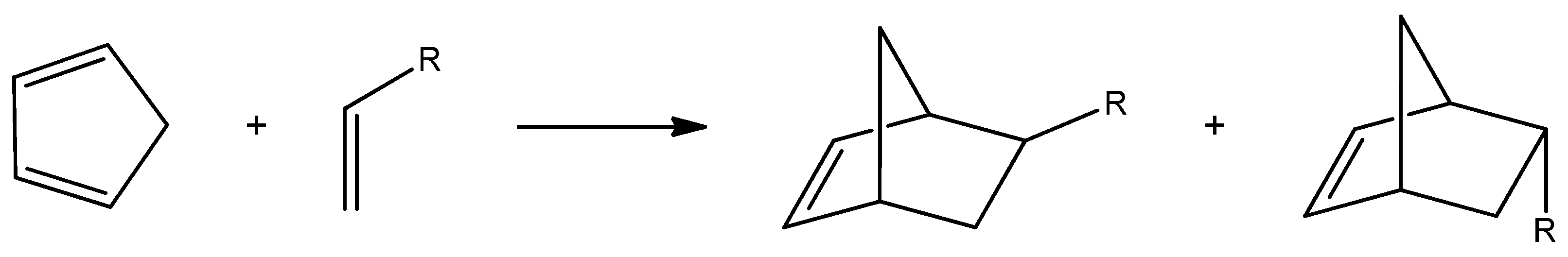
2.1. The Electron Localization Function (ELF)
The ELF [
20] serves as a powerful method for analyzing the electronic structure of organic molecules, offering both qualitative and quantitative insights. Therefore, based on the known structure–reactivity relationships of
CNA, a topological ELF analysis was carried out for
CNA (
2a–
h,
3a–
h,
4a–
h) and
Cp (
1a–
h) to examine their electronic structures and gain insights into their potential reactivity in DA reactions.
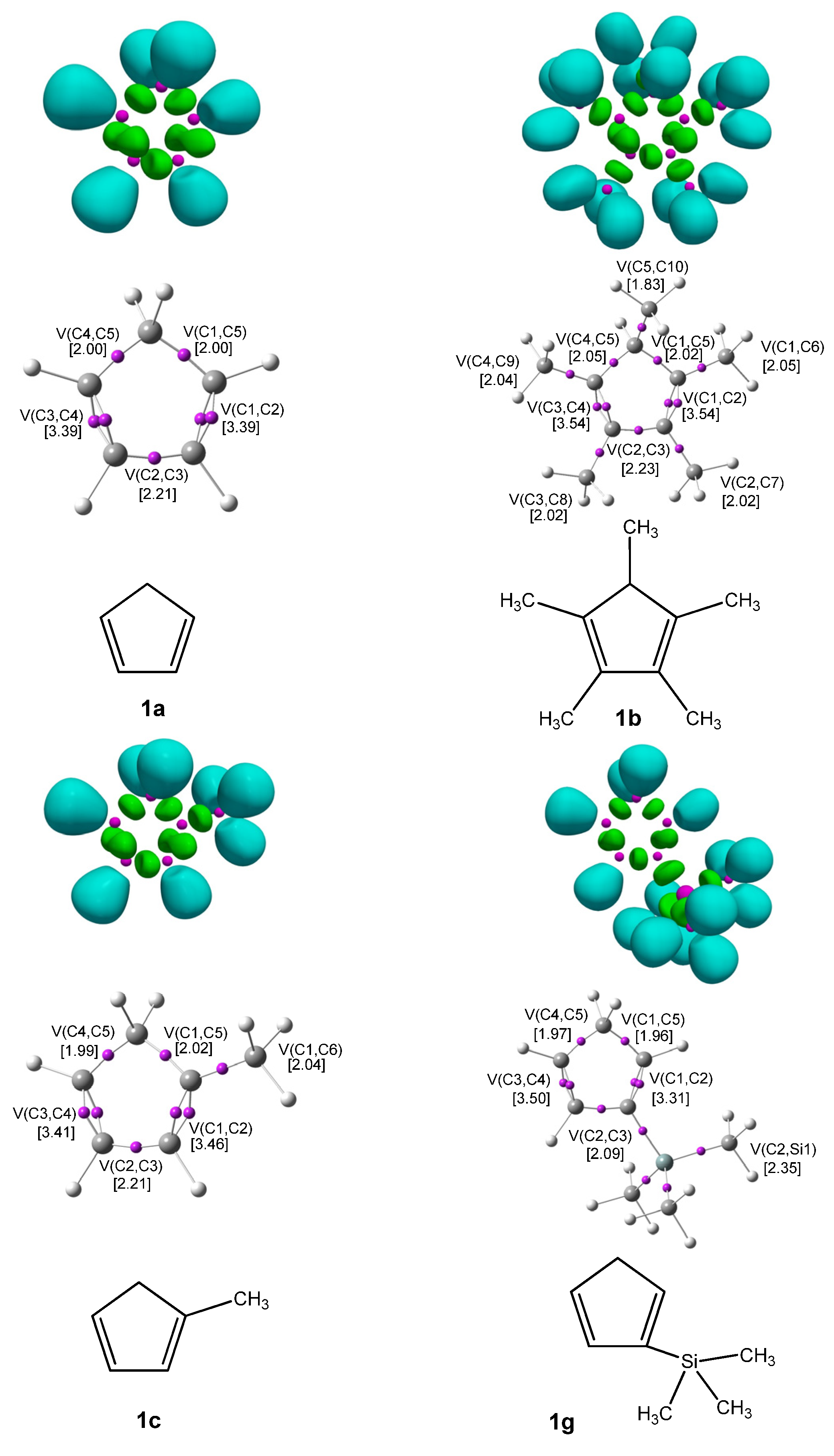
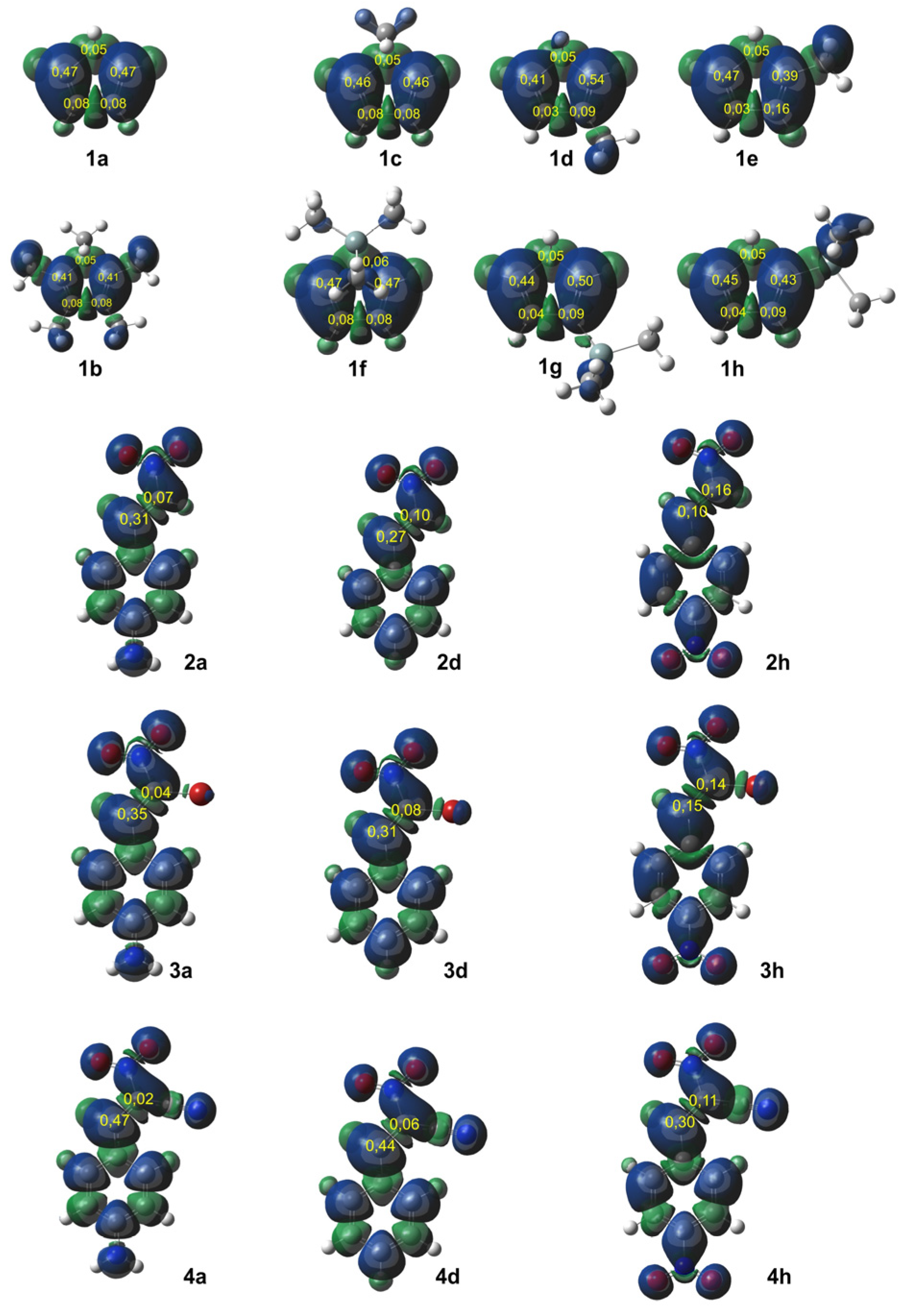
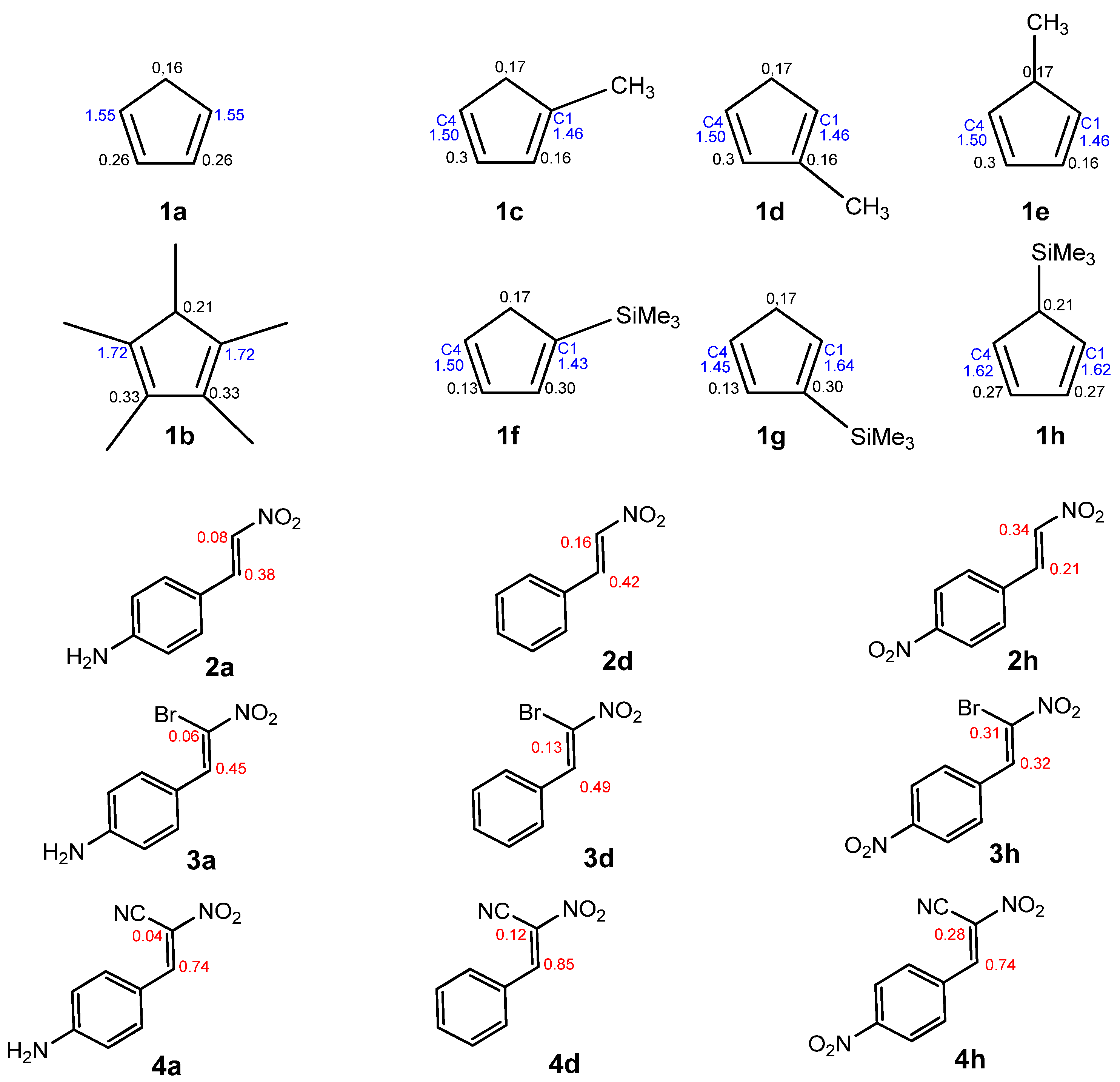
Figure 1 displays the ELF localization domains, the ELF basin attractor positions, and corresponding proposed Lewis-like structures for
