
A Straightforward Approach Towards Phosphadecalones by Microwave-Assisted Diels–Alder Reaction


Abstract

1. Introduction
2. Results and Discussion
2.1. Synthesis of Bicyclic Phenylphosphin-2-en-4-one Derivatives
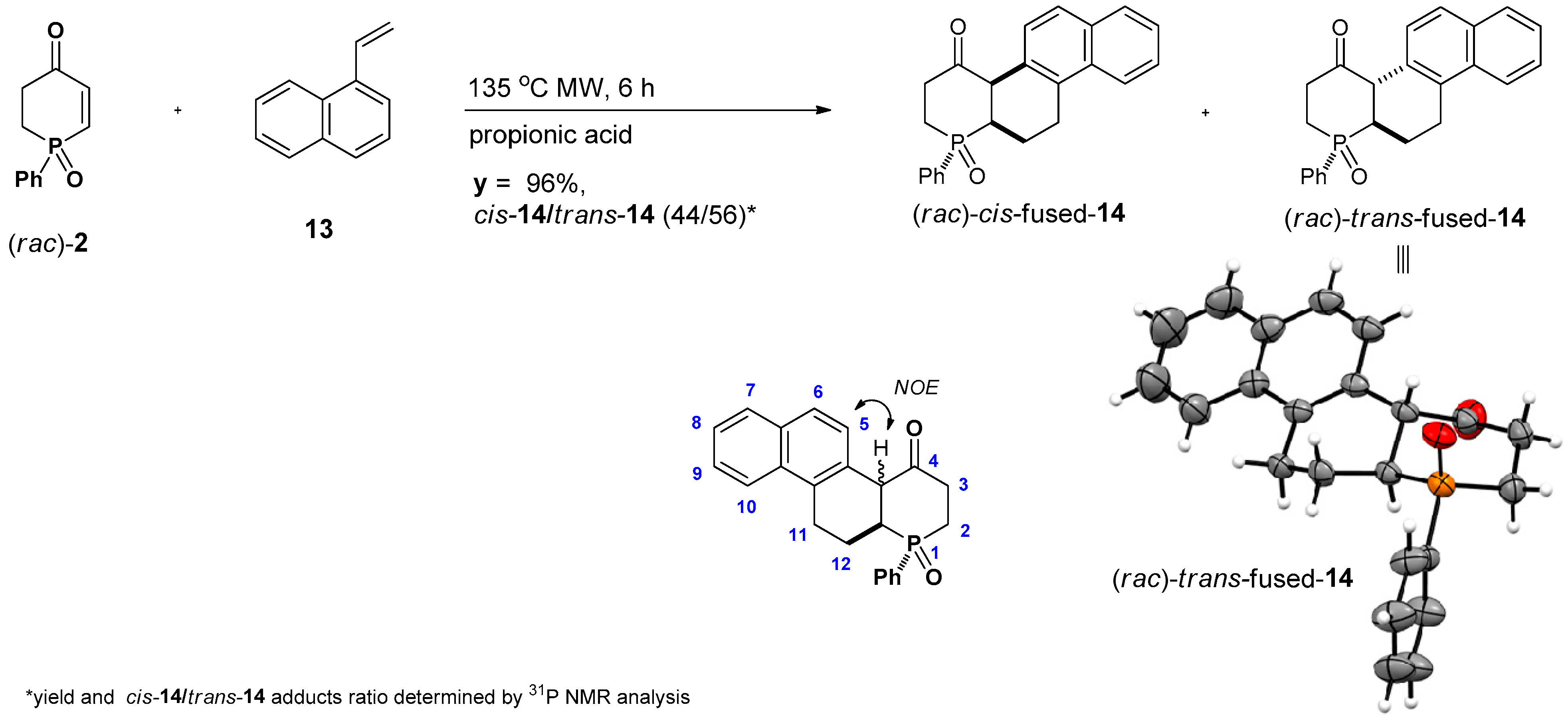
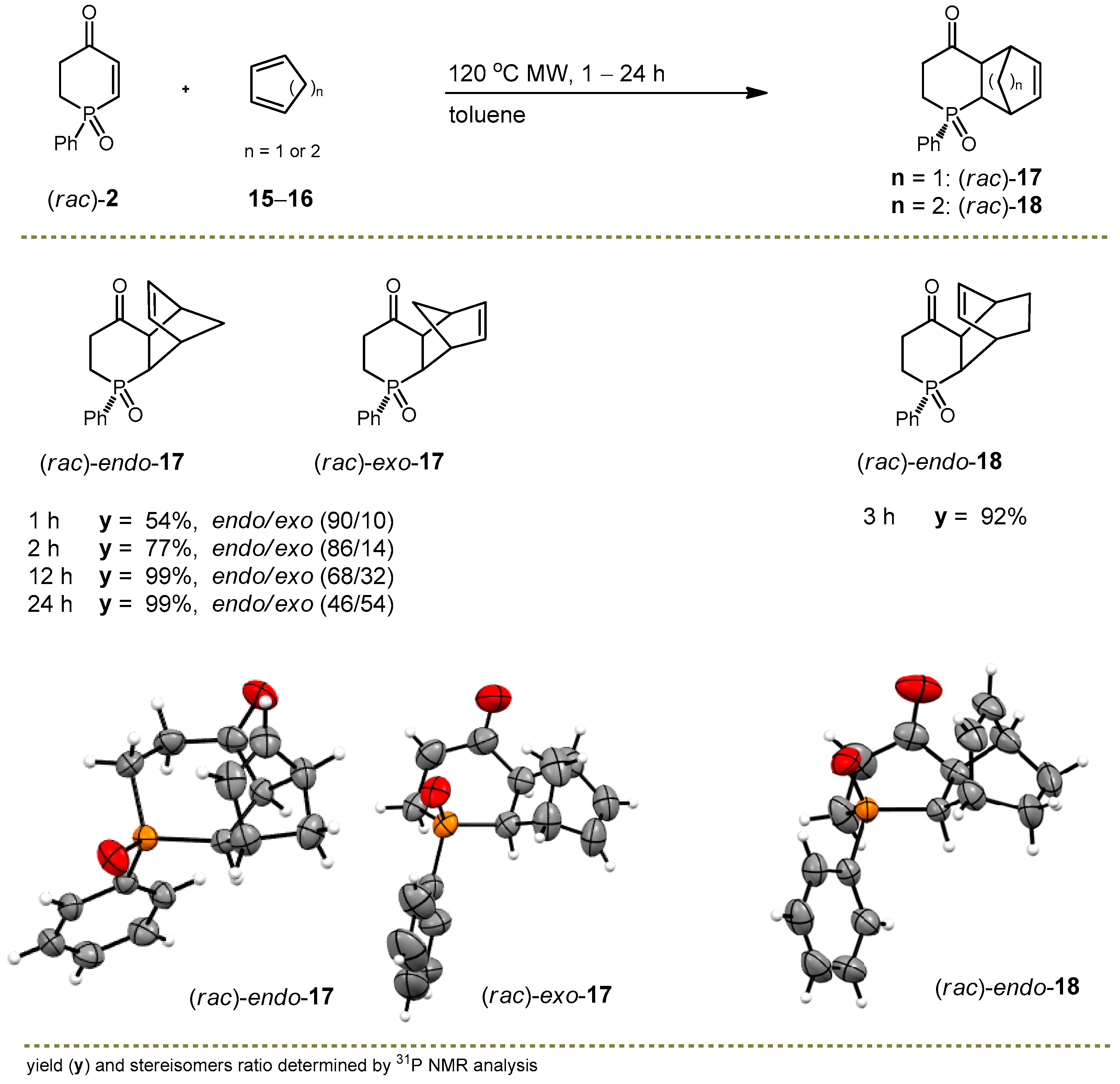
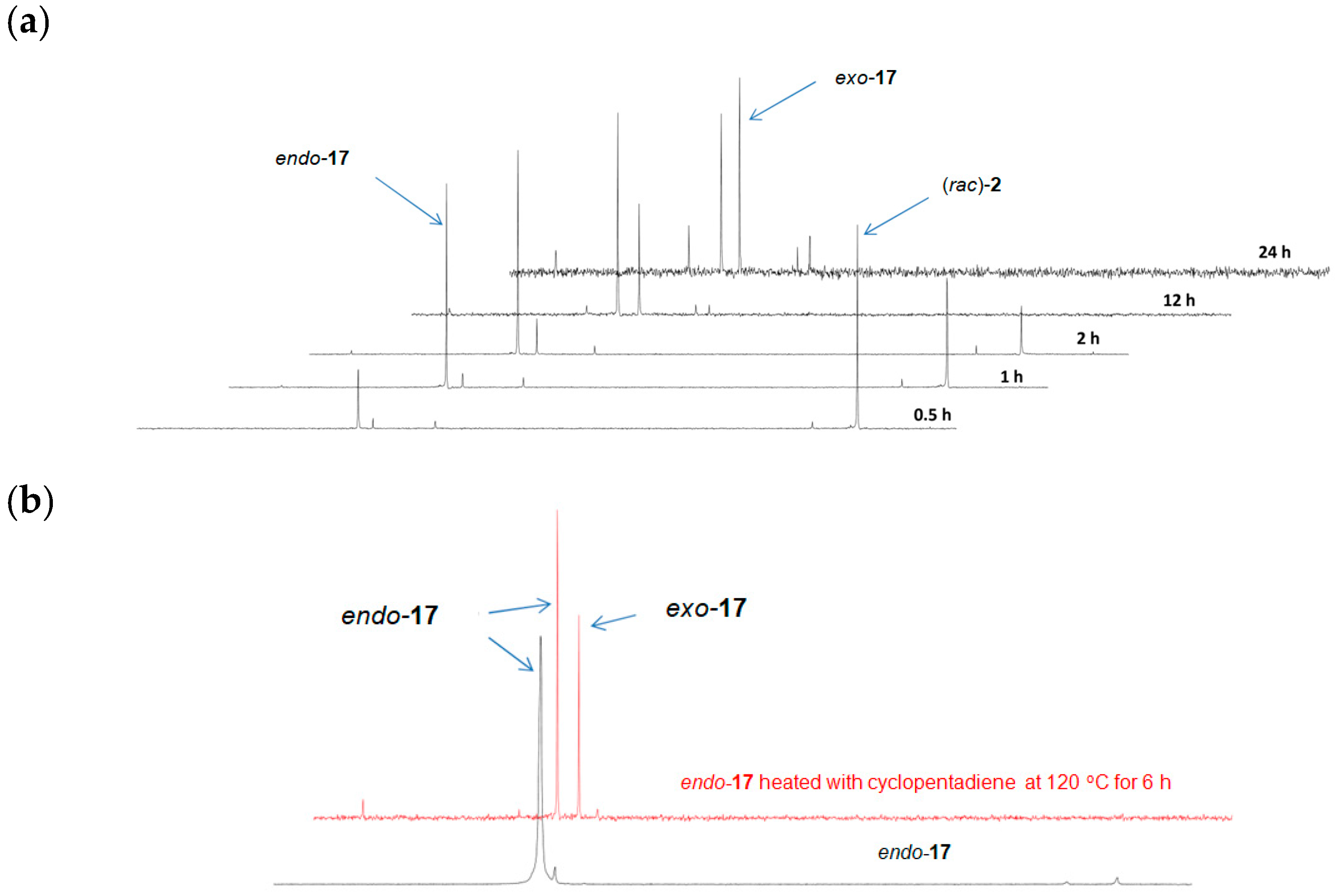
2.2. Synthesis of Tricyclic Phenylphosphin-2-en-4-one Derivatives
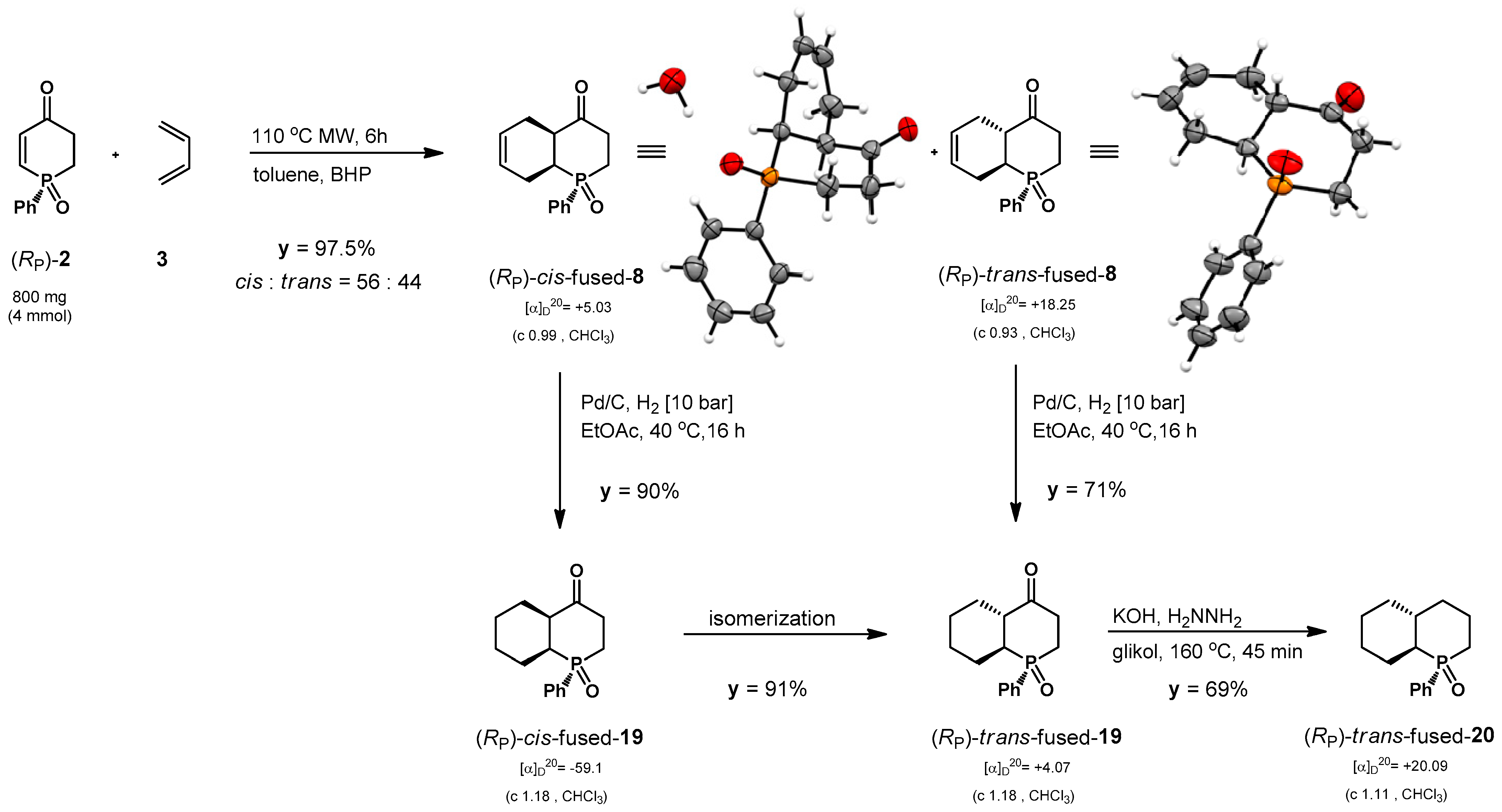
2.3. Synthesis of Bicyclic P-Stereogenic Phosphadecalones
3. Materials and Methods
3.1. General Information
3.2. X-Ray Crystallography
3.3. Synthesis and Spectral Data
3.3.1. Synthesis of rel-(SP,1R,6S)-2-Phenyl 2-phosphabicyclo[4.4.0]dec-8-en-5-one 2-Oxide ((rac)-cis-8) and rel-(SP,1R,6R)-2-Phenyl 2-phosphabicyclo[4.4.0]dec-8-en-5-one 2-Oxide ((rac)-trans-8): General Procedure
3.3.2. Synthesis of rel-(SP,1R,6S)-8,9-Dimethyl-2-phenyl-2-phosphabicyclo[4.4.0]dec-8-en-5-one 2-Oxide ((rac)-cis-9): General Procedure
3.3.3. Synthesis of rel-(SP,1R,6S)-8-Methyl-2-phenyl-2-phosphabicyclo[4.4.0]dec-8-en-5-one 2-Oxide ((rac)-cis-10a) and rel-(SP,1R,6S)-9-Methyl-2-phenyl-2-phosphabicyclo[4.4.0]dec-8-en-5-one 2-Oxide ((rac)-cis-10b)
3.3.4. Synthesis of rel-(SP,1R,6S,7S)-7-Methyl-2-phenyl-2-phosphabicyclo[4.4.0]dec-8-en-5-one 2-Oxide ((rac)-cis-11) and rel-(SP,1R,6R,7S)-7-Methyl-2-phenyl-2-phosphabicyclo[4.4.0]dec-8-en-5-one 2-Oxide ((rac)-trans-11)
3.3.5. Synthesis of rel-(SP,1R,6S,7S)-7-Methoxy-2-phenyl-2-phosphabicyclo[4.4.0]dec-8-en-5-one 2-Oxide ((rac)-cis-12)
3.3.6. Synthesis of rel-(SP,4aR,12aR)-1-Phenyl-1-phospha-2,3,11,12,12a-hexahydrochrysen-4-one 1-Oxide ((rac)-cis-14) and rel-(SP,4aS,12aR)-1-Phenyl-1-phospha-2,3,11,12,12a-hexahydrochrysen-4-one 1-Oxide ((rac)-trans-14)
3.3.7. Synthesis of rel-(SP,1S,2R,7S,8R)-3-Phenyl-3-phosphatricyclo[6.2.1.02,7]undec-9-en-6-one 3-Oxide ((rac)-endo-17) and rel-(SP,1R,2R,7S,8S)-3-Phenyl-3-phosphatricyclo[6.2.1.02,7]undec-9-en-6-one 3-Oxide ((rac)-exo-17)
