



In Silico Identification and Characterization of Spiro[1,2,4]triazolo[1,5-c]quinazolines as Diacylglycerol Kinase α Modulators

 , ,
, ,  ,
,  ,
,  and
and 
Abstract

1. Introduction
2. Results and Discussion
2.1. Molecular Docking Studies
2.1.1. Structural and Functional Basis of DGK-α as a Therapeutic Target
- Calcium binding triggers conformational changes that promote membrane translocation;
- The N-terminal recoverin homology domain works synergistically with EF-hand motifs to regulate enzyme activation;
- Calcium-independent basal activity can be supported by specific lipid environments, including phosphatidylethanolamine and cholesterol.
- Immune cell function, particularly T-cell responses;
- Cell proliferation and migration;
- Modulation of Rac activation and actin cytoskeleton remodeling.
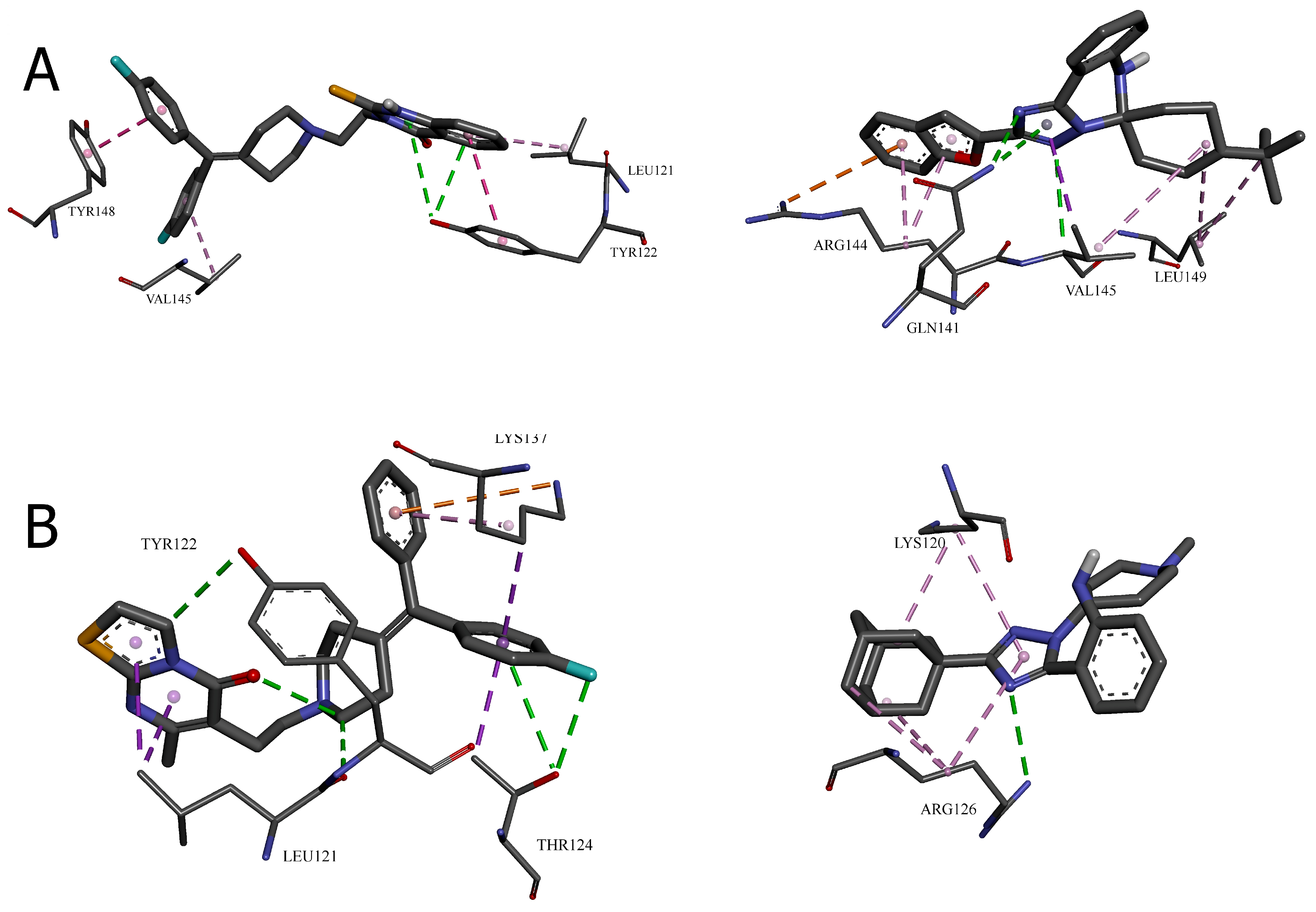
