
Organophotoredox-Catalyzed Stereoselective Synthesis of Bicyclo[3.2.0]heptanes via [2+2] Photocycloaddition

 , ,
, ,  and
and 
Abstract

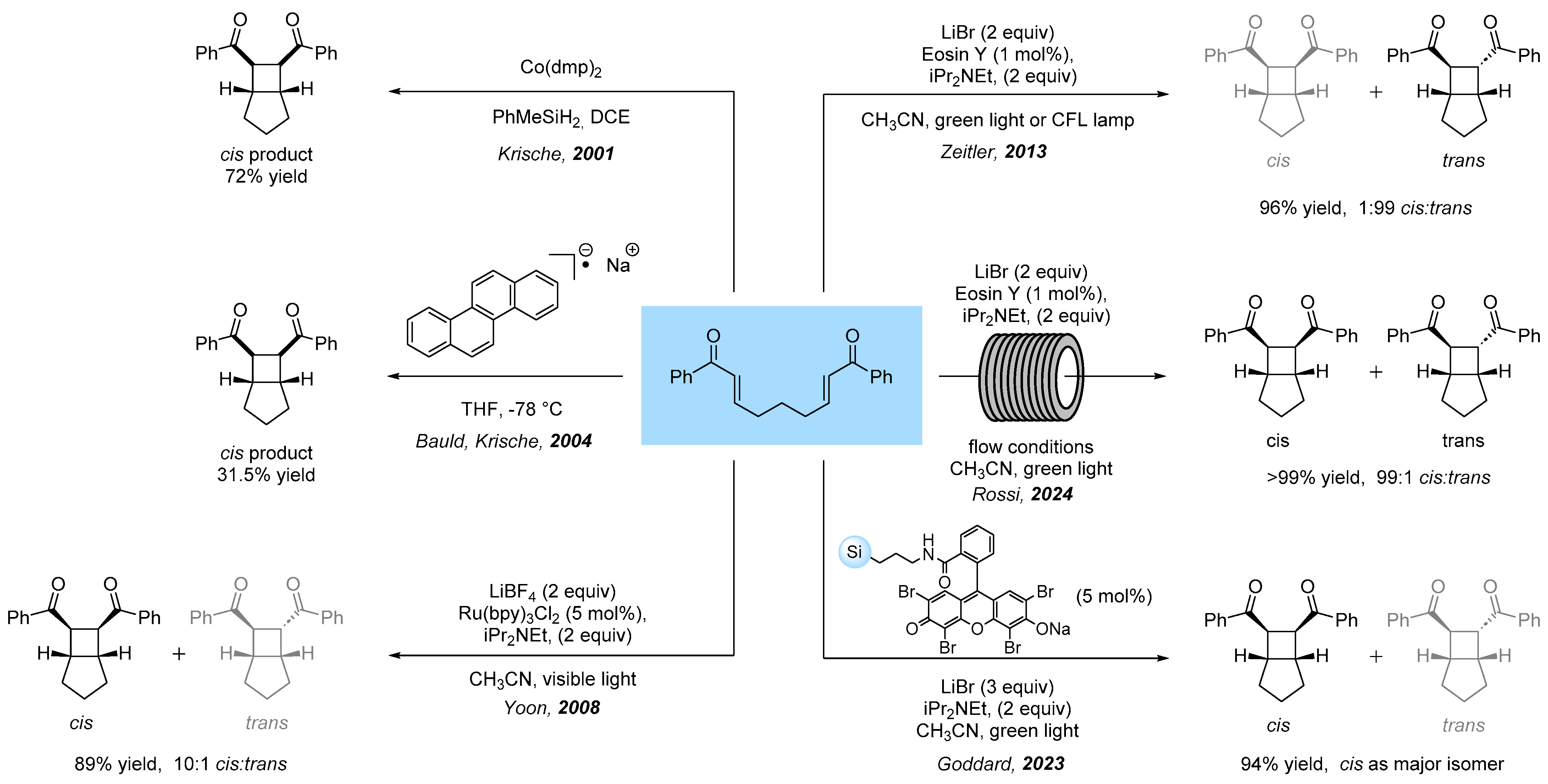
1. Introduction
2. Results
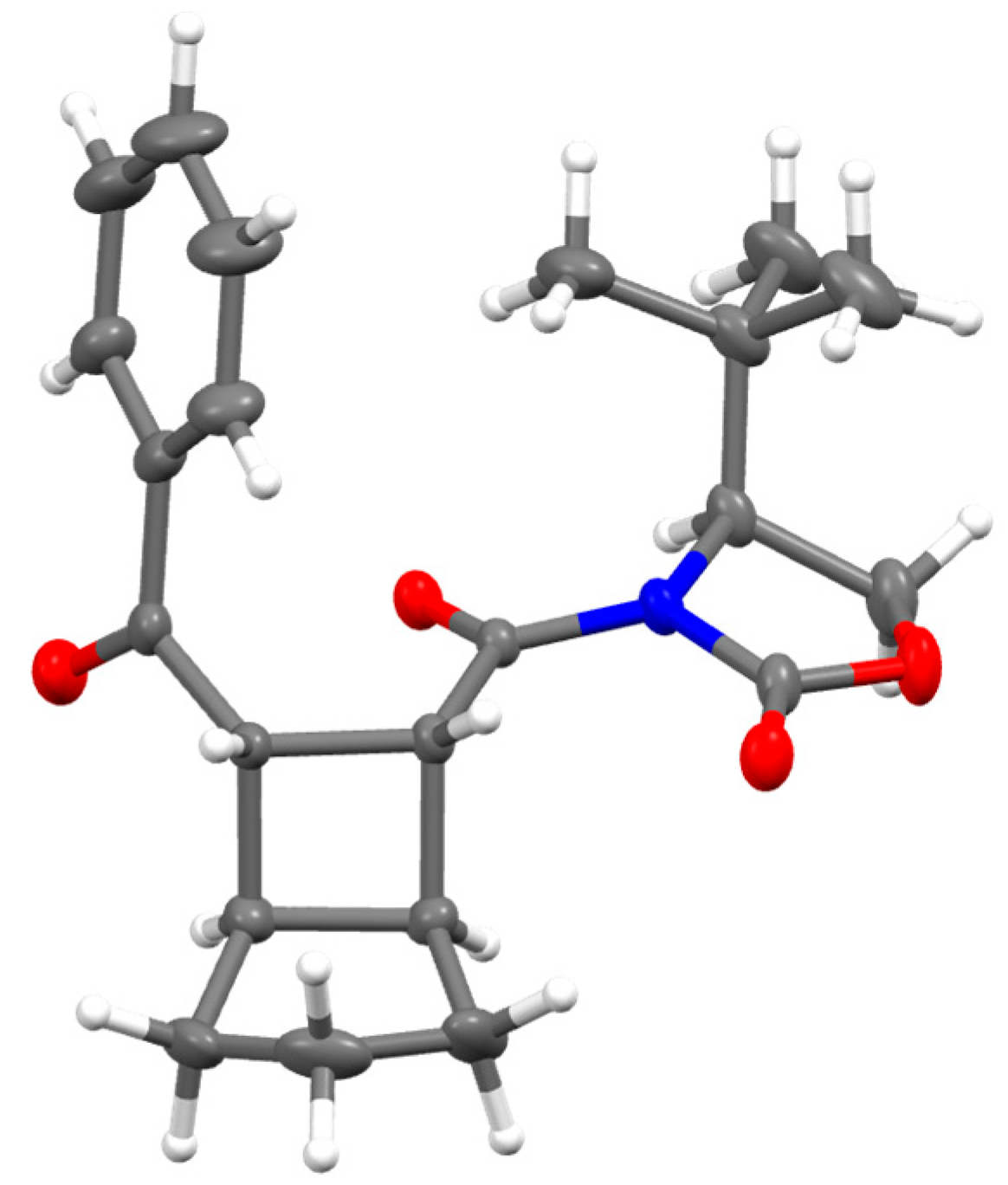
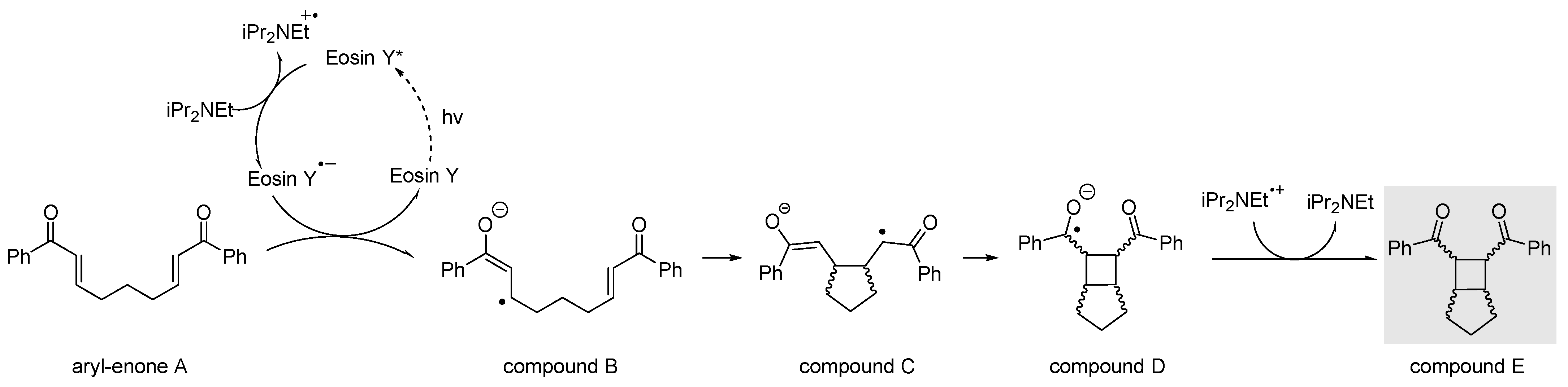
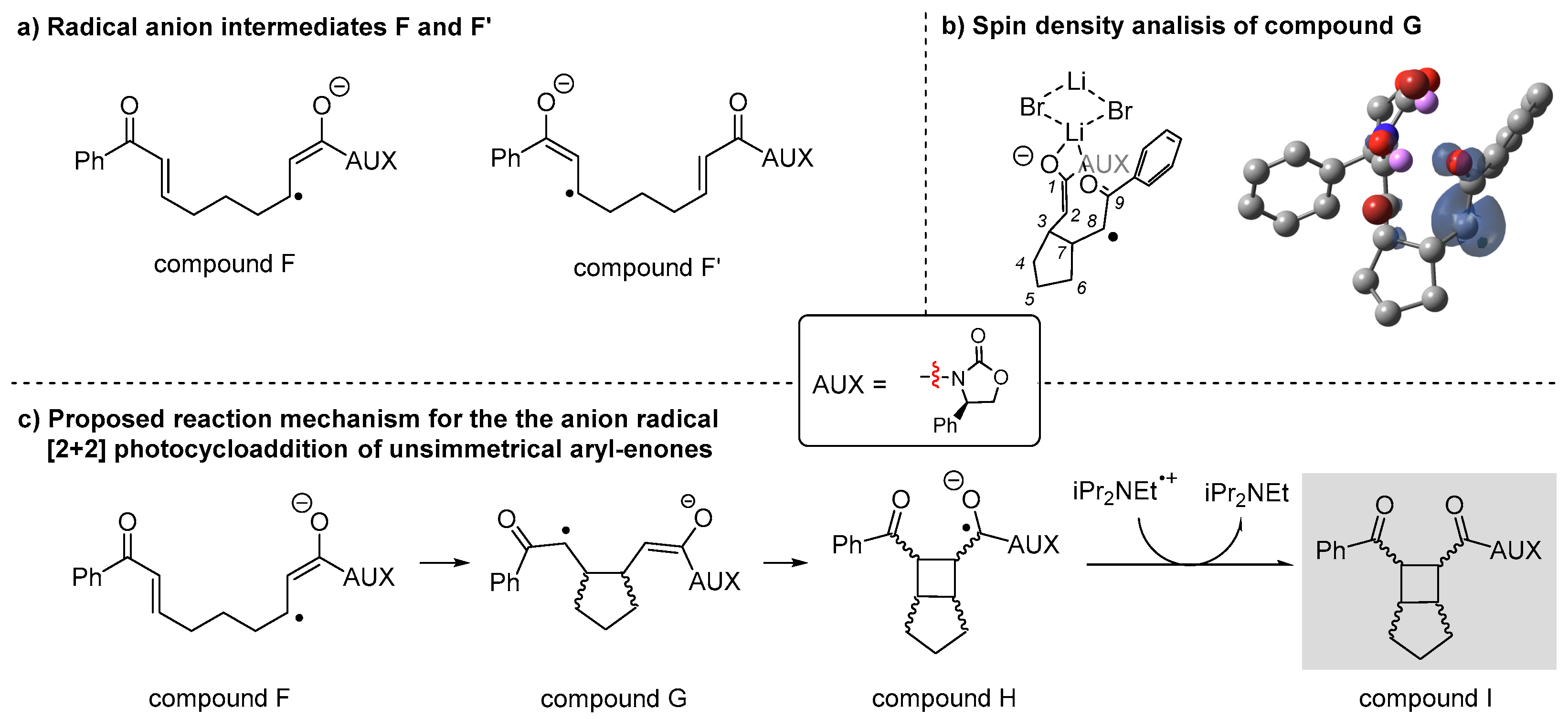
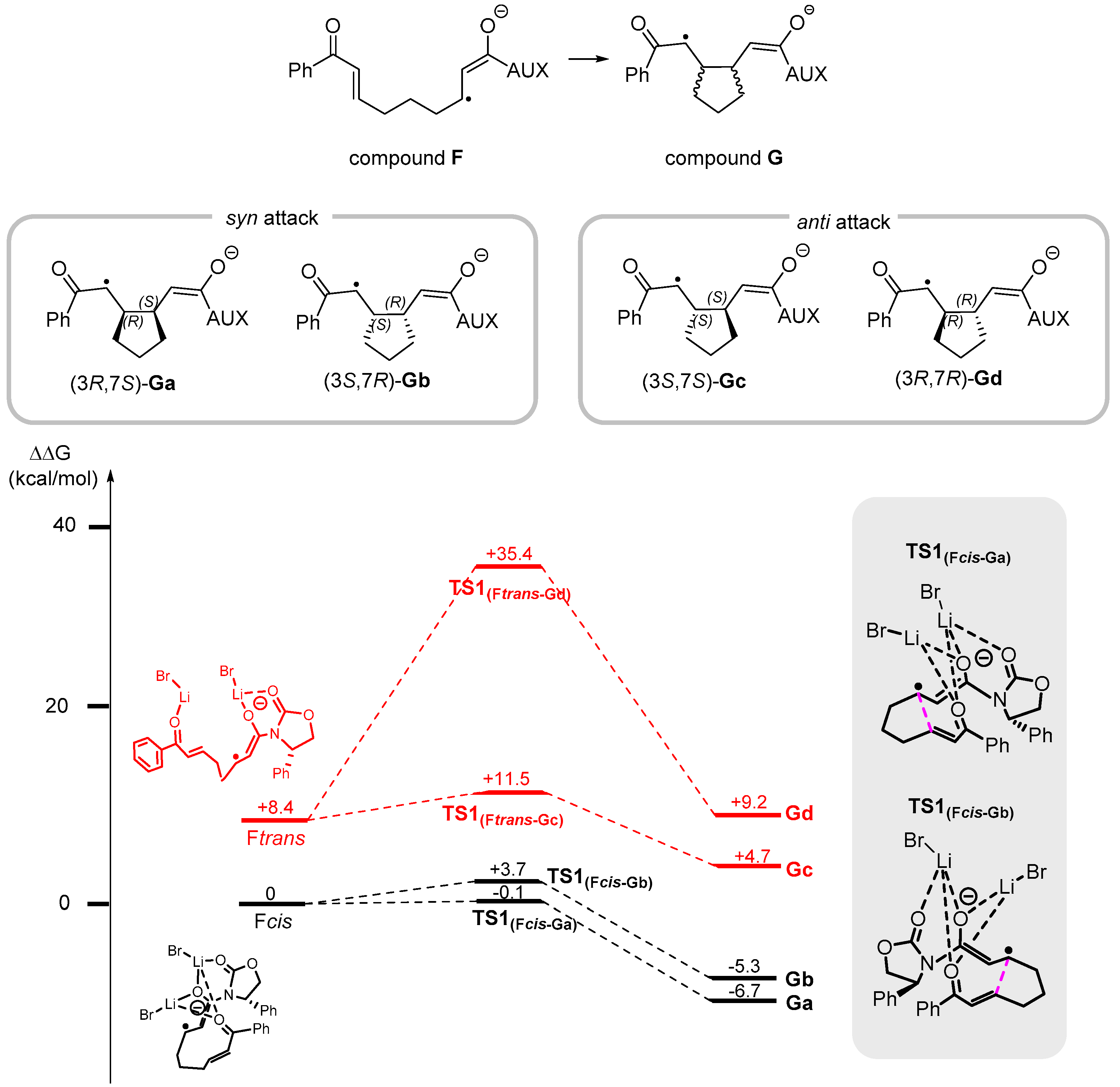
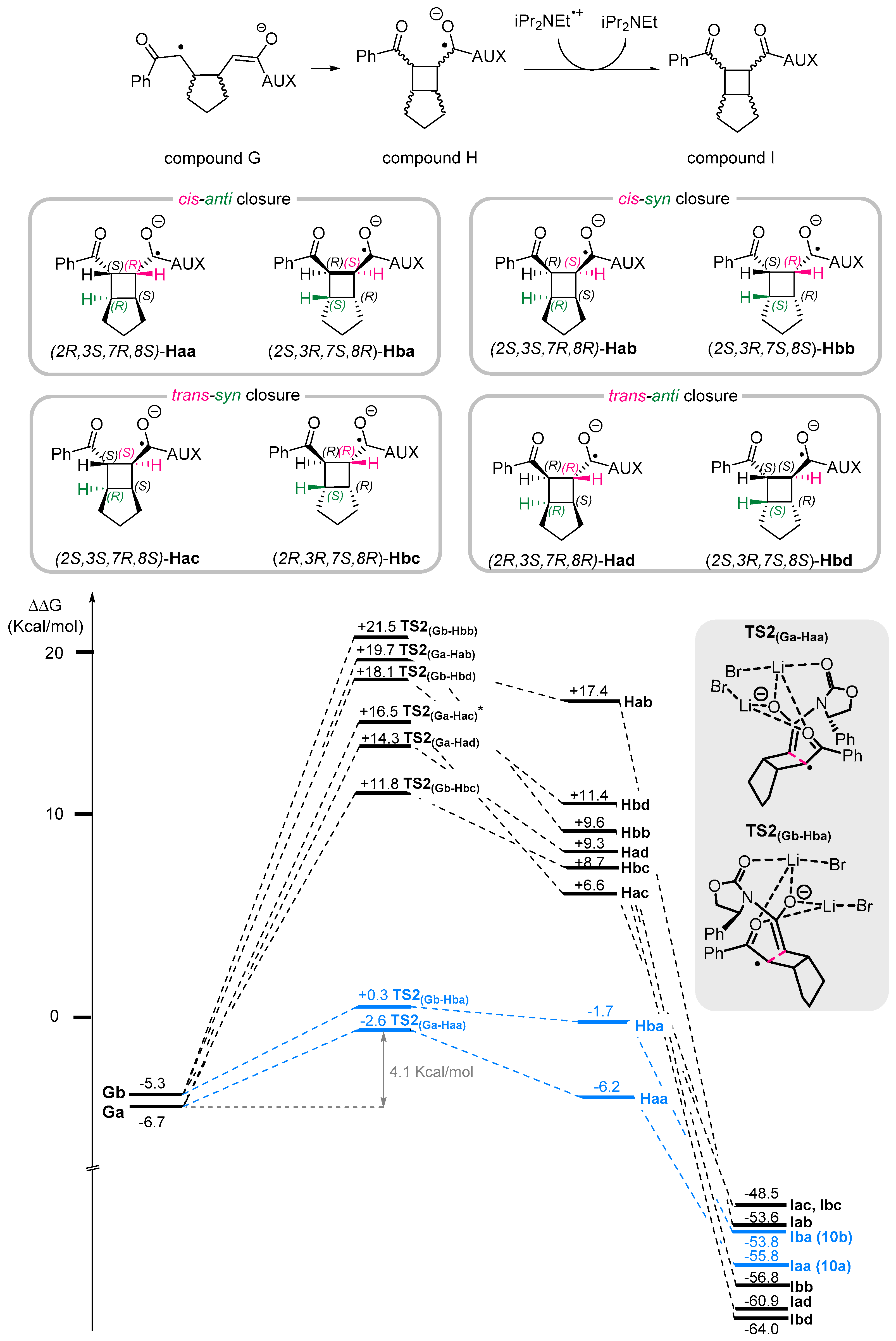
3. Reaction Mechanism
4. Conclusions
5. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References and Note
- Skubi, K.L.; Blum, T.R.; Yoon, T.P. Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev. 2016, 116, 10035–10074. [Google Scholar] [CrossRef] [PubMed]
