

A PI3K Inhibitor with Low Cardiotoxicity and Its Synergistic Inhibitory Effect with Gilteritinib in Acute Myelogenous Leukemia (AML) Cells

 and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
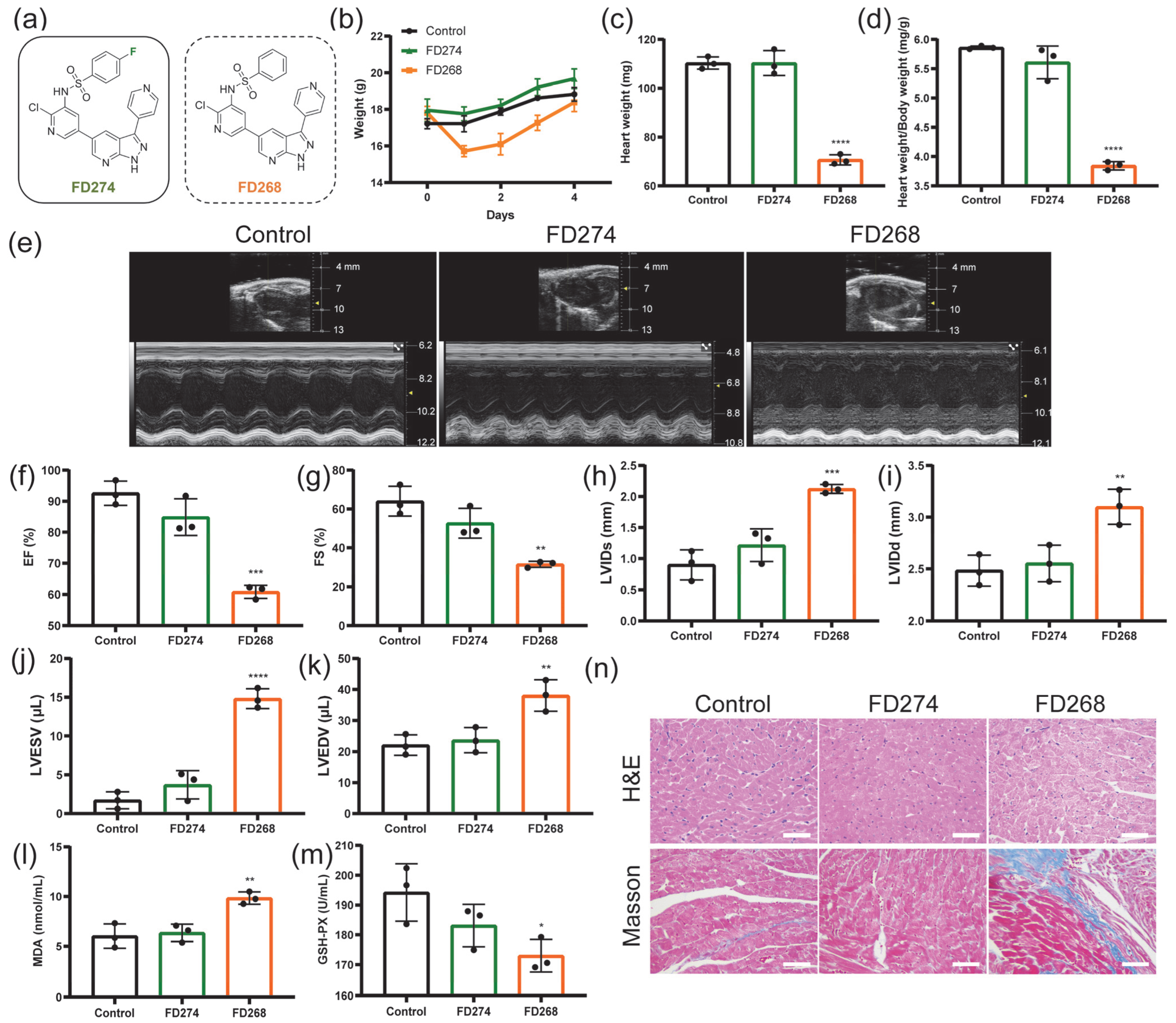
2.1. The Cardiotoxicity of FD274 and FD268 in C57BL/6 Mouse Model
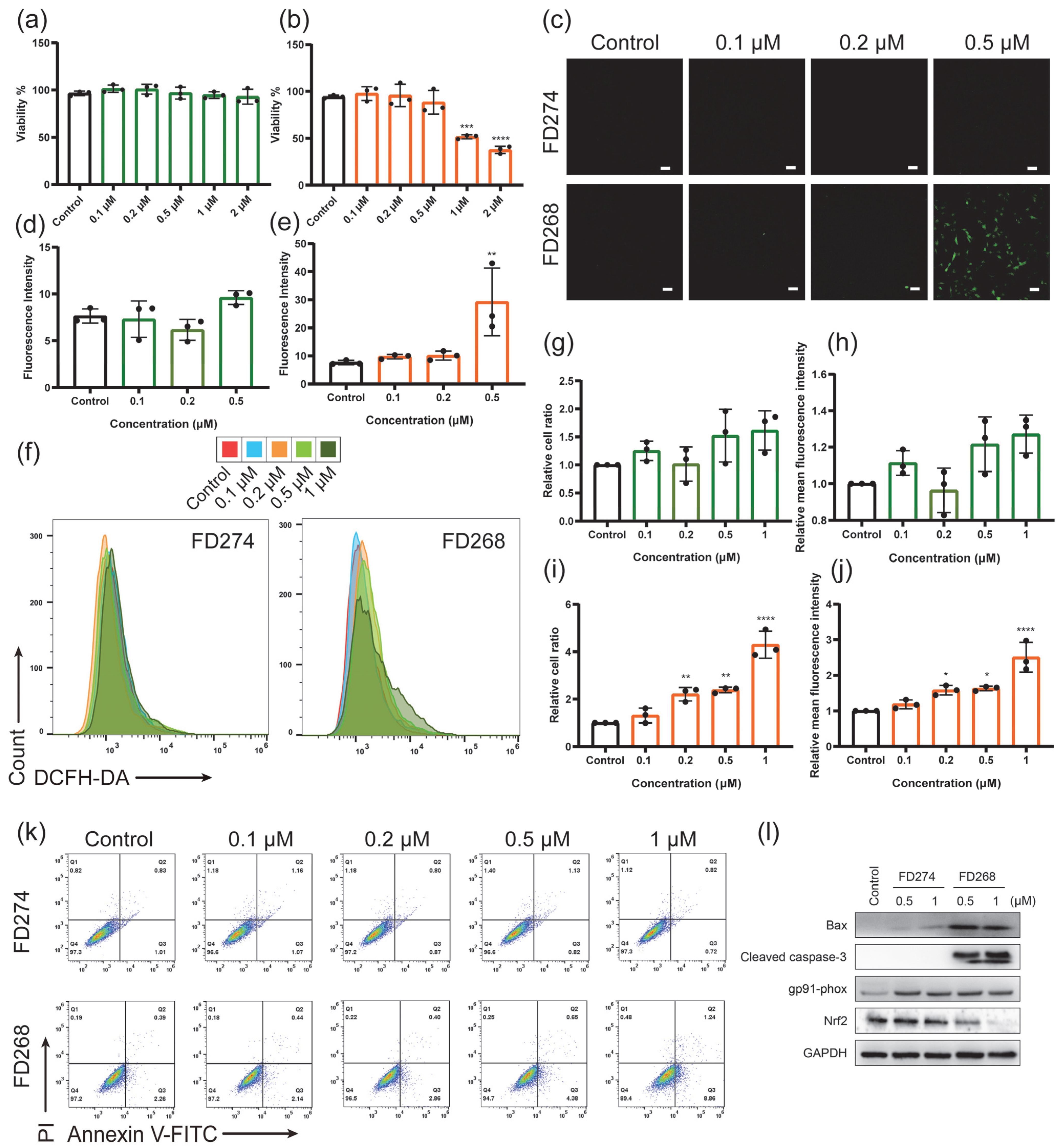
2.2. The Cytotoxic Effects of FD274 and FD268 in H9C2 Cell Line
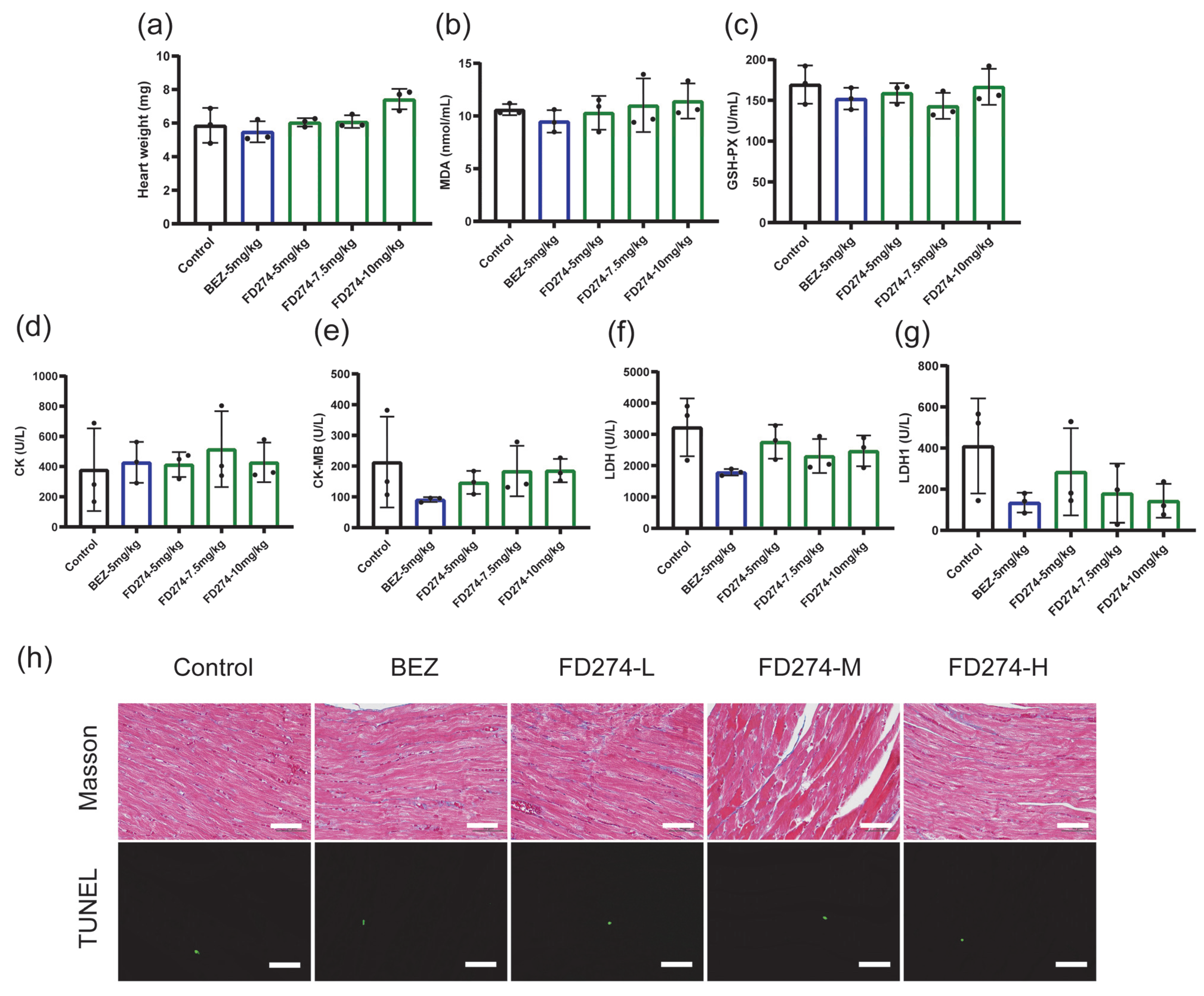
2.3. The Cardiotoxicity of FD274 in the HL-60 Xenograft Model After the Consecutive 20-Day Treatment Process
2.4. The Synergistic Inhibitory Effects of FD274 with Gilteritinib in HL-60 and MV-4-11 Cell Lines
3. Discussion
4. Materials and Methods
4.1. General
4.2. Drug Administration
4.3. Echocardiography
4.4. Serum Biochemical Analysis
4.5. Histopathological Analysis
4.6. Cell Viability Analysis
4.7. Determination of Intracellular ROS Level
4.8. Determination of Apoptosis Level
4.9. Western Blot
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | acute myelogenous leukemia |
| PI3K | Phosphoinositide-3-kinases |
| ROS | reactive oxygen species |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate Hydrogen |
| FLT3 | Fms-like tyrosine kinase 3 |
| ITD | internal tandem duplication |
| MDA | malondialdehyde |
| GSH-PX | glutathione peroxidase |
| LVIDs | left ventricular internal dimension systole |
| LVIDd | left ventricular internal dimension diastole |
| LVESV | left ventricular end-systolic volume |
| LVEDV | left ventricular end-diastolic volume |
| EF | ejection fraction |
| FS | fractional shortening |
| TUNEL | terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling |
| MFI | mean fluorescence intensity |
| DCFH-DA | 2′,7′-Dichlorofluorescein diacetate |
| FCM | flow cytometry |
| CLSM | confocal laser scanning microscopy |
| H&E | hematoxylin–eosin |
| CK | Creatine Kinase |
| CK-MB | Creatine Kinase, MB form |