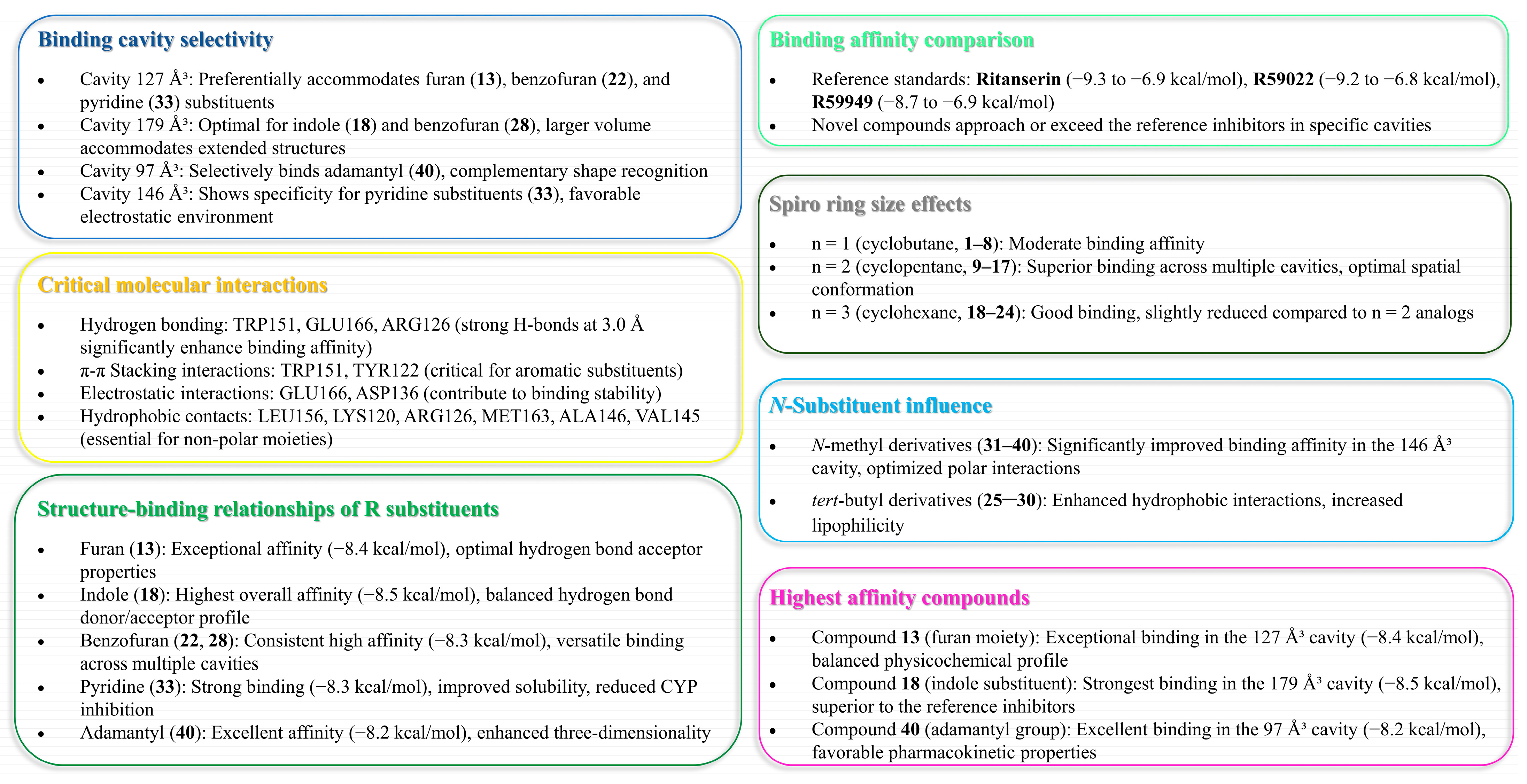
2.1.2. Molecular Visualization and Key Interaction Analysis
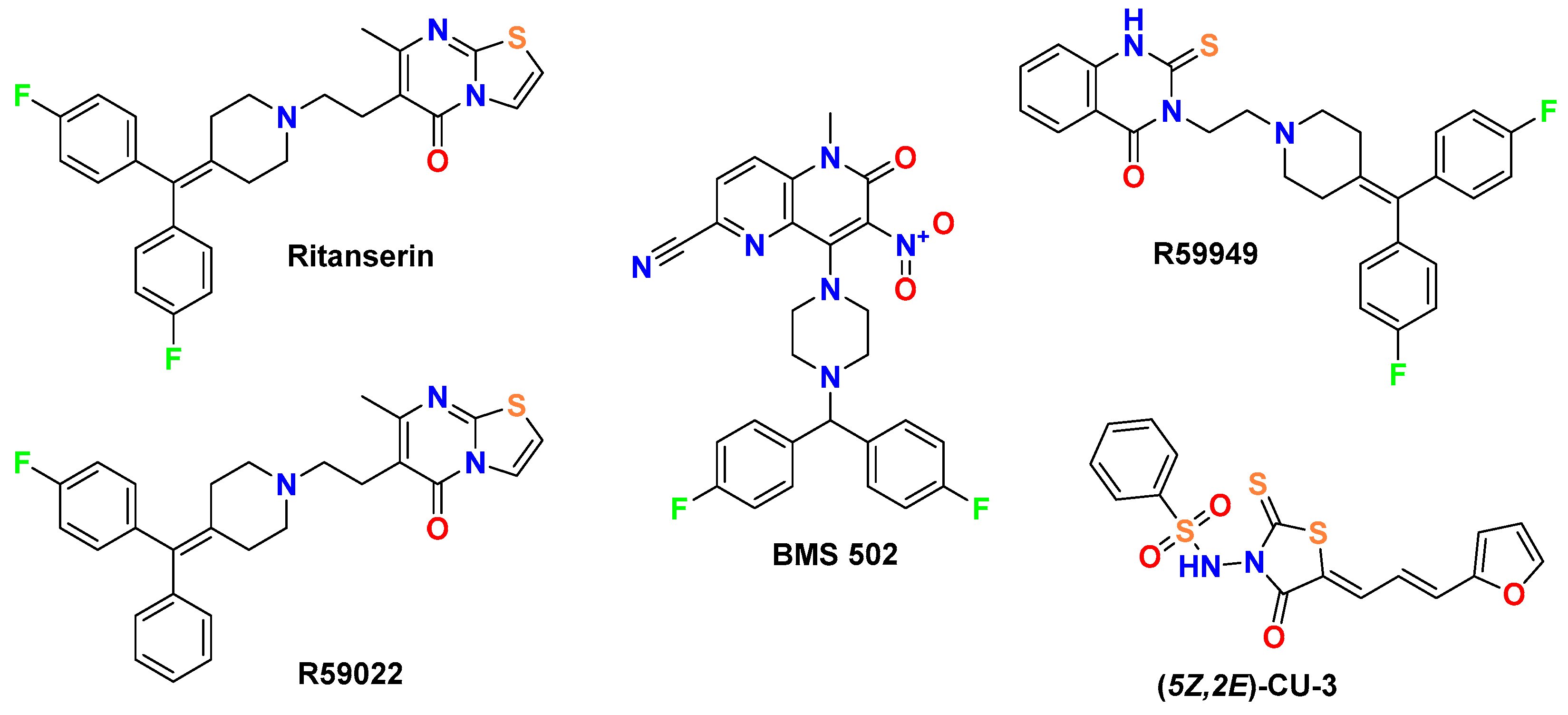
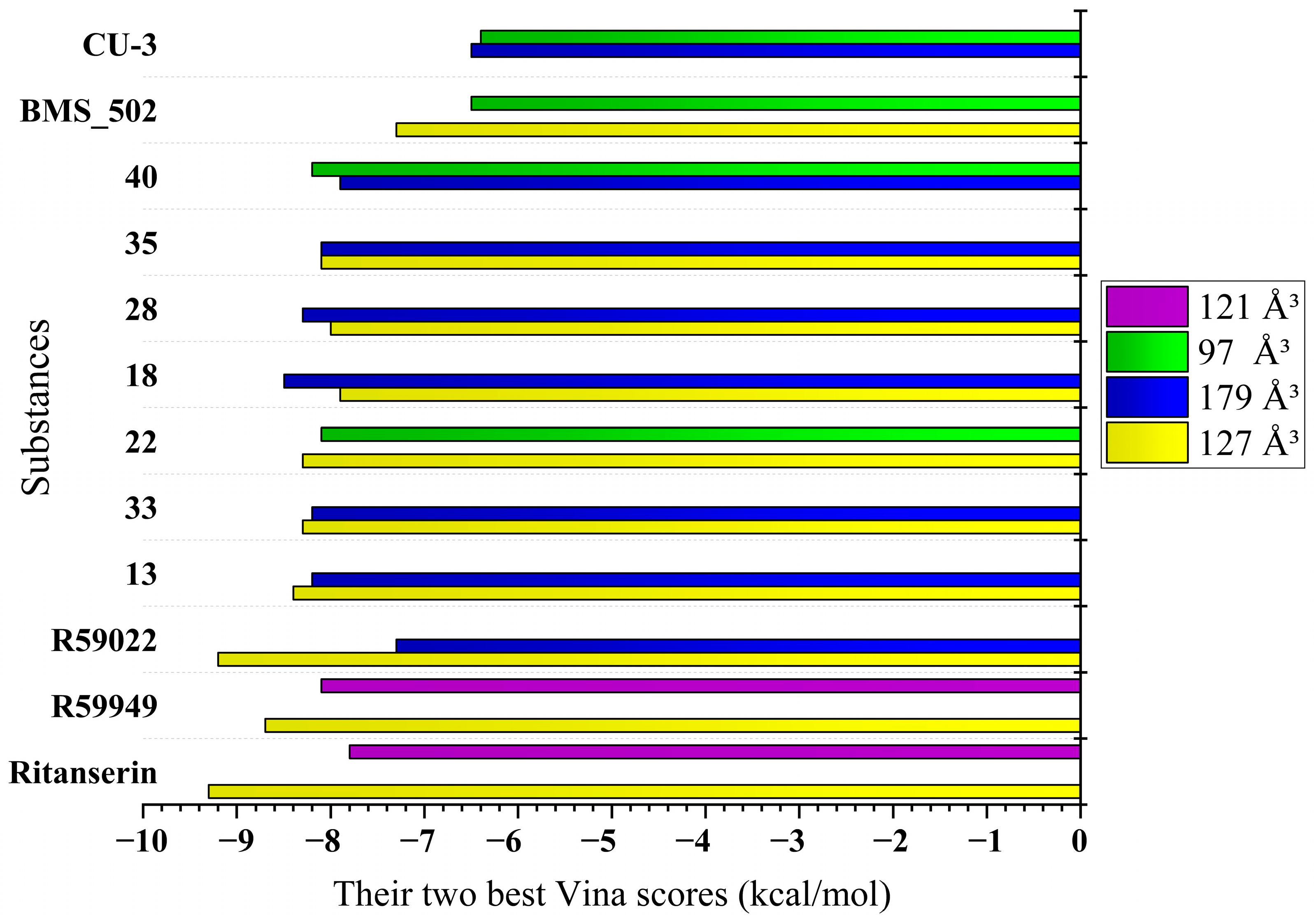
- Cavity 127 Å3: Ritanserin vs. Compound 3
- Cavity 179 Å3: R59949 vs. Compound 18
- Cavity 97 Å3: R59949 vs. Compound 40
- Cavity 121 Å3: R59949 vs. Compound 28
- Cavity 146 Å3: R59022 vs. Compound 33
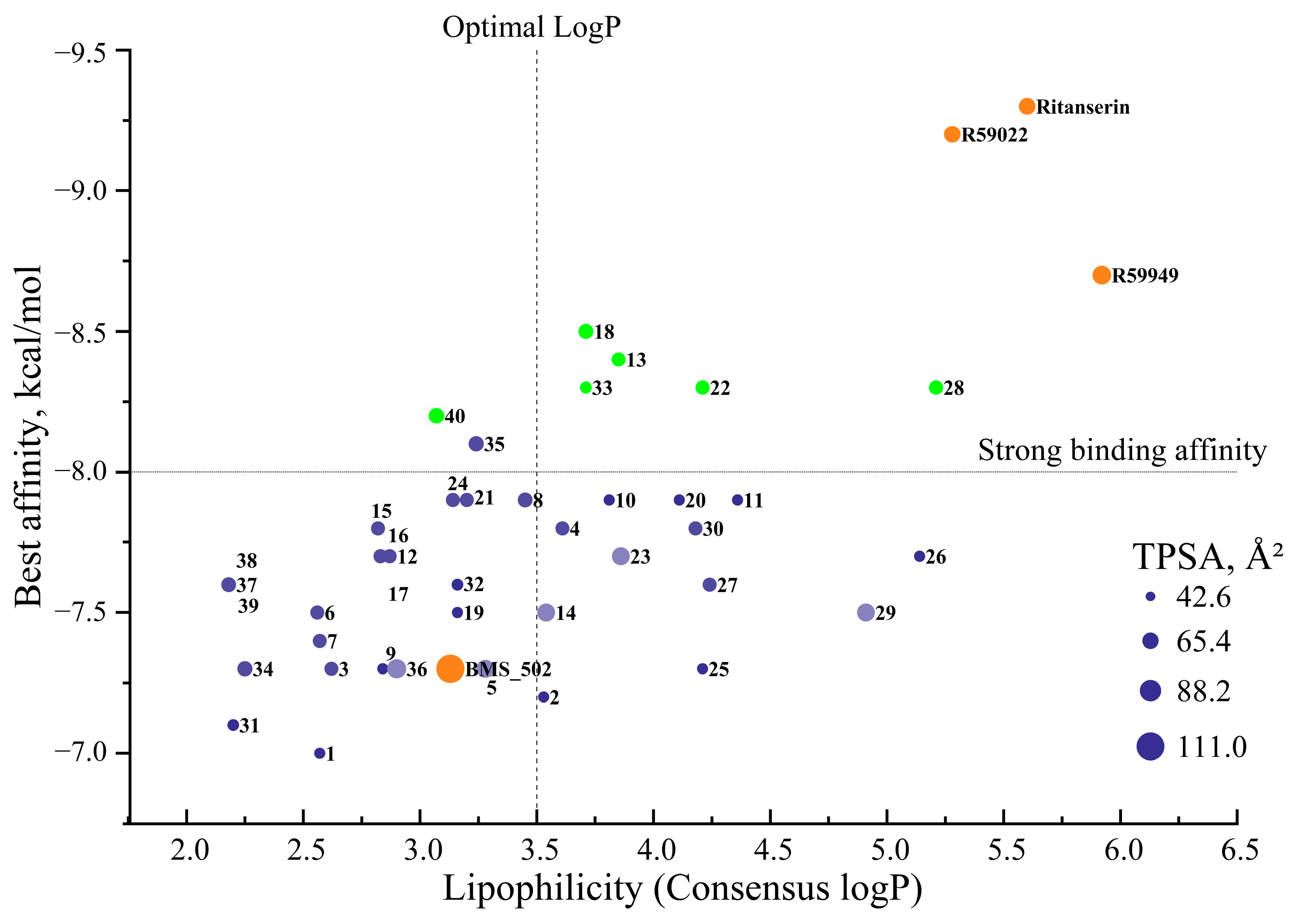
2.1.3. Correlation Between Interaction Patterns and Binding Affinities
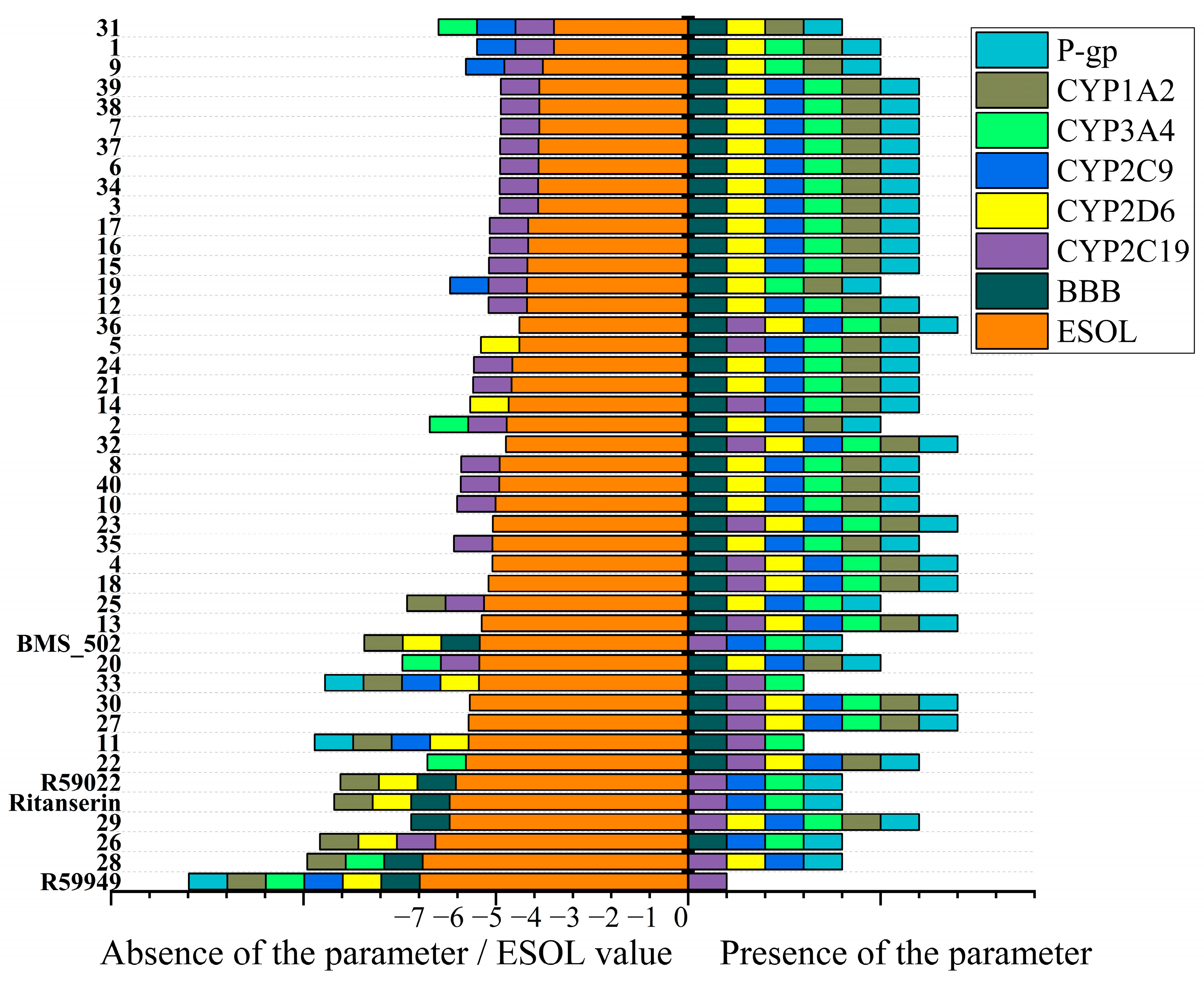
2.2. In Silico ADME Property Analysis
2.2.1. Molecular Descriptors and Fundamental Property Analysis
2.2.2. Toxicity
2.2.3. Physicochemical Drug-Likeness
2.2.4. Drug Development Considerations
2.3. Comparative Analysis with the Reference DGK Inhibitors
2.3.1. Comparison with Established DGK Inhibitors