- Candish, L.; Collins, K.D.; Cook, G.C.; Douglas, J.J.; Gomez-Suarez, A.; Jolit, A.; Keess, S. Photocatalysis in the Life Science Industry. Chem. Rev. 2022, 122, 2907–2980. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef]
- Resta, S.; Benettin, T.; Puglisi, A.; Raimondi, L.; Rossi, S. Immobilized eosin Y as heterogeneous photoredox catalyst in light-driven stereoselective aryl-enones cyclization. Tetrahedron Lett. 2024, 138, 154976. [Google Scholar] [CrossRef]
- Medici, F.; Resta, S.; Presenti, P.; Caruso, L.; Puglisi, A.; Raimondi, L.; Rossi, S.; Benaglia, M. Stereoselective Visible-Light Catalyzed Cyclization of Bis(enones): A Viable Approach to the Synthesis of Enantiomerically Enriched Cyclopentane Rings. Eur. J. Org. Chem. 2021, 2021, 4521–4524. [Google Scholar] [CrossRef]
- Baik, T.G.; Luis, A.L.; Wang, L.C.; Krische, M.J. A diastereoselective metal-catalyzed [2+2] cycloaddition of bis-enomes. J. Am. Chem. Soc. 2001, 123, 6716–6717. [Google Scholar] [CrossRef]
- Yang, J.; Felton, G.A.N.; Bauld, N.L.; Krische, M.J. Chemically Induced Anion Radical Cycloadditions: Intramolecular Cyclobutanation of Bis(enones) via Homogeneous Electron Transfer. J. Am. Chem. Soc. 2004, 126, 1634–1635. [Google Scholar] [CrossRef]
- Ischay, M.A.; Anzovino, M.E.; Du, J.; Yoon, T.P. Efficient visible light photocatalysis of [2+2] enone cycloadditions. J. Am. Chem. Soc. 2008, 130, 12886–12887. [Google Scholar] [CrossRef]
- Du, J.; Yoon, T.P. Crossed intermolecular [2+2] cycloadditions of acyclic enones via visible light photocatalysis. J. Am. Chem. Soc. 2009, 131, 14604–14605. [Google Scholar] [CrossRef]