3.3.8. Synthesis of rel-(SP,1S,2R,7S,8R)-3-Phenyl-3-phosphatricyclo[6.2.2.02,7]dodeca-9-en-6-one 3-Oxide ((rac)-endo-18)
3.3.9. Synthesis of (RP,1S,6R)-2-Phenyl-2-phosphabicyclo[4.4.0]dec-8-en-5-one 2-Oxide ((RP)-cis-8) and (RP,1S,6S)-2-Phenyl-2-phosphabicyclo[4.4.0]dec-8-en-5-one 2-Oxide ((RP)-trans-8): General Procedure
3.3.10. Synthesis of (RP,1S,6R)-2-Phenyl-2-phosphabicyclo[4.4.0]dec-5-one 2-Oxide ((RP)-cis-19) and (RP,1S,6S)-2-Phenyl-2-phosphabicyclo[4.4.0]dec-5-one 2-Oxide ((RP)-trans-19)
3.3.11. Synthesis of (RP,1S,6R)-2-Phenyl-2-phosphabicyclo[4.4.0]decane 2-Oxide ((RP)-trans-20)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Paton, K.F.; Kumar, N.; Crowley, R.S.; Harper, J.L.; Prisinzano, T.E.; Kivell, B.M. The Analgesic and Anti-Inflammatory Effects of Salvinorin A Analogue β-Tetrahydropyran Salvinorin B in Mice. Eur. J. Pain 2017, 21, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Madasu, C.; Karri, S.; Sangaraju, R.; Sistla, R.; Uppuluri, M.V. Synthesis and biological evaluation of some novel 1,2,3-triazole hybrids of myrrhanone B isolated from Commiphora mukul gum resin: Identification of potent antiproliferative leads active against prostate cancer cells (PC-3). Eur. J. Med. Chem. 2020, 188, 111974. [Google Scholar] [CrossRef]
- Tomioka, N.; Kishimoto, C.; Matsumori, A.; Kawai, C. Effects of Prednisolone on Acute Viral Myocarditis in Mice. J. Am. Coll. Cardiol. 1986, 7, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Yousef, I.M.; Mignault, D.; Weber, A.M.; Tuchweber, B. Influence of Dehydrocholic Acid on the Secretion of Bile Acids and Biliary Lipids in Rats. Digestion 1990, 45, 40–51. [Google Scholar] [CrossRef]