- R59949 shows higher mutagenicity probability (0.75) compared to most novel compounds (typically <0.61);
- Ritanserin and R59022 both exhibit cytotoxicity probabilities (0.67) that exceed those of the novel derivatives (0.53–0.60);
- BMS502 uniquely demonstrates a higher mutagenicity probability (0.91/yes) compared to all novel compounds, which consistently showed “no” predictions for mutagenicity.
2.3.2. Comparison with Representatives of Patent CN 115362003 B
- The spiro fusion creates a unique three-dimensional architecture that optimizes the spatial orientation within the binding pockets, which is particularly evident in the 179 Å3 cavity, where compound 18 achieves a remarkable −8.5 kcal/mol binding score.
- The introduction of conformational constraint through spiro-ring systems appears to position key pharmacophore elements for optimal interaction with critical protein residues, as demonstrated by compound 13’s excellent binding (−8.2 kcal/mol) in the 179 Å3 cavity.
- The incorporation of heterocyclic substituents in the spiro-derivatives provides additional hydrogen bonding capabilities and π–electron interactions that are absent in the basic scaffold, contributing to compound 33’s enhanced binding (−8.2 kcal/mol) in the 179 Å3 cavity.
- The adamantyl-substituted compound 33 demonstrates particularly strong binding across multiple cavities, suggesting that its unique spatial geometry and lipophilic character create favorable interactions within the hydrophobic regions of the binding sites.
- Compound 40 shows exceptional versatility with strong binding across all cavities, including an optimal interaction (−8.2 kcal/mol) in the 127 Å3 cavity, indicating its balanced pharmacophore arrangement.