| LDH | lactate dehydrogenase |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| CI | combination index |
References
- MacDonald, J.S.; Robertson, R.T. Toxicity Testing in the 21st Century: A View from the Pharmaceutical Industry. Toxicol. Sci. 2009, 110, 40–46. [Google Scholar] [CrossRef]
- Ziegler, R.; Häusermann, F.; Kirchner, S.; Polonchuk, L. Cardiac Safety of Kinase Inhibitors—Improving Understanding and Prediction of Liabilities in Drug Discovery Using Human Stem Cell-Derived Models. Front. Cardiovasc. Med. 2021, 8, 639824. [Google Scholar] [CrossRef] [PubMed]
- Sadasivan, C.; Zhabyeyev, P.; Labib, D.; White, J.A.; Paterson, D.I.; Oudit, G.Y. Cardiovascular toxicity of PI3Kα inhibitors. Clin. Sci. 2020, 134, 2595–2622. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu. Rev. Med. 2016, 67, 11–28. [Google Scholar] [CrossRef]
- Curigliano, G.; Shah, R.R. Safety and Tolerability of Phosphatidylinositol-3-Kinase (PI3K) Inhibitors in Oncology. Drug Safety 2019, 42, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Meoli, D.F.; Moslehi, J.; Roden, D.M. Inhibition of the α-Subunit of Phosphoinositide 3-Kinase in Heart Increases Late Sodium Current and Is Arrhythmogenic. J. Pharmacol. Exp. Ther. 2018, 365, 460–466. [Google Scholar] [CrossRef]
- Kong, C.Y.; Guo, Z.; Song, P.; Zhang, X.; Yuan, Y.P.; Teng, T.; Yan, L.; Tang, Q.Z. Underlying the Mechanisms of Doxorubicin-Induced Acute Cardiotoxicity: Oxidative Stress and Cell Death. Int. J. Biol. Sci. 2022, 18, 760–770. [Google Scholar] [CrossRef]
- Bouitbir, J.; Panajatovic, M.V.; Kraehenbuehl, S. Mitochondrial Toxicity Associated with Imatinib and Sorafenib in Isolated Rat Heart Fibers and the Cardiomyoblast H9c2 Cell Line. Int. J. Mol. Sci. 2022, 23, 2282. [Google Scholar] [CrossRef]
- Angsutararux, P.; Luanpitpong, S.; Issaragrisil, S. Chemotherapy-Induced Cardiotoxicity: Overview of the Roles of Oxidative Stress. Oxid. Med. Cell. Longev. 2015, 2015, 795602. [Google Scholar] [CrossRef]