Cp 1, while
Figure 2 shows the same data for
CNA. The most relevant valence basin populations are summarized in
Table 1 and
Table 2.
The topological ELF analysis of Cp (1a–f) reveals the presence of two disynaptic basins in the C1–C2 and C3–C4 bonding regions, denoted as V(C–C) and V′(C–C), with a total electron population ranging from 3.24 to 3.54 e, depending on the substituents present on the cyclopentadienyl ring. In symmetric compounds, these values are equal. These results suggest a double bond character between the C1–C2 and C3–C4 carbon atoms. The remaining bonds in the ring correspond to single V(C–C) bonds with populations ranging from 1.99 to 2.24 e. The C–R bonding region (where the substituent is attached) is characterized by the presence of V(C, R) disynaptic basins, with populations ranging from 2.02 to 2.44 e when the substituents are in positions 1 or 2, and slightly lower values of 1.81 to 1.89 e when the substituent is attached to the C5 atom of the ring. These values indicate that the C–R bond exhibits a single bond character.
The ELF topological analysis of the most relevant fragment in the
CNA (
Figure 2) reveals the presence of two pairs of disynaptic basins, V(C1,C2) and V′(C1,C2), with a combined electron population of approximately 3.5 e, depending on the substituent R in the phenyl ring (
Table 2). This indicates a double bond character between C1 and C2. The nitro group is bonded to C1 through a single bond, with a total electron population of 2.31–2.32 e for compounds
2b–
h, and a slightly higher population of 2.58 e for
2a.
Both oxygen atoms in the nitro group are connected to the nitrogen atom via single bonds, with electron populations ranging from 1.84 to 1.89 e. The analysis also shows the presence of two monosynaptic basins, V(O1) and V′(O1), located at the O1 atom, integrating a total electron population of 5.62–5.67 e, and one monosynaptic basin, V(O2), at the O2 atom, with a population of 2.82–2.90 e. Compounds 2b–h exhibit two monosynaptic basins at the N1 atom, V(N1) and V′(N1), with a combined electron population of 0.30–0.32 e. In contrast, compound 2a shows no monosynaptic basin associated with the N1 atom in the ELF analysis.