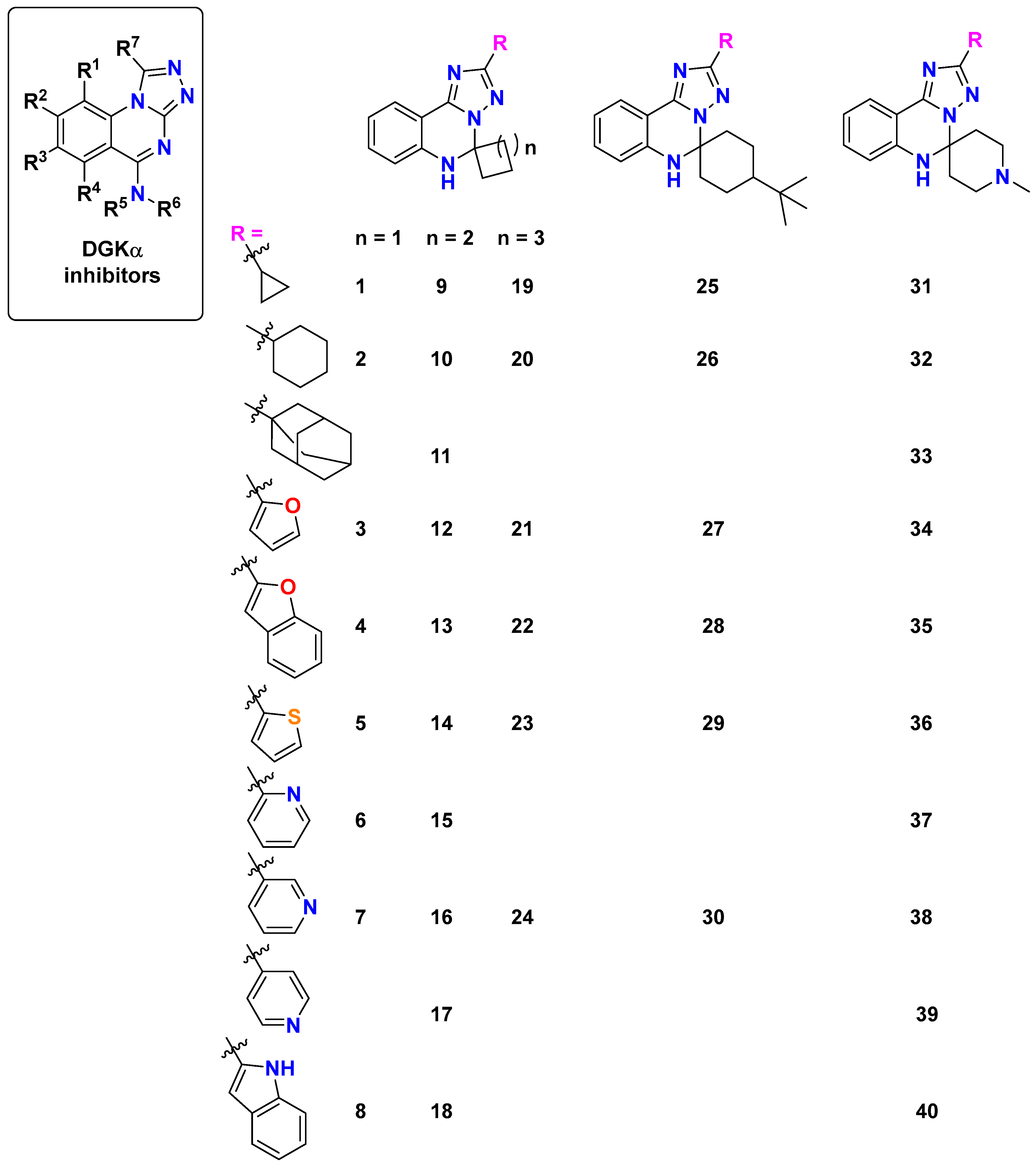
2.4. Structure–Activity Relationships
2.5. Limitations
- Methodological
- Absence of experimental validation. The most significant limitation is the lack of experimental validation through biochemical assays. An experimental determination of IC50 values, binding constants, and enzyme inhibition profiles would be essential to confirm the predicted activities of these compounds.
- Comprehensive structural modeling. The integration of the complete protein structure may reveal additional interaction sites, particularly within the catalytic domain, where inhibitors like R59949 demonstrate binding affinity.
- Implementation of dynamic models. The application of molecular dynamic simulations could potentially capture protein flexibility and conformational changes during ligand binding, offering more nuanced insights than static docking approaches.
- 2.
- Mechanistic Validation Possibilities
- Biochemical assay development. To address the validation gap, the development of biochemical assays could provide critical validation parameters (IC50, Ki values, and binding kinetics) that may confirm or refine computational predictions.
- Cellular activity assessment. The absence of cell-based evaluations limits our understanding of the compounds’ abilities to penetrate cellular membranes and modulate DGK-α activity in physiologically relevant environments.
- Isoform selectivity assessment. Comprehensive profiling across DGK isoforms might identify compounds with optimal selectivity profiles, potentially minimizing off-target effects that commonly limit kinase inhibitor utility.
- Structure–activity relationship development. Systematic structural modifications could potentially establish clear correlations between molecular features and both binding affinity and selectivity, guiding rational design iterations.
- 3.
- Translational research opportunities
- Expanded pharmacokinetic evaluation. A detailed investigation of physiological stability, metabolic pathways, and bioavailability profiles could identify candidates with favorable drug-like properties.
- Enhanced CNS penetration analysis. Advanced modeling and experimental validation of blood–brain barrier permeability might identify compounds suitable for neurological applications, particularly through refined QSPR analyses.
- Comprehensive interaction profiling. A broader assessment of potential drug–drug interactions, including transporter effects and pharmacodynamic consequences, could identify compounds with favorable clinical profiles.
- 4.
- Technical advancement possibilities
- Scoring function diversification. The application of multiple complementary evaluation methods might yield more robust binding predictions by mitigating algorithm-specific biases.
- Advanced simulation implementation. Extended molecular dynamics with enhanced sampling techniques could potentially provide deeper insights into the stability and kinetics of predicted protein–ligand complexes.
- Quantum mechanical modeling integration. The incorporation of QM/MM approaches might more accurately represent electronic effects on critical binding interactions, particularly for compounds with complex electronic distributions.