- Feng, Q.; Xu, M.; Yu, Y.Y.; Hou, Y.; Mi, X.; Sun, Y.X.; Ma, S.; Zuo, X.Y.; Shao, L.L.; Hou, M.; et al. High-Dose Dexamethasone or All-Trans-Retinoic Acid Restores the Balance of Macrophages towards M2 in Immune Thrombocytopenia. J. Thromb. Haemost. 2017, 15, 1845–1858. [Google Scholar] [CrossRef]
- Hirano, T.; Horigome, A.; Takatani, M.; Oka, K. Cortisone Counteracts Apoptosis-Inducing Effect of Cortisol in Human Peripheral-Blood Mononuclear Cells. Int. Immunopharmacol. 2001, 1, 2109–2115. [Google Scholar] [CrossRef]
- Hwang, N.; Ban, H.; Wu, S.; McGuire, K.; Hernandez, E.; Chen, J.; Zhao, Q.; Suresh, M.; Blass, B.; Viswanathan, U.; et al. 4-Oxooctahydroquinoline-1(2H)-carboxamides as Hepatitis B Virus (HBV) Capsid Core Protein Assembly Modulators. Bioorg. Med. Chem. Lett. 2022, 58, 128518. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cao, L.; Gao, H.; Wu, Y.; Wang, Y.; Fang, F.; Lan, T.; Lou, Z.; Rao, Y. Discovery, Optimization, and Target Identification of Novel Potent Broad-Spectrum Antiviral Inhibitors. J. Med. Chem. 2019, 62, 4056–4073. [Google Scholar] [CrossRef]
- Robbins, J.D.; Boring, D.L.; Tang, W.-J.; Shank, R.; Seamon, K.B. Forskolin Carbamates: Binding and Activation Studies with Type I Adenylyl Cyclase. J. Med. Chem. 1996, 39, 2745–2752. [Google Scholar] [CrossRef]
- Kashman, Y.; Benary, E. The synthesis of the 9-phosphabicyclo [3.3.1]nonanic and 2-phospha-6-oxa- adamantanic systems. Tetrahedron 1972, 28, 4091–4098. [Google Scholar] [CrossRef]
- Kashman, Y.; Ronen, H. The synthesis of a phosphadecalone system. Tetrahedron 1973, 29, 4275–4278. [Google Scholar] [CrossRef]
- Bosyakov, Y.G.; Shiganakova, O.V.; Revenko, G.P.; Logunov, A.P. Michael Reaction of P(IV)-Phosphabicyclodecanones. J. Gen. Chem. USSR 1990, 60, 2211–2215, [Zh. Obshch. Khim. 1990, 60, 2473–2478]. [Google Scholar]
