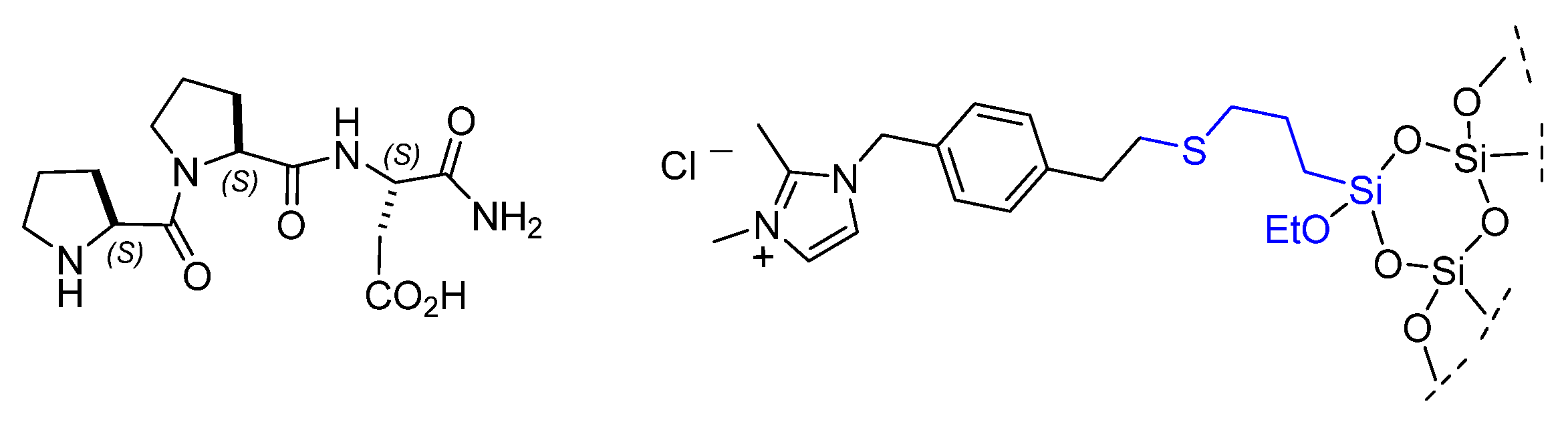
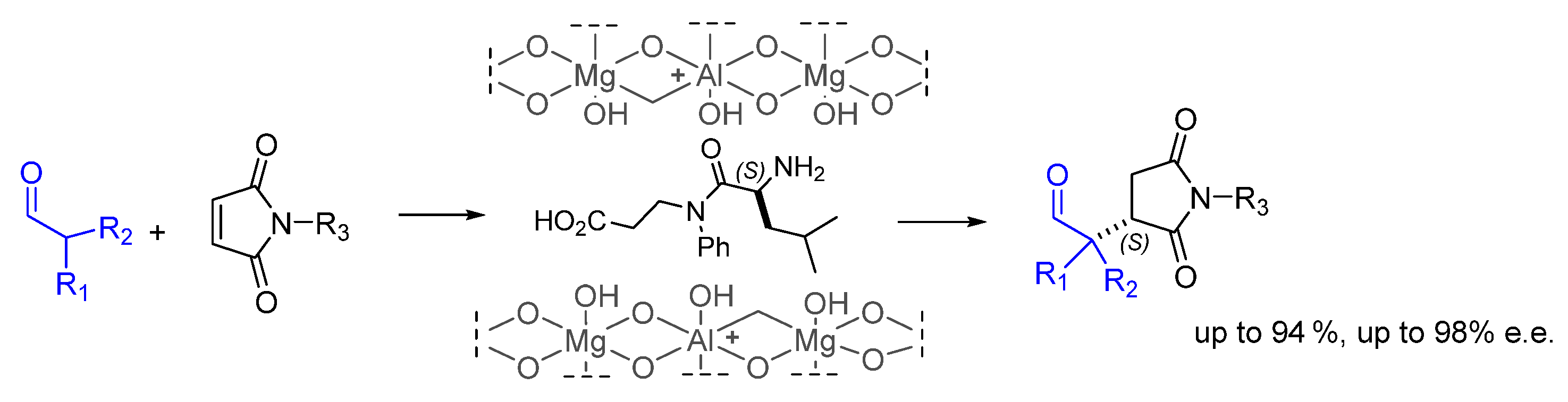
- Miranda, R.-A.; Llorca, J.; Medina, F.; Sueiras, J.E.; Segarra, A.M. Asymmetric epoxidation of chalcone catalyzed by reusable poly-L-leucine immobilized on hydrotalcite. J. Catal. 2011, 282, 65–73. [Google Scholar] [CrossRef]
- Landeros, J.M.; Cruz-Hernández, C.; Juaristi, E. α-Amino acids and α,β-dipeptides intercalated into