2.6. Future Directions
- Direct binding assessment possibilities. Radioligand binding assays using [3H]phorbol 12-myristate 13-acetate displacement might provide valuable data on direct interactions with the DGK-α catalytic domain for promising compounds such as 13, 18, 33, and 40. Surface plasmon resonance experiments could potentially measure binding kinetics and affinity constants, which would offer quantitative validation of predicted binding affinities.
- Enzyme inhibition evaluation options. In vitro DGK-α inhibition assays using purified recombinant human DGK-α could determine IC50 values and the inhibitory mechanisms. Thermal shift assays might confirm physical interactions with the target protein and provide insights into binding-induced conformational changes.
- Selectivity investigation avenues. Compounds showing promising activity could be screened against various DGK isoforms to assess their selectivity profiles. Additionally, counter-screening against related kinases might evaluate potential off-target activities.
- Comparative analysis considerations. Competitive binding studies with established inhibitors like R59949 could help determine binding site overlap and provide context for the novel compounds’ inhibitory mechanisms, complementing the computational comparison presented here.
- Ex vivo organoid models to evaluate the compounds’ efficacy in more complex tissue environments;
- Animal models of cancer and inflammatory disorders to validate in vivo efficacy [9].
- For compounds showing the highest binding affinity (e.g., 18 and 40), introduce modifications to improve solubility while maintaining target engagement;
- For compounds with balanced pharmacokinetic profiles (e.g., 33), explore bioisosteric replacements of the adamantyl group to maintain binding while reducing synthetic complexity;
- Develop hybrid structures incorporating the indole moiety from compound 18 with the spiro-piperidine scaffold from compound 33 to potentially combine superior binding with improved pharmacokinetics;
- Neurodegenerative disorders [2], where abnormal lipid signaling contributes to disease progression;
- Autoimmune disorders [4], where the modulation of T-cell responses could restore immune homeostasis;
- Cardiac hypertrophy and heart failure [3], where DGK-α plays a role in pathological remodeling;
- Metabolic disorders, particularly type 2 diabetes [2], where altered diacylglycerol signaling affects the insulin response.
3. Materials and Methods
3.1. Molecular Docking Studies
Computational Framework of the Structure-Based Blind Docking Analysis
3.2. Physicochemical and ADME Property Predictions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DGK-α | Diacylglycerol kinase alpha |
| PDB | Protein Data Bank |
| EF | EF-hand (calcium-binding motif) |
| DAG | Diacylglycerol |
| PA | Phosphatidic acid |
| PKC | Protein kinase C |
| PH | Pleckstrin homology (domain) |
| SAM | Sterile alpha motif |
| MARCKS | Myristoylated alanine-rich C-kinase substrate |
| PDZ | Postsynaptic density protein, Drosophila disc large tumor suppressor, Zonula occludens-1 protein (domain) |
| HIV | Human immunodeficiency virus |
| HBV | Hepatitis B virus |
| 5-HT2R | Serotonin 2 receptor |
| IC50 | Half maximal inhibitory concentration |
| Kd | Dissociation constant |
| ADME | Absorption, eistribution, metabolism, excretion |
| TPSA | Topological polar surface area |
| ESOL | Estimated aqueous solubility |
| MW | Molecular weight |
| MR | Molecular refractivity |
| logP | Partition coefficient (octanol–water) |
| PAINS | Pan-Assay Interference Compounds |
| BBB | Blood–brain barrier |
| P-gp | P-glycoprotein |
| CYP | Cytochrome P450 |
| LD50 | Median lethal dose |
| HT | Hepatotoxicity |
| CG | Carcinogenicity |
| IT | Immunotoxicity |
| MG | Mutagenicity |
| CT | Cytotoxicity |
| SAR | Structure–activity relationship |
| CNS | Central nervous system |
| PMA | Phorbol 12-myristate 13-acetate |
| GHS | Globally Harmonized System (of Classification and Labeling of Chemicals) |
References
- Dominguez, C.L.; Floyd, D.H.; Xiao, A.; Mullins, G.R.; Kefas, B.A.; Xin, W.; Yacur, M.N.; Abounader, R.; Lee, J.K.; Wilson, G.M.; et al. Diacylglycerol kinase α is a critical signaling node and novel therapeutic target in glioblastoma and other cancers. Cancer Discov. 2013, 3, 782–797. [Google Scholar] [CrossRef] [PubMed]
- Topham, M.K.; Epand, R.M. Mammalian diacylglycerol kinases: Molecular interactions and biological functions of selected isoforms. Biochim. Biophys. Acta 2009, 1790, 416–424. [Google Scholar] [CrossRef]
- Sakane, F.; Hoshino, F.; Murakami, C. New era of diacylglycerol kinase, phosphatidic acid and phosphatidic acid-binding protein. Int. J. Mol. Sci. 2020, 21, 6794. [Google Scholar] [CrossRef]
- Riese, M.J.; Moon, E.K.; Johnson, B.D.; Albelda, S.M. Diacylglycerol kinases (DGKs): Novel targets for improving T cell activity in cancer. Front. Cell Dev. Biol. 2016, 4, 108. [Google Scholar] [CrossRef] [PubMed]
- de Courcelles, D.D.C.; Roevens, P.; Van Belle, H. R 59022, a diacylglycerol kinase inhibitor. Its effect on diacylglycerol and thrombin-induced C kinase activation in the intact platelet. J. Biol. Chem. 1985, 260, 15762–15770. [Google Scholar] [CrossRef]
- Gómez-Merino, F.C.; Arana-Ceballos, F.A.; Trejo-Téllez, L.I.; Skirycz, A.; Brearley, C.A.; Dörmann, P.; Mueller-Roeber, B. Arabidopsis AtDGK7, the smallest member of plant diacylglycerol kinases (DGKs), displays unique biochemical features and saturates at low substrate concentration: The DGK inhibitor R59022 differentially affects AtDGK2 and AtDGK7 activity in vitro and alters plant growth and development. J. Biol. Chem. 2005, 280, 34888–34899. [Google Scholar]
- Tu-Sekine, B.; Goldschmidt, H.; Petro, E.; Raben, D.M. Diacylglycerol kinase θ: Regulation and stability. Adv. Biol. Regul. 2013, 53, 118–126. [Google Scholar] [CrossRef]
- Jiang, Y.; Sakane, F.; Kanoh, H.; Walsh, J.P. Selectivity of the diacylglycerol kinase inhibitor 3-[2-[4-[bis-(4-fluorophenyl)methylene]-1-piperidinyl]ethyl]-2,3-dihydro-2-thioxo-4(1H)quinazolinone (R59949) among diacylglycerol kinase subtypes. Biochem. Pharmacol. 2000, 59, 763–772. [Google Scholar] [CrossRef]
- Boroda, S.; Niccum, M.; Raje, V.; Purow, B.W.; Harris, T.E. Dual activities of ritanserin and R59022 as DGK-α inhibitors and serotonin receptor antagonists. Biochem. Pharmacol. 2017, 123, 29–39. [Google Scholar] [CrossRef]
- Kalna Bioscience and Gilead Sciences. Diacylglycerol Kinase Modulating Compounds. CN Patent 115362003 B, 22 March 2024.