- Franisal, N.; Gallagher, M.J. Organophosphorus Intermediates. X. The Synthesis and Properties of the 1,4-Dioxo- -2,3,4a,5,6,7,8,8a-Octahydro-1-λ-5-Phosphinoline System. Aust. J. Chem. 1987, 40, 1353–1363. [Google Scholar] [CrossRef]
- Ananthnag, G.S.; Balakrishna, M.S. Six-Membered Rings with Two or More Heteroatoms with at Least One Phosphorus. In Comprehensive Heterocyclic Chemistry IV; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier: Oxford, UK, 2022; Volume 9, pp. 735–767. [Google Scholar] [CrossRef]
- Thakur, D.; Sushmita; Meena, S.A.; Verma, A.K. Advancement in Synthetic Strategies of Phosphorus Heterocycles: Recent Progress from Synthesis to Emerging Class of Optoelectronic Materials. Chem. Rec. 2024, 24, e202400058. [Google Scholar] [CrossRef]
- Ostermeier, M.; Prieß, J.; Helmchen, G. Mono- and Bidentate Phosphinanes-New Chiral Ligands and Their Application in Catalytic Asymmetric Hydrogenations. Angew. Chem. Int. Ed. 2002, 41, 612–614. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, X. Six-membered bis(azaphosphorinane), readily available ligand for highly enantioselective asymmetric hydrogenations. Tetrahedron Lett. 2006, 47, 1567–1569. [Google Scholar] [CrossRef]
- Kräh, S.; Kachel, I.; Trapp, O. Electron-Rich Silicon Containing Phosphinanes for Rapid Pd-Catalyzed C−X Coupling Reactions. ChemCatChem 2022, 14, e202200734. [Google Scholar] [CrossRef]
- Edwards, P.G.; Kariuki, B.M.; Limon, M.; Ooi, L.-L.; Platts, J.A.; Newman, P.D. Metal Complexes of a Structurally Embellished Phosphinane Ligand: An Assessment of Stereoelectronic Effects. Eur. J. Inorg. Chem. 2011, 2011, 1230–1239. [Google Scholar] [CrossRef]
- Kumar, A.; Mukhopadhyay, J.; Bhagat, S. Phosphorus Heterocycles and Their Biological Applications. Chem. Sel. 2024, 9, e202404258. [Google Scholar] [CrossRef]