- Schramm, A.; Matusik, P.; Osmenda, G.; Guzik, T.J. Targeting NADPH oxidases in vascular pharmacology. Vasc. Pharmacol. 2012, 56, 216–231. [Google Scholar] [CrossRef]
- Xu, Y.X.; Wang, W.T.; Jin, K.K.; Zhu, Q.F.; Lin, H.Z.; Xie, M.Y.; Wang, D.X. Perillyl alcohol protects human renal tubular epithelial cells from hypoxia/reoxygenation injury via inhibition of ROS, endoplasmic reticulum stress and activation of PI3K/Akt/eNOS pathway. Biomed. Pharmacother. 2017, 95, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Xu, C.; Liu, J.; Li, Y.; Wang, H.; Shan, D.; Wainer, I.W.; Hu, X.; Zhang, Y.; Woo, A.Y.; et al. Heterodimerization with 5-HT2BR Is Indispensable for β2AR-Mediated Cardioprotection. Circ. Res. 2021, 128, 262–277. [Google Scholar] [CrossRef]
- Ye, Y.C.; Cai, Y.Y.; Xia, E.J.; Shi, K.J.; Jin, Z.S.; Chen, H.F.; Xia, F.F.; Xia, Y.; Papadimos, T.J.; Xu, X.Z.; et al. Apelin-13 Reverses Bupivacaine-Induced Cardiotoxicity via the Adenosine Monophosphate-Activated Protein Kinase Pathway. Anesth. Analg. 2021, 133, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Kovács, I.A.; Barabási, A.-L. Network-based prediction of drug combinations. Nat. Commun. 2019, 10, 1197. [Google Scholar] [CrossRef]
- Zheng, S.; Wang, W.; Aldahdooh, J.; Malyutina, A.; Shadbahr, T.; Tanoli, Z.; Pessia, A.; Tang, J. SynergyFinder Plus: Toward Better Interpretation and Annotation of Drug Combination Screening Datasets. Genom. Proteom. Bioinf. 2022, 20, 587–596. [Google Scholar] [CrossRef]
- Pratz, K.W.; Levis, M. How I treat FLT3-mutated AML. Blood 2017, 129, 565–571. [Google Scholar] [CrossRef]
- Darici, S.; Zavatti, M.; Braglia, L.; Accordi, B.; Serafin, V.; Horne, G.A.; Manzoli, L.; Palumbo, C.; Huang, X.; Jørgensen, H.G.; et al. Synergistic cytotoxicity of dual PI3K/mTOR and FLT3 inhibition in FLT3-ITD AML cells. Adv. Biol. Regul. 2021, 82, 100830. [Google Scholar] [CrossRef]