- Du, J.; Espelt, L.R.; Guzei, I.A.; Yoon, T.P. Photocatalytic Reductive Cyclizations of Enones: Divergent Reactivity of Photogenerated Radical and Radical Anion Intermediates. Chem. Sci. 2011, 2, 2115–2119. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Zeitler, K. A cooperative hydrogen-bond-promoted organophotoredox catalysis strategy for highly diastereoselective, reductive enone cyclization. Chemistry 2013, 19, 6950–6955. [Google Scholar] [CrossRef] [PubMed]
- Benettin, T.; Resta, S.; Puglisi, A.; Rossi, S. Synthesis of Bicyclo[3.2.0]heptanes by Organophotoredox Catalytic Diastereoselective Anion Radical [2+2] Photocycloadditions of Arylenones under Batch and Flow Conditions. Eur. J. Org. Chem. 2024, 27, e202400816. [Google Scholar] [CrossRef]
- Mahmoud, N.; Awassa, J.; Toufaily, J.; Lebeau, B.; Daou, T.J.; Cormier, M.; Goddard, J.P. Heterogeneous Photoredox Catalysis Based on Silica Mesoporous Material and Eosin Y: Impact of Material Support on Selectivity of Radical Cyclization. Molecules 2023, 28, 549. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T.P. Photochemical Stereocontrol Using Tandem Photoredox-Chiral Lewis Acid Catalysis. Acc. Chem. Res. 2016, 49, 2307–2315. [Google Scholar] [CrossRef]
- Yoon, T.P. Stereocontrolled Photochemical Synthesis. ChemPhotoChem 2019, 3, 1201–1202. [Google Scholar] [CrossRef]
- Chen, L.Y.; Huang, P.Q. Evans’ Chiral Auxiliary-Based Asymmetric Synthetic Methodology and Its Modern Extensions. Eur. J. Org. Chem. 2023, 27, e202301131. [Google Scholar] [CrossRef]