- Schaffner, A.-P.; Sansilvestri-Morel, P.; Despaux, N.; Ruano, E.; Persigand, T.; Rupin, A.; Mennecier, P.; Vallez, M.-O.; Raimbaud, E.; Desos, P.; et al. Phosphinanes and Azaphosphinanes as Potent and Selective Inhibitors of Activated Thrombin-Activatable Fibrinolysis Inhibitor (TAFIa). J. Med. Chem. 2021, 64, 3897–3910. [Google Scholar] [CrossRef] [PubMed]
- Stowasser, B.; Budt, K.-H.; Jian-Qi, L.; Peyman, A.; Ruppert, D. New hybrid transition state analog inhibitors of HIV protease with peripheric C2-symmetry. Tetrahedron Lett. 1992, 33, 6625–6628. [Google Scholar] [CrossRef]
- Monbrun, J.; Dayde, B.; Cristau, H.-J.; Volle, J.-N.; Virieux, D.; Pirat, J.-L. Diastereoselective Michael addition of 2H-2-oxo-1, 4, 2-oxaza phosphinanes to olefins. Tetrahedron 2011, 67, 540–545. [Google Scholar] [CrossRef]
- Butin, B.M.; Logunov, A.P.; Maishinova, G.T.; Ugarina, I.G. Oximes, Semicarbazones, and α-Hydroxy- phosphonates Derived from 9-Oxa-2λ5-Phosphabicyclo[4.4.0]decan-5-one 2-Oxides. Russ. J. Gen. Chem. 1996, 66, 567–569. [Google Scholar]
- Yamashita, M. Preparation, Structure, and Biological Properties of Phosphorus Heterocycles with a C–P Ring System. In Bioactive Heterocycles II; Eguchi, S., Ed.; Topics in Heterocyclic Chemistry; Springer: Berlin/Heidelberg, Germany, 2007; Volume 8. [Google Scholar] [CrossRef]
- Chen, C.H.; Brighty, K.E.; Michaels, F.M. Synthesis of Phosphalilolidine and Phosphajulolidine. J. Org. Chem. 1981, 46, 361–367. [Google Scholar] [CrossRef]
- Laurenco, C.; Villien, L.; Kaufmann, G. Experimentation du Plan de Synthese Etabli avec l’Aide de PASCOP: Synthese Assistee par Ordinateur de la Phosphacarnegine-II. Tetrahedron 1984, 40, 2731–2740. [Google Scholar] [CrossRef]
- Kruszynski, R.; Czubacka, E.; Trzesowska-Kruszynska, A.; Bartczak, T.J.; Bruzik, K.S.; Knopik, P.; Kudzin, Z.; Stec, W.J.; Wolf, W.M. Conformation of Sterically Hindered 4-Methyl-2-oxo-2-trityl-1,3,2-dioxaphosphorinane in the Solid State and the Solution. J. Chem. Crystallogr. 2011, 41, 908–918. [Google Scholar] [CrossRef]