- Zhao, J.; Song, Y.; Liu, D. Gilteritinib: A novel FLT3 inhibitor for acute myeloid leukemia. Biomark. Res. 2019, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.D.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia. Cancer Discov. 2019, 9, 1050–1063. [Google Scholar] [CrossRef]
- Yang, C.B.; Zhang, X.; Wang, Y.; Yang, Y.T.; Liu, X.F.; Deng, M.L.; Jia, Y.; Ling, Y.; Meng, L.H.; Zhou, Y.M. Discovery of a Novel Series of 7-Azaindole Scaffold Derivatives as PI3K Inhibitors with Potent Activity. ACS Med. Chem. Lett. 2017, 8, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, Y.; Wu, T.; Gao, Y.; Liu, X.; Yang, Y.; Ling, Y.; Jia, Y.; Deng, M.; Wang, J.; et al. Discovery of N-(2-chloro-5-(3-(pyridin-4-yl)-1H-pyrazolo[3,4-b]pyridin-5-yl)pyridin-3-yl)-4-fluorobenzenesulfonamide (FD274) as a highly potent PI3K/mTOR dual inhibitor for the treatment of acute myeloid leukemia. Eur. J. Med. Chem. 2023, 258, 115543. [Google Scholar] [CrossRef]
- Wu, T.; Chen, Y.; Yang, C.; Lu, M.; Geng, F.; Guo, J.; Pi, Y.; Ling, Y.; Xu, J.; Cai, T.; et al. Systematical Evaluation of the Structure-Cardiotoxicity Relationship of 7-Azaindazole-based PI3K Inhibitors Designed by Bioisosteric Approach. Cardiovasc. Toxicol. 2023, 23, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Lenneman, C.G.; Sawyer, D.B. Cardio-Oncology an Update on Cardiotoxicity of Cancer-Related Treatment. Circ. Res. 2016, 118, 1008–1020. [Google Scholar] [CrossRef]
- Morelli, M.B.; Bongiovanni, C.; Da Pra, S.; Miano, C.; Sacchi, F.; Lauriola, M.; D’Uva, G. Cardiotoxicity of Anticancer Drugs: Molecular Mechanisms and Strategies for Cardioprotection. Front. Cardiovasc. Med. 2022, 9, 847012. [Google Scholar] [CrossRef]