hydrotalcite: Efficient catalysts in the asymmetric Michael addition reaction of aldehydes to N-substituted maleimides. Eur. J. Org. Chem. 2021, 37, 5117–5126. [Google Scholar] [CrossRef]
- Szőllősi, G.; Gombkötő, D.; Mogyorós, A.Z.; Fülöp, F. Surface-improved asymmetric Michael addition catalyzed by amino acids adsorbed on Laponite. Adv. Synth. Catal. 2018, 360, 1992–2004. [Google Scholar] [CrossRef]
- Anselmi, M.; Stavole, P.; Boanini, E.; Bigi, A.; Juaristi, E.; Gentilucci, L. Green synthesis of bioactive oligopeptides promoted by recyclable nanocrystalline hydroxyapatite. Future Med. Chem. 2020, 12, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Santino, F.; Petruzzelli, R.; Zhao, J.; Boanini, E.; Gentilucci, L. Peptide bond formation using unprotected N-carboxyanhydrides under green chemistry conditions. Sustain. Chem. Pharm. 2021, 24, 100540. [Google Scholar] [CrossRef]
- He, T.; Valagussa, B.; Boanini, E.; Gentilucci, L. Expedient recycling of peptide organocatalysts using a nanocrystalline hydroxyapatite catching system. Sus. Chem. Pharm. 2024, 37, 101383. [Google Scholar] [CrossRef]



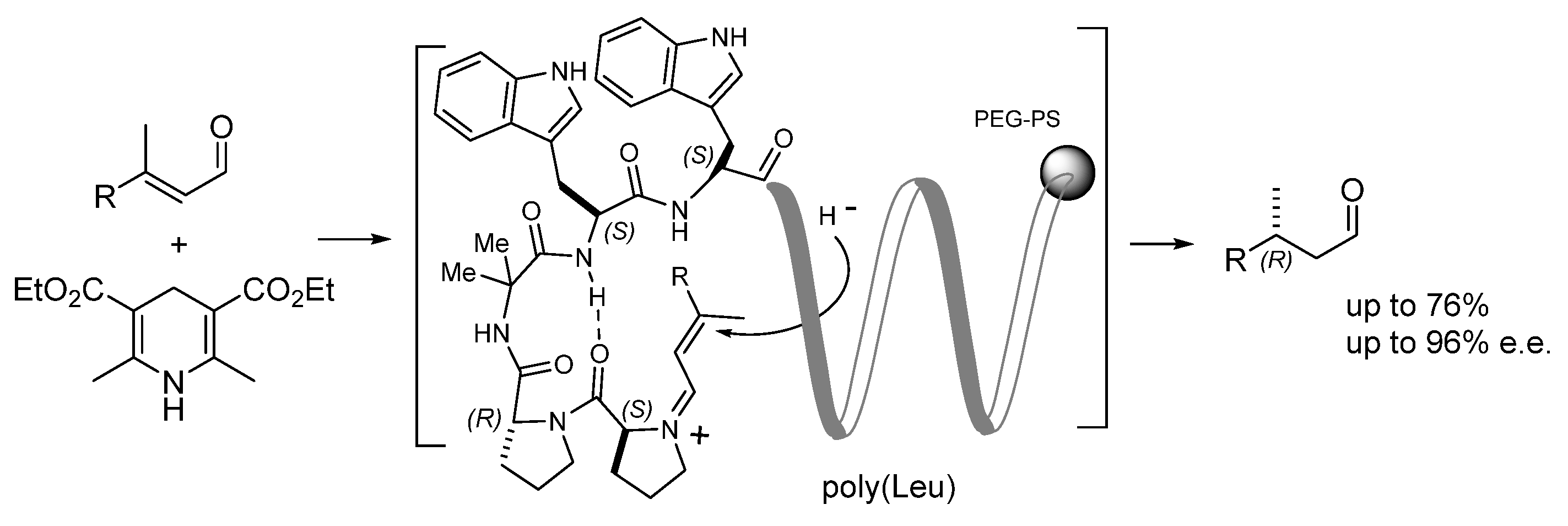
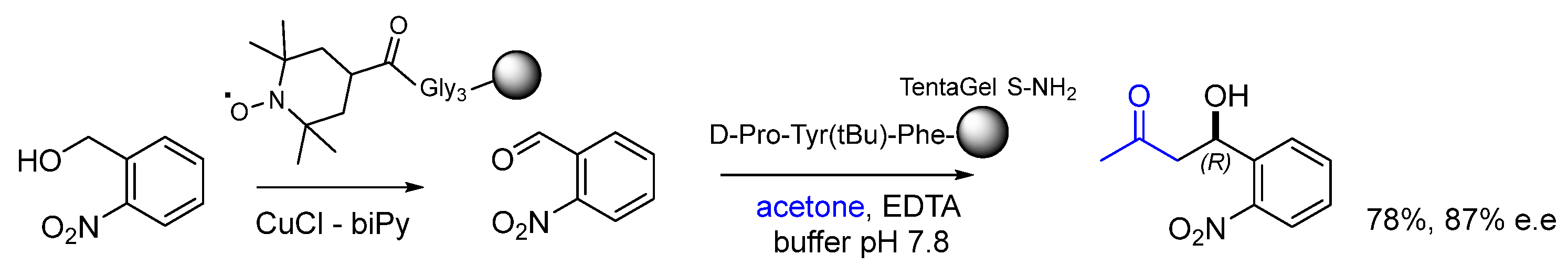

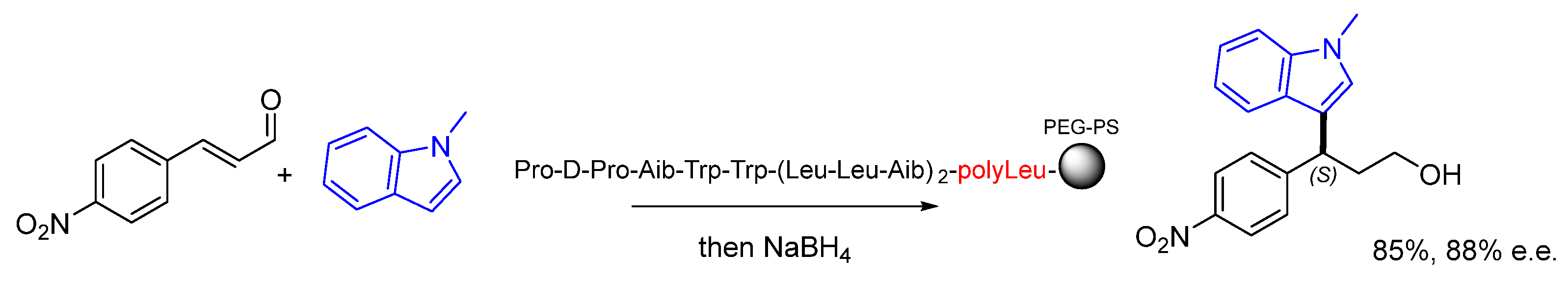