- Felton, G.A.N.; Bauld, N.L. Dramatic effects of the electrolyte cation on the selectivity of electroreductive cycloaddition reactions of bis(enones). Tetrahedron Lett. 2004, 45, 8465–8469. [Google Scholar] [CrossRef]
- Zhang, X.N.; Shi, M. A Highly Nucleophilic Multifunctional Chiral Phosphane-Catalyzed Asymmetric Intramolecular Rauhut–Currier Reaction. Eur. J. Org. Chem. 2012, 2012, 6271–6279. [Google Scholar] [CrossRef]
- Bosveli, A.; Montagnon, T.; Kalaitzakis, D.; Vassilikogiannakis, G. Eosin: A versatile organic dye whose synthetic uses keep expanding. Org. Biomol. Chem. 2021, 19, 3303–3317. [Google Scholar] [CrossRef]
- For improved readability, the first descriptor (cis or trans) indicates the relative position of the hydrogen atoms on carbons 8 and 2, while the second descriptor (syn or anti) specifies the relative position of the hydrogen atoms on carbons 8 and 7.
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Schrödinger Release, 2024–1: MacroModel, Schrödinger, LLC, New York 2024. Available online: https://www.schrodinger.com/platform/products/macromodel/ (accessed on 30 April 2025).
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2007, 120, 215–241. [Google Scholar]
- Frisch, M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 16, Revision C.02; Gaussian: Wallingford CT, USA, 2019; Available online: https://gaussian.com/gaussian16/ (accessed on 30 April 2025).