- Kitakata, H.; Endo, J.; Ikura, H.; Moriyama, H.; Shirakawa, K.; Katsumata, Y.; Sano, M. Therapeutic Targets for DOX-Induced Cardiomyopathy: Role of Apoptosis vs. Ferroptosis. Int. J. Mol. Sci. 2022, 23, 1414. [Google Scholar] [CrossRef]
- Zhao, Y.Y.; McLaughlin, D.; Robinson, E.; Harvey, A.P.; Hookham, M.B.; Shah, A.M.; McDermott, B.J.; Grieve, D.J. Nox2 NADPH Oxidase Promotes Pathologic Cardiac Remodeling Associated with Doxorubicin Chemotherapy. Cancer Res. 2010, 70, 9287–9297. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, Q.Q.; Li, W.F.; Li, H.Y.; Bao, J.; Yang, C.P.; Wang, A.P.; Wei, J.F.; Chen, S.Z.; Jin, H.T. Role of Nrf2 in the antioxidation and oxidative stress induced developmental toxicity of honokiol in zebrafish. Toxicol. Appl. Pharmacol. 2019, 373, 48–61. [Google Scholar] [CrossRef]
- Hu, X.P.; Liu, H.G.; Wang, Z.W.; Hu, Z.P.; Li, L.C. miR-200a Attenuated Doxorubicin-Induced Cardiotoxicity through Upregulation of Nrf2 in Mice. Oxid. Med. Cell. Longev. 2019, 2019, 1512326. [Google Scholar] [CrossRef]
- Yang, C.; Xu, C.; Li, Z.; Chen, Y.; Wu, T.; Hong, H.; Lu, M.; Jia, Y.; Yang, Y.; Liu, X.; et al. Bioisosteric replacements of the indole moiety for the development of a potent and selective PI3Kδ inhibitor: Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2021, 223, 113661. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, T.Z.; Yang, C.B.; Lu, M.Z.; Chen, Z.X.; Deng, M.L.; Jia, Y.; Yang, Y.T.; Liu, X.F.; Wang, H.Y.; et al. A pyridinesulfonamide derivative FD268 suppresses cell proliferation and induces apoptosis via inhibiting PI3K pathway in acute myeloid leukemia. PLoS ONE 2022, 17, e0277893. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, T.; Engelman, R.M.; Maulik, N.; Das, D.K. Role of Akt signaling in mitochondrial survival pathway triggered by hypoxic preconditioning. Circulation 2004, 109, 3042–3049. [Google Scholar] [CrossRef] [PubMed]