- Gallagher, M.J. Six-membered rings: Phosphinanes, Dihydro- and Tetrahydro-phosphinines. In Phosphorus-Carbon Heterocyclic Chemistry. The Rise of New Domain; Mathey, F., Ed.; Elsevier Science Ltd.: Amsterdam, The Netherlands, 2001; Volume 5.1, pp. 463–483. [Google Scholar] [CrossRef]
- Quin, L.D. The Heterocyclic Chemistry of Phosphorus. Systems Based on Phosphorus-Carbon Bond; Wiley-Interscience: New York, NY, USA, 1981; Volume 10, ISBN 978-0471064619. [Google Scholar]
- Rammal, F.; Magné, V.; Berionni, G.; Lakhdar, S. Six-Membered Rings with One Phosphorus Atom. In Comprehensive Heterocyclic Chemistry IV; Maulide, N., Ed.; Elsevier: Oxford, UK, 2022; Volume 9, pp. 685–717. [Google Scholar] [CrossRef]
- Hewitt, D.G. Six-Membered Rings with One Phosphorus Atom. In Comprehensive Heterocyclic Chemistry II; Katritzky, A.R.C., Rees, W., Scriven, E.F.V., Eds.; Pergamon: Oxford, UK, 1996; Volume 5, pp. 639–668. [Google Scholar] [CrossRef]
- Pearce, K.G.; Simenok, V.; Crossley, I.R. Phosphacycloalkyldiones: Synthesis and coordinative behaviour of 6- and 7-member cyclic diketophosphanyls. Dalton Trans. 2020, 49, 5482–5492. [Google Scholar] [CrossRef] [PubMed]
- Quin, L.D.; Hughes, A.N.; Kisalus, J.C.; Pete, B. Synthesis and NMR Spectral Properties of Phosphines in the 2-Phosphabicyclo[2.2.2]oct-5-ene and 2-Phosphabicyclo[2.2.2]octa-5,7-diene Systems. J. Org. Chem. 1988, 53, 1722–1729. [Google Scholar] [CrossRef]
- Keglevich, G.; Töke, L.; Steinhauser, K.; Novák, T.; Ludányi, K. Synthesis and Use of 6-, 7- and 8-Membered P-Heterocycles. Phosphorus Sulfur Silicon Relat. Elem. 1999, 144, 593–596. [Google Scholar] [CrossRef]