In
CNA (
3a–
h) (
Table 3), similarly to the previous cases, a double bond is observed between the C1 and C2 atoms. A disynaptic basin V(C1, Br) with an electron population of approximately 1.6e can be observed. On the bromine atom, monosynaptic basins with a total electron population of 6.5e are noted. In the case of conjugated nitroalkenes (
4a–h), the topological analysis of the ELF revealed two disynaptic basins, V(C1,C2) and V′(C1,C2), with a combined electron population of approximately 3.5 e, characteristic of a localized C=C double bond. The nitro group is connected to the carbon framework via a single bond, represented by a disynaptic basin V(C1,N1) with an electron population ranging from 2.25 to 2.30 electrons, indicating a typical single bond character. The presence of disynaptic basins V(N1,O1) and V(N1,O2), each integrating between 1.84 and 2.24 electrons, confirms the single bond nature of the N–O interactions. Each oxygen atom (O1 and O2) exhibits two monosynaptic basins, V(O) and V′(O), with a combined electron population of approximately 5.63 electrons per atom. This electronic distribution reflects the presence of three lone pairs on each oxygen atom, consistent with their high electronegativity and the resonance delocalization characteristic of the nitro group.
Within the case of
CNA (
4a–
h) (
Table 4), the topological analysis of the ELF revealed two disynaptic basins, V(C1,C2) and V′(C1,C2), with a combined electron population of approximately 3.5 e, characteristic of a localized C=C double bond. A disynaptic basin V(C1,C
CN) with an electron population of 2.39 e was also detected between the C1 carbon atom and the nitrile substituent, corresponding to a single C–C bond. Furthermore, two additional disynaptic basins were observed, integrating 4.43 e and 4.54 e, respectively, suggesting a highly polarized double bond, possibly influenced by conjugation EW groups. A monosynaptic basin V(N2), located at the nitrogen atom of the nitro group, was identified with an electron population ranging from 3.16 e to 3.21 e, indicative of significant lone pair localization. The structure of the nitro group is consistent with the characteristics observed in the previous compounds.
The Lewis structures of the investigated compounds were elucidated based on the results of ELF topological analysis (