- Takahashi, D.; Suzuki, K.; Sakamoto, T.; Iwamoto, T.; Murata, T.; Sakane, F. Crystal structure and calcium-induced conformational changes of diacylglycerol kinase α EF-hand domains. Protein Sci. 2019, 28, 694–706. [Google Scholar] [CrossRef]
- Chupak, L.; Wichroski, M.; Zheng, X.; Ding, M.; Martin, S.; Allard, C.; Shi, J.; Gentles, R.; Meanwell, N.A.; Fang, J.; et al. Discovery of potent, dual-inhibitors of diacylglycerol kinases alpha and zeta guided by phenotypic optimization. ACS Med. Chem. Lett. 2023, 14, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Martinez, K.; Shyamasundar, S.; Kennedy, A.; Chuang, C.C.; Marsh, A.; Kincaid, J.; Reid, T.; McIntosh, M. Diacylglycerol kinase inhibitor R59022 attenuates conjugated linoleic acid-mediated inflammation in human adipocytes. J. Lipid Res. 2013, 54, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, X.; Gan, J.; Chen, S.; Xiao, Z.X.; Cao, Y. CB-Dock2: Improved protein-ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res. 2022, 50, W284–W293. [Google Scholar] [CrossRef]
- Yang, X. CB-Dock2: Cavity Detection Guided Blind Docking. Available online: https://cadd.labshare.cn/cb-dock2/index.php (accessed on 7 January 2025).
- RCSB PDB—6IIE: Crystal Structure of Human Diacylglycerol Kinase Alpha EF-Hand Domains Bound to Ca2+. RCSB PDB: Homepage. Available online: https://www.rcsb.org/structure/6IIE (accessed on 17 February 2025).
- Shityakov, S.; Förster, C. In silico predictive model to determine vector-mediated transport properties for the blood–brain barrier choline transporter. Adv. Appl. Bioinform. Chem. 2014, 7, 23–36. [Google Scholar] [CrossRef] [PubMed]
- García-Ortegón, M.; Simm, G.N.C.; Tripp, A.J.; Hernández-Lobato, J.M.; Bender, A.; Bacallado, S. DOCKSTRING: Easy molecular docking yields better benchmarks for ligand design. J. Chem. Inf. Model. 2022, 62, 3486–3502. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- SwissADME. Available online: http://www.swissadme.ch/ (accessed on 16 May 2024).
- Banerjee, P.; Kemmler, E.; Dunkel, M.; Preissner, R. ProTox 3.0: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2024, 52, W440–W445. [Google Scholar] [CrossRef]
- ProTox-2 and Protox-3—Prediction of Toxicity of Chemicals. Available online: https://tox-new.charite.de/protox_II/index.php?site=home.-do (accessed on 11 May 2024).
- Janssen, P.A. Pharmacology of potent and selective S2-serotonergic antagonists. J. Cardiovasc. Pharmacol. 1985, 7 (Suppl 7), S2–S11. [Google Scholar] [CrossRef]
- Liu, K.; Kunii, N.; Sakuma, M.; Yamaki, A.; Mizuno, S.; Sato, M.; Sakai, H.; Kado, S.; Kumagai, K.; Kojima, H.; et al. A novel diacylglycerol kinase α-selective inhibitor, CU-3, induces cancer cell apoptosis and enhances immune response. J. Lipid Res. 2016, 57, 368–379. [Google Scholar] [CrossRef]
- Gupta, R.S.; Epand, R.M. Phylogenetic analysis of the diacylglycerol kinase family of proteins and identification of multiple highly-specific conserved inserts and deletions within the catalytic domain that are distinctive characteristics of different classes of DGK homologs. PLoS ONE 2017, 12, e0182758. [Google Scholar] [CrossRef]
- Zheng, Y.; Tice, C.M.; Singh, S.B. The use of spirocyclic scaffolds in drug discovery. Bioorg. Med. Chem. Lett. 2014, 24, 3673–3682. [Google Scholar] [CrossRef] [PubMed]
- Batista, V.F.; Pinto, D.C.G.A.; Silva, A.M.S. Recent in vivo advances of spirocyclic scaffolds for drug discovery. Expert Opin. Drug Discov. 2022, 17, 603–618. [Google Scholar] [CrossRef] [PubMed]
- Shabelnyk, K.; Antypenko, L.; Bohdan, N.; Ryzhenko, V.; Belenichev, I.; Kamyshnyi, O.; Oksenych, V.; Kovalenko, S. Search for potential neuroprotectors for correction of cognitive and behavioral disorders after ketamine anesthesia among 2’-R-6’H-spiro(cycloalkyl-, heterocyclyl)[1,2,4]triazolo[1,5-c]quinazolines: Fragment-oriented design, molecular docking, ADMET, synthesis and in vivo study. Front. Pharmacol. 2025; under submission. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Volume, Å3 | Center (x, y, z) | Docking Size (x, y, z) | Ritanserin | R59022 | R59949 | BMS502 | (5Z,2E)-CU-3 |