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Mennucci, B. Polarizable continuum model. WIREs Comput. Mol. Sci. 2012, 2, 386–404. [Google Scholar] [CrossRef]
- Bursch, M.; Mewes, J.M.; Hansen, A.; Grimme, S. Best-Practice DFT Protocols for Basic Molecular Computational Chemistry. Angew. Chem. Int. Ed. Engl. 2022, 61, e202205735. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
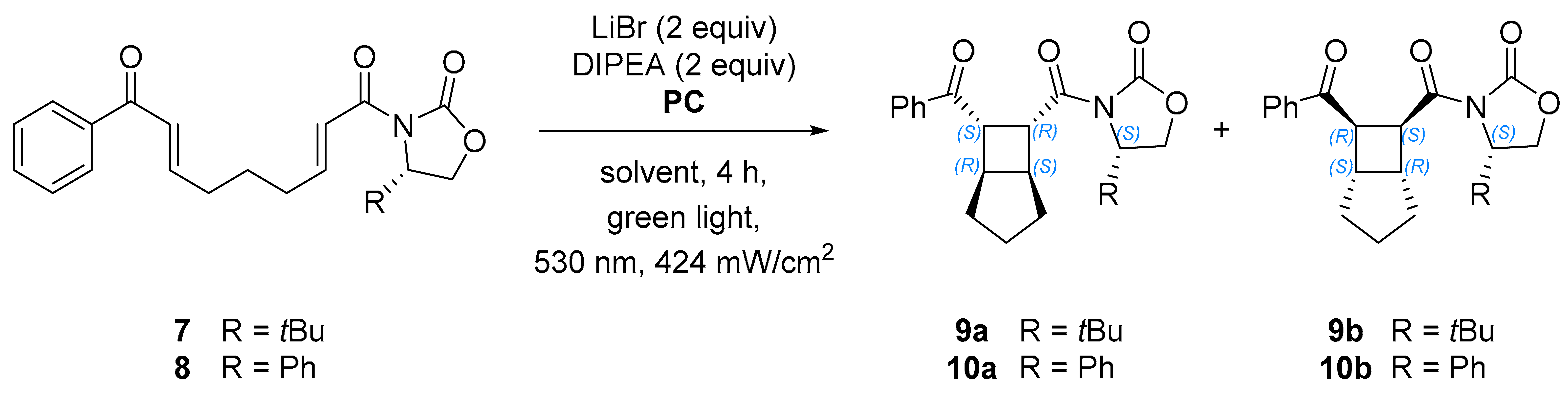 | |||||||
| Entry | Substrate | Solvent | PC | PC Loading (mol%) | Product | a:b Ratio | Yield (%) |
| 1 | 7 | CH3OH | Eosin Y | 1 | 9 | - | - |
| 2 | 7 | CH2Cl2 | Eosin Y | 1 | 9 | - | - |
| 3 | 7 | DMF | Eosin Y | 1 | 9 | 58:42 | 35 |
| 4 | 7 | THF | Eosin Y | 1 | 9 | 53:47 | 45 |
| 5 | 7 | CH3CN | Eosin Y | 1 | 9 | 60:40 | 76 |
| 6 | 7 | CH3CN | Eosin Y | 0.5 | 9 | 62:38 | 62 |
| 7 | 7 | CH3CN | Na2Eosin Y | 0.5 | 9 | 57:43 | 23 |
| 8 | 8 | CH3CN | Eosin Y | 0.5 | 10 | 74:26 | 43 |
| 9 | 8 | CH3CN | Eosin Y | 1 | 10 | 78:22 | 57 |
| 10 a | 8 | CH3CN | Eosin Y | 1 | 10 | 77:23 | 31 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benettin, T.; Resta, S.; Forni, A.; Raimondi, L.; Puglisi, A.; Rossi, S. Organophotoredox-Catalyzed Stereoselective Synthesis of Bicyclo[3.2.0]heptanes via [2+2] Photocycloaddition. Molecules 2025, 30, 2090. https://doi.org/10.3390/molecules30102090
Benettin T, Resta S, Forni A, Raimondi L, Puglisi A, Rossi S. Organophotoredox-Catalyzed Stereoselective Synthesis of Bicyclo[3.2.0]heptanes via [2+2] Photocycloaddition. Molecules. 2025; 30(10):2090. https://doi.org/10.3390/molecules30102090
Chicago/Turabian StyleBenettin, Tommaso, Simonetta Resta, Alessandra Forni, Laura Raimondi, Alessandra Puglisi, and Sergio Rossi. 2025. "Organophotoredox-Catalyzed Stereoselective Synthesis of Bicyclo[3.2.0]heptanes via [2+2] Photocycloaddition" Molecules 30, no. 10: 2090. https://doi.org/10.3390/molecules30102090
APA StyleBenettin, T., Resta, S., Forni, A., Raimondi, L., Puglisi, A., & Rossi, S. (2025). Organophotoredox-Catalyzed Stereoselective Synthesis of Bicyclo[3.2.0]heptanes via [2+2] Photocycloaddition. Molecules, 30(10), 2090. https://doi.org/10.3390/molecules30102090