Figure 2).
2.3. Reactivity of Cp toward CNA
Based on the data gathered so far, we can now examine the reactivity of
Cp toward
CNA in DA reactions. The chemical potential is a useful parameter for determining the direction of electron density flow between the reactants [
28]. The chemical potential of
CNA (
2–
4) ranges from −5.79 to −4.25 eV, while for
Cp (
1a–
h), this value is consistently higher in each case (
Table 5). Based on these values, we can conclude that the electron density will flow from the cyclopentadiene to the conjugated nitroalkenes, indicating a forward electron density flux (FEDF) [
29] from the diene to the dienophile (
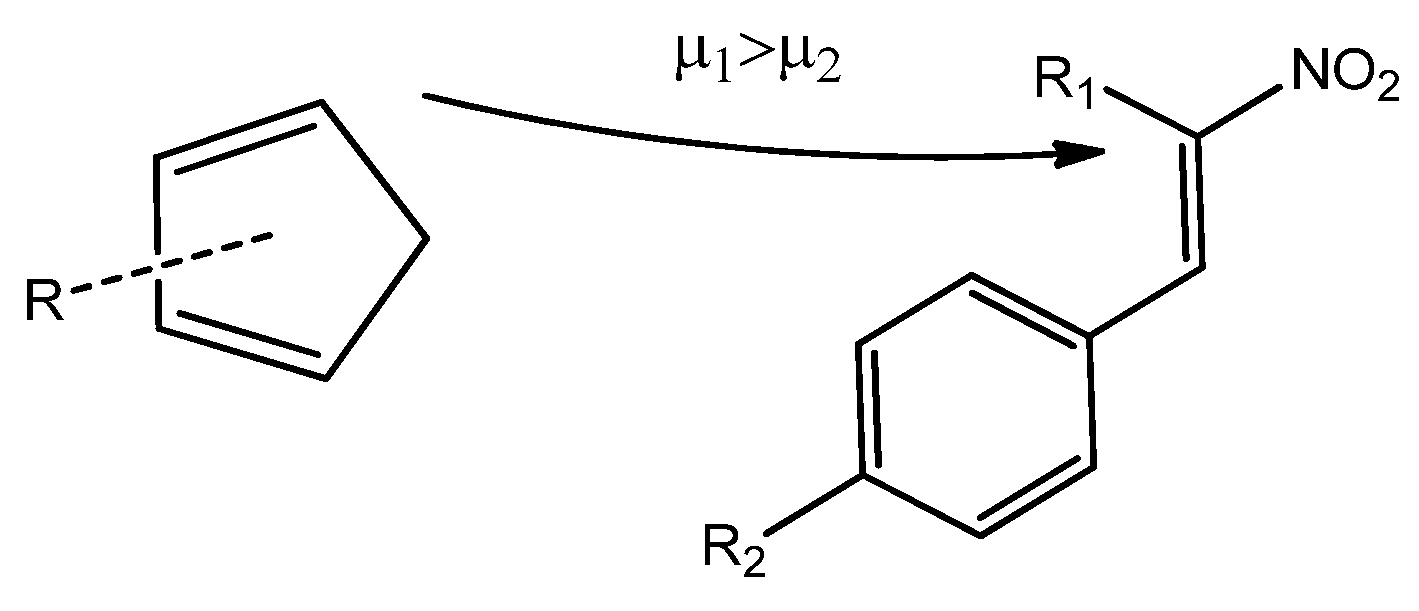
Scheme 5).
Determining which reactant acts as the electrophile and which as the nucleophile—along with identifying the most electrophilic and nucleophilic centers—is essential in reactions that can yield multiple isomeric products [
30,
31]. In addition, a thorough analysis of the local electronic characteristics of the atoms involved in bond formation is necessary to theoretically predict the most favorable reaction pathway during the formation of the new ring system [
32].
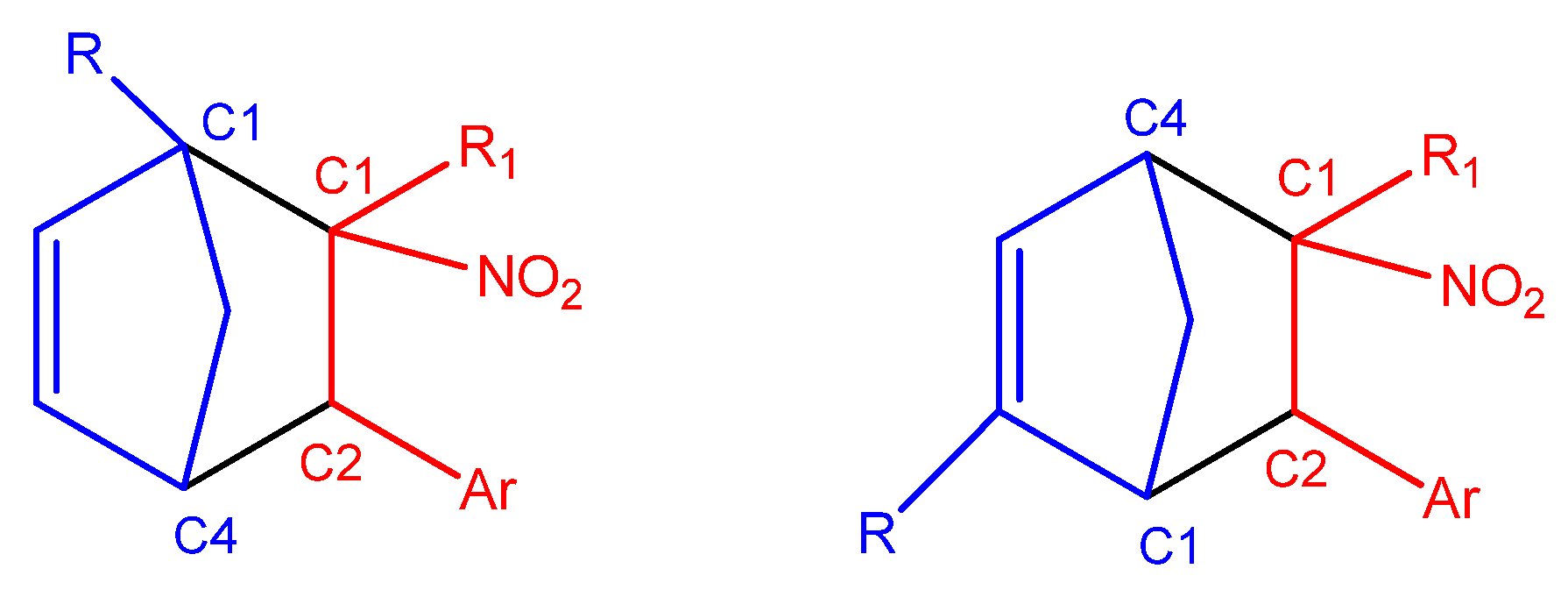
Due to the higher local electrophilicity of the β-carbon atom (C2, adjacent to the phenyl group in CNA), certain reaction pathways of CNA (2a–g, 3a–g, 4a–g) with unsymmetrical Cp are expected to be favored. However, in the case of conjugated nitroalkenes substituted with a nitro group in a phenyl ring (2–4h), the reaction selectivity shifts, as the C1 carbon atom becomes the most electrophilic center in these derivatives.
In the case of asymmetrically substituted
Cp, the nucleophilicity of the carbon atoms is strongly influenced by the position of the substituent. For 1-substituted cyclopentadienyl derivatives, the more nucleophilic carbon C4 preferentially forms a bond with the more electrophilic β-carbon (C2) of the conjugated alkenes (
2a–
g,
3a–
g,
4a–
g) during the DA reaction. In 2-substituted systems, the C1 carbon exhibits the highest nucleophilicity and is, therefore, most likely to engage in bond formation with the β-carbon (C2) of the nitroalkene. For
CNA 2–4h, the presence of EW substituents at the α-position may lead to the formation of alternative regioisomeric products (
Scheme 6).