- Keglevich, G.; Kovács, J.; Körtvélyesi, T.; Parlagh, G.; Imre, T.; Ludányi, K.; Hegedűs, L.; Hanusz, M.; Simon, K.; Márton, A.; et al. Novel 2-phosphabicyclo[2.2.2]oct-5-ene derivatives and their use in phosphinylations. Heteroat. Chem. 2004, 15, 97–106. [Google Scholar] [CrossRef]
- Łastawiecka, E.; Frynas, S.; Pietrusiewicz, K.M. Desymmetrization Approach to the Synthesis of Optically Active P-Stereogenic Phosphin-2-en-4-ones. J. Org. Chem. 2021, 86, 6195–6206. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.H.; Wong, L.T.L. Benzo[b]phosphole and derivatives. Can. J. Chem. 1971, 49, 530–531. [Google Scholar] [CrossRef]
- Nief, F.; Charrier, C.; Mathey, F.; Simalty, M. On Some Chemical Properties of 1-Phenylphosphindole. Phosphorus Sulfur 1982, 13, 259. [Google Scholar] [CrossRef]
- Pietrusiewicz, K.M.; Koprowski, M.; Drzazga, Z.; Parcheta, R.; Łastawiecka, E.; Demchuk, O.M.; Justyniak, I. Efficient Oxidative Resolution of 1-Phenylphosphol-2-Ene and Diels–Alder Synthesis of Enantiopure Bicyclic and Tricyclic P-Stereogenic C-P Heterocycles. Symmetry 2020, 12, 346. [Google Scholar] [CrossRef]
- Łastawiecka, E.; Włodarczyk, A.; Kozioł, A.E.; Małuszyńska, H.; Pietrusiewicz, K.M. Resolution of P-Stereogenic 1-Phenylphosphin-2-en-4-one 1-Oxide into Two Enantiomers by (R,R)-TADDOL and Conformational Diversity of the Phosphinenone Ring and TADDOL in the Crystal State. Molecules 2021, 26, 6873. [Google Scholar] [CrossRef] [PubMed]
- Bodalski, R.; Koszuk, J.; Krawczyk, H.; Pietrusiewicz, K.M. An Efficient Synthesis of the Enantiomeric 17-Phosphasteroid System. J. Org. Chem. 1982, 47, 2219–2220. [Google Scholar] [CrossRef]
- Vinokurov, N.; Pietrusiewicz, K.M.; Butenschön, H. Asymmetric Diels–Alder Cycloaddition of a di-P-Stereogenic Dienophile with Cyclopentadiene. Tetrahedron Asymmetry 2009, 20, 1081–1085. [Google Scholar] [CrossRef]