- Arslan, F.; Lai, R.C.; Smeets, M.B.; Akeroyd, L.; Choo, A.; Aguor, E.N.E.; Timmers, L.; van Rijen, H.V.; Doevendans, P.A.; Pasterkamp, G.; et al. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. 2013, 10, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Guenancia, C.; Gudjoncik, A.; Hachet, O.; Zeller, M.; Cottin, Y.; Vergely, C. Anthracyclines/trastuzumab: New aspects of cardiotoxicity and molecular mechanisms. Trends Pharmacol. Sci. 2015, 36, 326–348. [Google Scholar] [CrossRef]
- Huang, A.; Zeng, P.; Li, Y.; Lu, W.; Lai, Y. LY294002 Is a Promising Inhibitor to Overcome Sorafenib Resistance in FLT3-ITD Mutant AML Cells by Interfering with PI3K/Akt Signaling Pathway. Front. Oncol. 2021, 11, 782065. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, T.; Chen, Y.; Gong, Y.; Lu, M.; Yang, C.; Yang, Y.; Ling, Y.; Zhou, Y. A PI3K Inhibitor with Low Cardiotoxicity and Its Synergistic Inhibitory Effect with Gilteritinib in Acute Myelogenous Leukemia (AML) Cells. Molecules 2025, 30, 2347. https://doi.org/10.3390/molecules30112347
Wu T, Chen Y, Gong Y, Lu M, Yang C, Yang Y, Ling Y, Zhou Y. A PI3K Inhibitor with Low Cardiotoxicity and Its Synergistic Inhibitory Effect with Gilteritinib in Acute Myelogenous Leukemia (AML) Cells. Molecules. 2025; 30(11):2347. https://doi.org/10.3390/molecules30112347
Chicago/Turabian StyleWu, Tianze, Yi Chen, Yimin Gong, Mingzhu Lu, Chengbin Yang, Yannan Yang, Yun Ling, and Yaming Zhou. 2025. "A PI3K Inhibitor with Low Cardiotoxicity and Its Synergistic Inhibitory Effect with Gilteritinib in Acute Myelogenous Leukemia (AML) Cells" Molecules 30, no. 11: 2347. https://doi.org/10.3390/molecules30112347
APA StyleWu, T., Chen, Y., Gong, Y., Lu, M., Yang, C., Yang, Y., Ling, Y., & Zhou, Y. (2025). A PI3K Inhibitor with Low Cardiotoxicity and Its Synergistic Inhibitory Effect with Gilteritinib in Acute Myelogenous Leukemia (AML) Cells. Molecules, 30(11), 2347. https://doi.org/10.3390/molecules30112347