By analyzing the difference in global electrophilicity (Δω) between the components involved in the cycloaddition reaction, it is possible to assess the polar character of the process. Cycloadditions between strong electrophiles and strong nucleophiles are expected to proceed via a polar mechanism when Δω > 1 eV. In contrast, when Δω < 1 eV, the reaction is predicted to follow a non-polar pathway.
A comparative analysis of the global electrophilicity indices of cyclopentadienes and conjugated nitroalkenes reveals that the presence of ER substituents on the phenyl ring of conjugated nitroalkenes favors a polar DA reaction. Moreover, for cyano-substituted conjugated nitroalkenes, the reaction is polar regardless of the nature of the substituent.
The global electrophilicity differences (Δω) for all investigated DA reactions were computed and are presented in the
Supplementary Materials.
To better understand the role of substituents in modulating the polarity and reactivity of the studied Diels–Alder reactions, we performed a classical Hammett-type correlation analysis. While global and local DFT-based reactivity descriptors provide mechanistic insight into the electronic properties of the reacting species, they do not directly quantify the influence of individual substituents in a chemically intuitive way. In contrast, Hammett σ constants are well-established empirical parameters that capture the electron-donating or electron-withdrawing nature of substituents through resonance and inductive effects. While not essential to understanding the mechanism per se, this correlation provides a valuable bridge between classical physical organic chemistry and modern theoretical approaches. By correlating the calculated reaction polarity index (Δω) with the Hammett parameters (σ⁺, σₚ, σᴿ, σᴵ) [
16], we aimed to establish a quantitative model linking substituent electronic properties to the global polarity of the reaction (
Figure 4). Among the tested constants, σ
p showed the strongest linear correlation (R = 0.991 for the
4+Cp reaction, R = 0.979 for compound 2 with
Cp, and R = 0.970 for compound 3 with
Cp (
Table 9)), suggesting that both inductive and resonance effects are transmitted from the substituents to the reaction center and influence the reaction’s polar character.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}