- Levandowski, B.J.; Houk, K.N. Theoretical Analysis of Reactivity Patterns in Diels–Alder Reactions of Cyclopentadiene, Cyclohexadiene, and Cycloheptadiene with Symmetrical and Unsymmetrical Dienophiles. J. Org. Chem. 2015, 80, 3530–3537. [Google Scholar] [CrossRef]
- Alder, K.; Stein, G. Untersuchungen Über den Verlauf der Diensynthese. Angew. Chem. 1937, 50, 510–519. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Snyder, S.A.; Montagnon, T.; Vassilikogiannakis, G. The Diels–Alder Reaction in Total Synthesis. Angew. Chem. Int. Ed. 2002, 41, 1668–1698. [Google Scholar] [CrossRef] [PubMed]
- Csende, F.; Stájer, G.; Fülöp, F. Retro Diels–Alder Reactions. In Comprehensive Organic Synthesis, 2nd ed.; Knochel, P., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 5.12, pp. 518–594. [Google Scholar] [CrossRef]
- CrysAlisPro Software System; SuperNova Diffractometer, Agilent Technologies Inc.: Yarnton, UK, 2013.
- CrysAlisPro 1.171.42.79a; Rigaku Oxford Diffraction: Tokyo, Japan, 2022.
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Łastawiecka, E.; Kozioł, A.E.; Pietrusiewicz, K.M. A Straightforward Approach Towards Phosphadecalones by Microwave-Assisted Diels–Alder Reaction. Molecules 2025, 30, 2338. https://doi.org/10.3390/molecules30112338
Łastawiecka E, Kozioł AE, Pietrusiewicz KM. A Straightforward Approach Towards Phosphadecalones by Microwave-Assisted Diels–Alder Reaction. Molecules. 2025; 30(11):2338. https://doi.org/10.3390/molecules30112338
Chicago/Turabian StyleŁastawiecka, Elżbieta, Anna E. Kozioł, and K. Michał Pietrusiewicz. 2025. "A Straightforward Approach Towards Phosphadecalones by Microwave-Assisted Diels–Alder Reaction" Molecules 30, no. 11: 2338. https://doi.org/10.3390/molecules30112338
APA StyleŁastawiecka, E., Kozioł, A. E., & Pietrusiewicz, K. M. (2025). A Straightforward Approach Towards Phosphadecalones by Microwave-Assisted Diels–Alder Reaction. Molecules, 30(11), 2338. https://doi.org/10.3390/molecules30112338