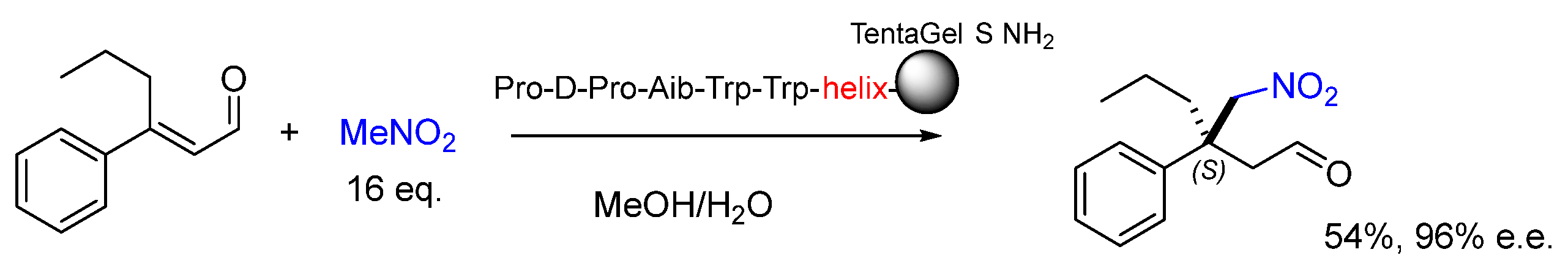
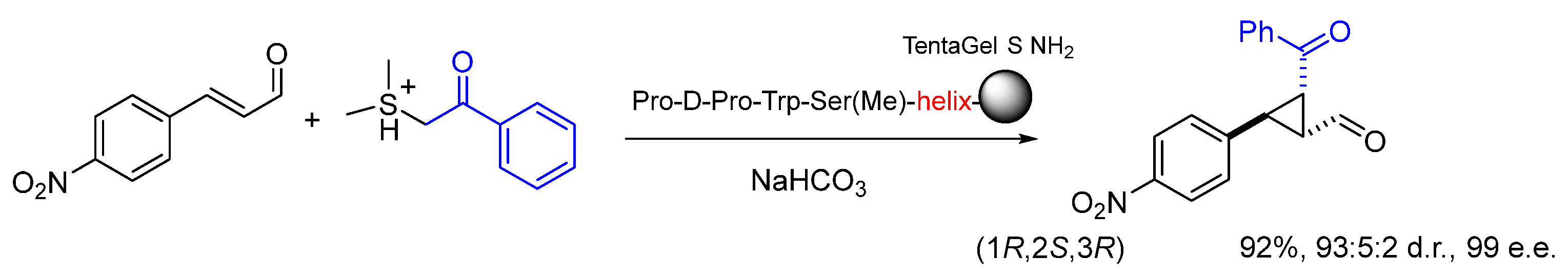

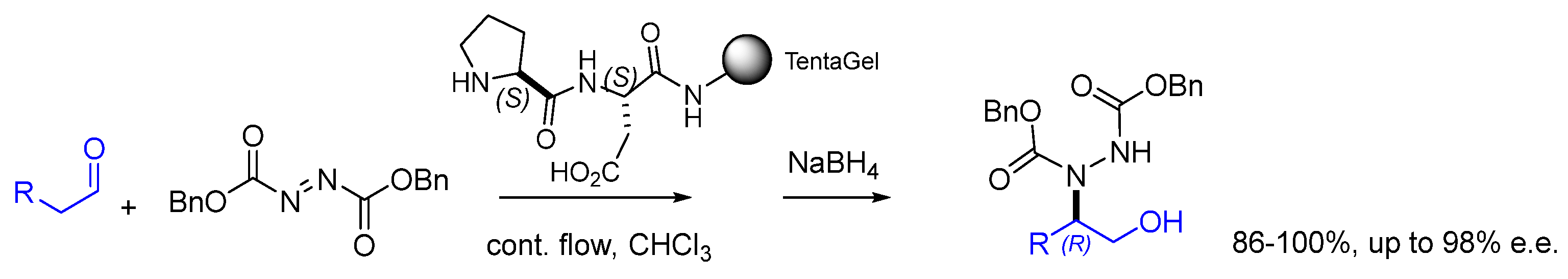
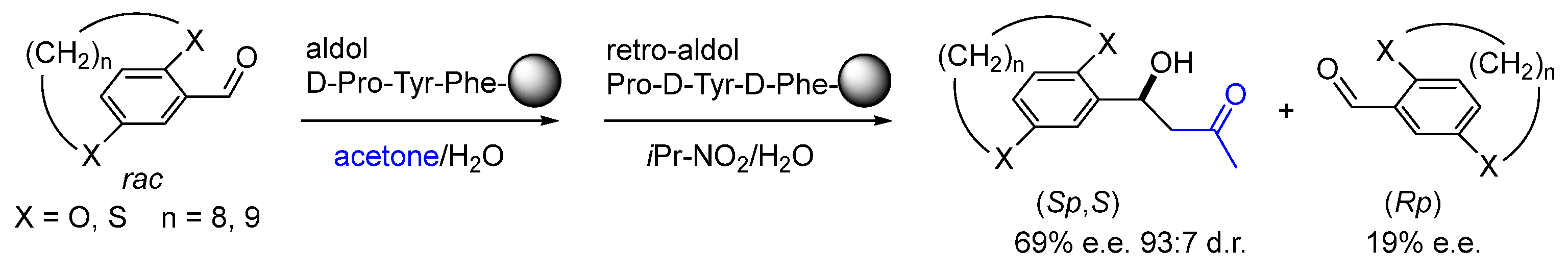
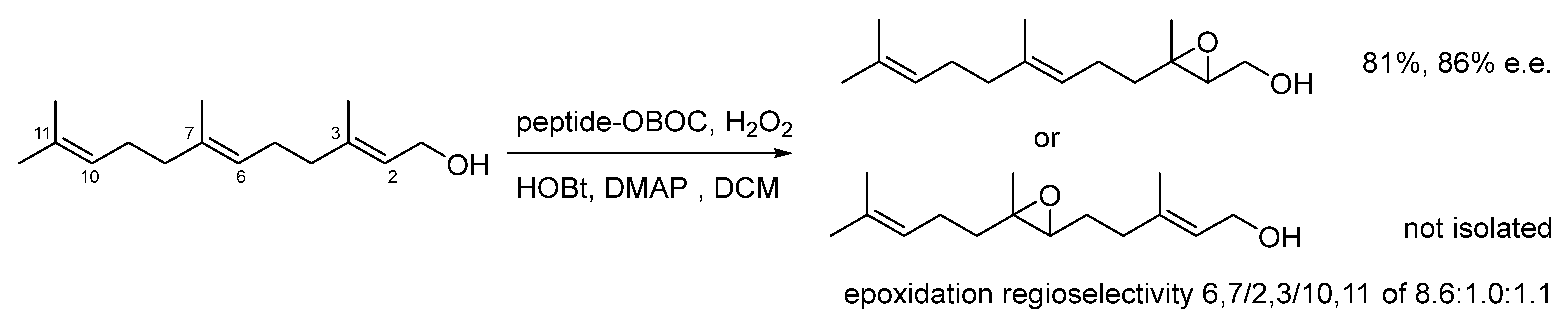
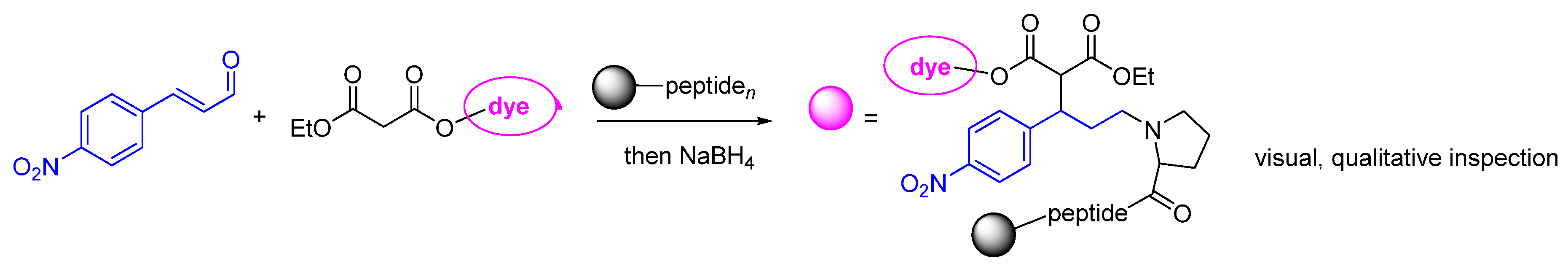
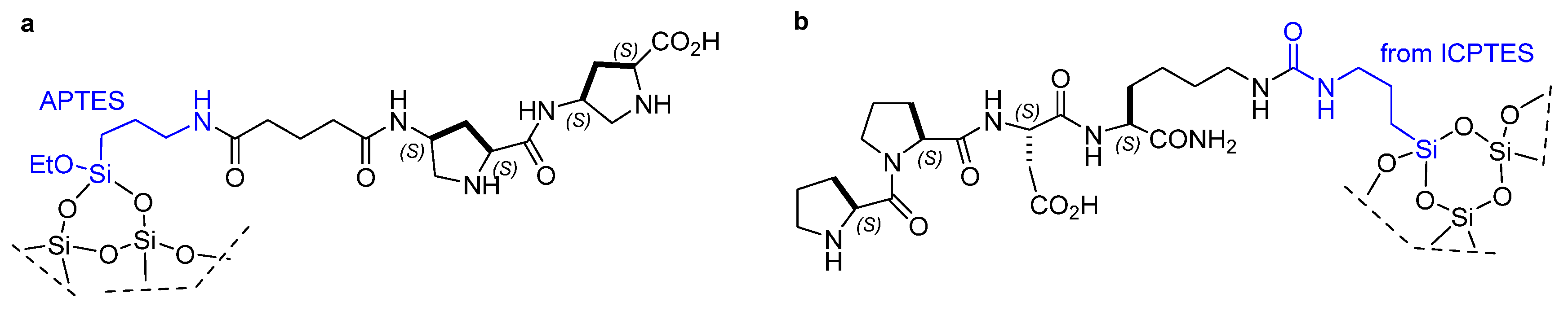
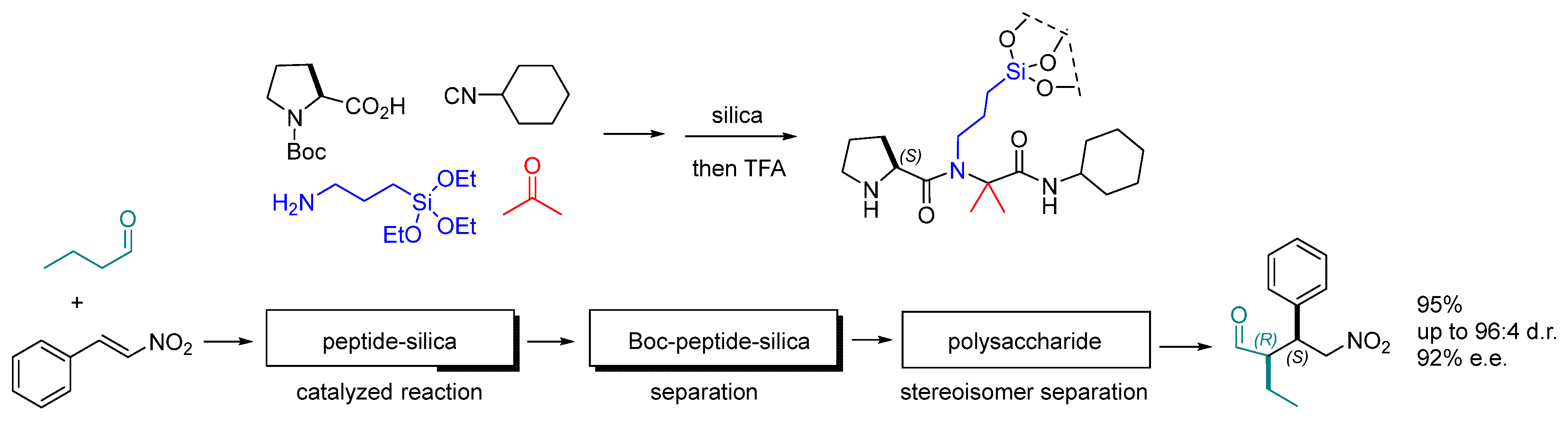


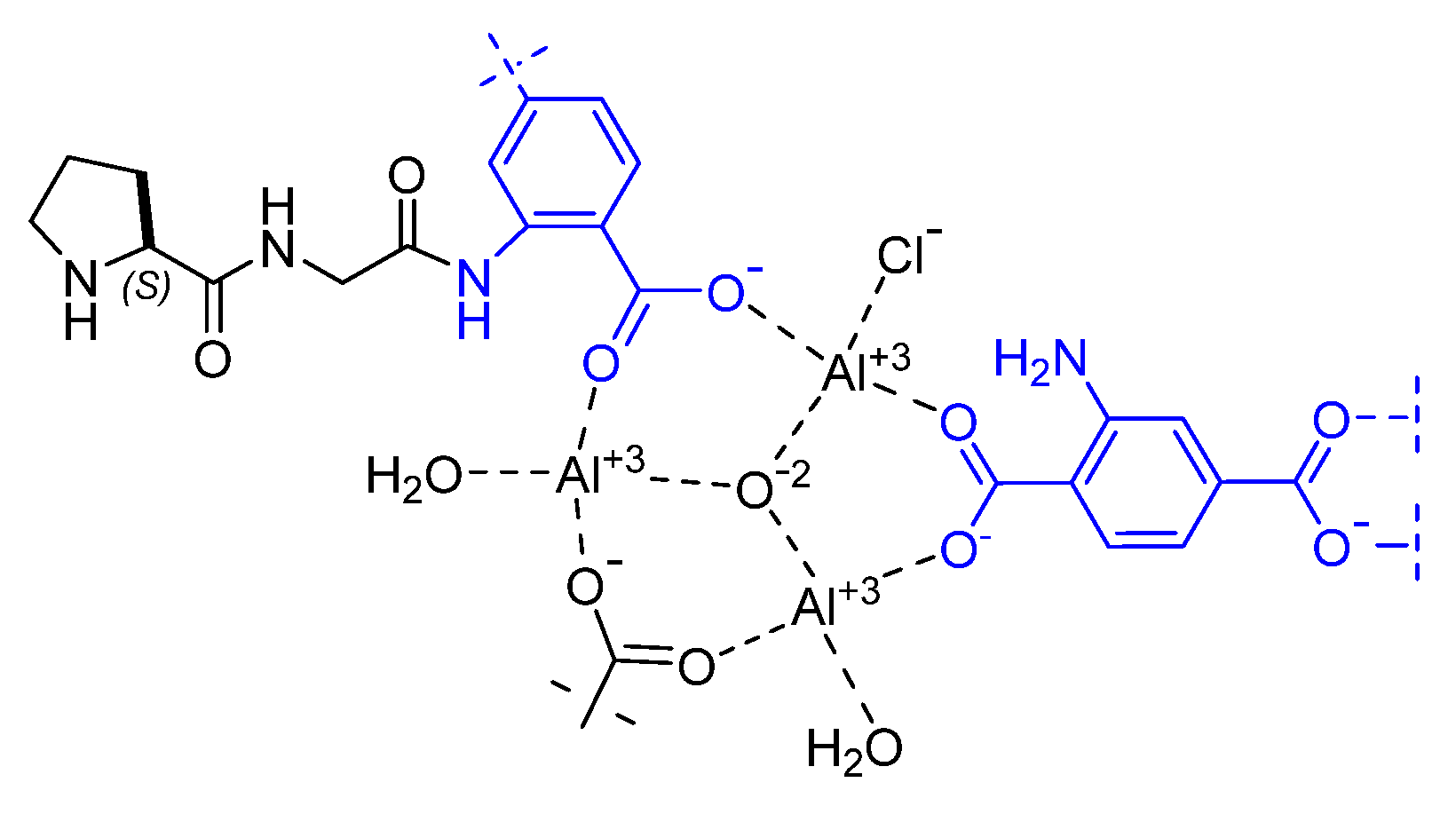

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francescato, M.; Liao, H.; Gentilucci, L. Engineering and Exploiting Immobilized Peptide Organocatalysts for Modern Synthesis. Molecules 2025, 30, 2517. https://doi.org/10.3390/molecules30122517
Francescato M, Liao H, Gentilucci L. Engineering and Exploiting Immobilized Peptide Organocatalysts for Modern Synthesis. Molecules. 2025; 30(12):2517. https://doi.org/10.3390/molecules30122517
Chicago/Turabian StyleFrancescato, Marco, Hang Liao, and Luca Gentilucci. 2025. "Engineering and Exploiting Immobilized Peptide Organocatalysts for Modern Synthesis" Molecules 30, no. 12: 2517. https://doi.org/10.3390/molecules30122517
APA StyleFrancescato, M., Liao, H., & Gentilucci, L. (2025). Engineering and Exploiting Immobilized Peptide Organocatalysts for Modern Synthesis. Molecules, 30(12), 2517. https://doi.org/10.3390/molecules30122517