|---|---|---|---|---|---|---|---|
| 127 | −27.971, 12.861, −7.859 | 21, 21, 21 | −9.3 | −9.2 | −8.7 | −7.3 | −6.3 |
| 179 | −16.001, 4.140, 1.890 | 21, 21, 21 | −7.1 | −7.3 | −7.7 | −6.4 | −6.5 |
| 97 | −12.904, 16.082, −0.064 | 21, 21, 21 | −7.5 | −7.3 | −7.6 | −6.5 | −6.4 |
| 121 | −29.580, 15.955, 7.175 | 21, 21, 21 | −7.8 | −6.8 | −8.1 | −6.2 | −5.7 |
| 146 | −21.667, 10.869, 16.559 | 21, 21, 21 | −6.9 | −7.1 | −6.9 | −6.2 | −6.2 |
| Cavity Volumes | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 127 Å3 | 179 Å3 | 97 Å3 | 121 Å3 | 146 Å3 | |||||
| Sub. | kcal/mol | Sub. | kcal/mol | Sub. | kcal/mol | Sub. | kcal/mol | Sub. | kcal/mol |
| Ritans. | −9.3 | 18 | −8.5 | 40 | −8.2 | R59949 | −8.1 | 33 | −7.5 |
| R59022 | −9.2 | 28 | −8.3 | 22 | −8.1 | Ritans. | −7.8 | 11 | −7.3 |
| R59949 | −8.7 | 13 | −8.2 | 8 | −7.9 | 28 | −7.2 | 28 | −7.3 |
| 13 | −8.4 | 33 | −8.2 | 18 | −7.9 | 33 | −7.2 | 22 | −7.1 |
| 22 | −8.3 | 35 | −8.1 | 28 | −7.8 | 40 | −7.1 | R59022 | −7.1 |
| 33 | −8.3 | 10 | −7.9 | 35 | −7.8 | 11 | −7.0 | 18 | −6.9 |
| 35 | −8.1 | 11 | −7.9 | 30 | −7.7 | 13 | −6.9 | Ritans. | −6.9 |
| 28 | −8.0 | 22 | −7.9 | R59949 | −7.6 | 35 | −6.8 | R59949 | −6.9 |
| 11 | −7.9 | 40 | −7.9 | 4 | −7.5 | R59022 | −6.8 | 4 | −6.8 |
| 18 | −7.9 | 4 | −7.8 | 13 | −7.5 | 30 | −6.7 | 13 | −6.8 |
| 20 | −7.9 | 8 | −7.8 | Ritans. | −7.5 | 18 | −6.6 | 35 | −6.8 |
| 21 | −7.9 | 15 | −7.8 | 29 | −7.4 | 22 | −6.6 | 40 | −6.8 |
| 24 | −7.9 | 30 | −7.8 | 24 | −7.3 | 26 | −6.6 | 8 | −6.7 |
| 10 | −7.8 | 12 | −7.7 | 27 | −7.3 | 4 | −6.4 | 24 | −6.6 |
| 40 | −7.8 | 16 | −7.7 | R59022 | −7.3 | 8 | −6.4 | 26 | −6.6 |
| 23 | −7.7 | 17 | −7.7 | 33 | −7.2 | 10 | −6.3 | 30 | −6.6 |
| 26 | −7.7 | R59949 | −7.7 | 11 | −7.1 | 29 | −6.3 | 27 | −6.5 |
| 4 | −7.6 | 20 | −7.6 | 15 | −7.1 | 24 | −6.2 | 29 | −6.5 |
| 8 | −7.6 | 26 | −7.6 | 21 | −7.1 | 32 | −6.2 | 32 | −6.5 |
| 32 | −7.6 | 27 | −7.6 | 25 | −7.0 | BMS502 | −6.2 | 15 | −6.3 |
| 37 | −7.6 | 37 | −7.6 | 37 | −7 | 2 | −6.1 | 20 | −6.3 |
| 6 | −7.5 | 38 | −7.6 | 12 | −6.9 | 16 | −6 | 23 | −6.3 |
| 19 | −7.5 | 39 | −7.6 | 26 | −6.9 | 27 | −6 | 16 | −6.2 |
| 7 | −7.4 | 14 | −7.5 | 31 | −6.9 | 38 | −6 | 17 | −6.2 |
| 15 | −7.4 | 29 | −7.5 | 34 | −6.9 | 15 | −5.9 | 21 | −6.2 |
| 30 | −7.4 | 6 | −7.4 | 16 | −6.8 | 17 | −5.9 | BMS502 | −6.2 |
| 27 | −7.3 | 7 | −7.4 | 3 | −6.6 | 20 | −5.9 | CU-3 | −6.2 |
| 29 | −7.3 | 19 | −7.4 | 6 | −6.6 | 37 | −5.9 | 6 | −6.1 |
| BMS502 | −7.3 | 24 | −7.4 | 7 | −6.6 | 39 | −5.9 | 10 | −6.1 |
| 16 | −7.2 | 32 | −7.4 | 9 | −6.6 | 7 | −5.8 | 12 | −6.1 |
| 17 | −7.2 | 3 | −7.3 | 14 | −6.6 | 34 | −5.8 | 37 | −6.1 |
| 3 | −7.1 | 5 | −7.3 | 17 | −6.6 | 6 | −5.7 | 38 | −6.1 |
| 5 | −7.1 | 9 | −7.3 | 23 | −6.6 | 21 | −5.7 | 39 | −6.1 |
| 12 | −7.1 | 21 | −7.3 | 38 | −6.6 | 25 | −5.7 | 14 | −6 |
| 25 | −7.1 | 25 | −7.3 | 10 | −6.5 | 36 | −5.7 | 25 | −5.9 |
| 38 | −7.1 | 34 | −7.3 | 36 | −6.5 | CU-3 | −5.7 | 31 | −5.9 |
| 1 | −7.0 | 36 | −7.3 | 39 | −6.5 | 9 | −5.6 | 34 | −5.9 |
| 2 | −7.0 | R59022 | −7.3 | BMS502 | −6.5 | 12 | −5.6 | 2 | −5.8 |
| 14 | −7 | 2 | −7.2 | 1 | −6.4 | 14 | −5.6 | 7 | −5.8 |
| 39 | −7 | 31 | −7.1 | 20 | −6.4 | 19 | −5.6 | 19 | −5.8 |
| 34 | −6.9 | Ritans. | −7.1 | CU-3 | −6.4 | 23 | −5.6 | 36 | −5.8 |
| 9 | −6.8 | 1 | −7.0 | 5 | −6.3 | 31 | −5.5 | 1 | −5.7 |
| 36 | −6.7 | 23 | −7.0 | 19 | −6.3 | 5 | −5.4 | 3 | −5.7 |
| 31 | −6.6 | CU-3 | −6.5 | 32 | −6.2 | 1 | −5.2 | 5 | −5.7 |
| CU-3 | −6.3 | BMS502 | −6.4 | 2 | −6 | 3 | −5.2 | 9 | −5.6 |
| Amino Acid Residue | Distance, Å3 | Bond Category | Bond Type |
|---|---|---|---|
| Ritanserin in cavity 127 Å3 | |||
| GLU166 | 5.29773 | Electrostatic | Attractive Charge |
| ALA146 | 3.65885 | Hydrogen Bond; Halogen | Conventional Hydrogen Bond; Halogen (Fluorine) |
| MET142 | 3.19895 | Halogen | Halogen (Fluorine) |
| ASP152 | 3.68042 | Halogen | Halogen (Fluorine) |
| LEU193 | 2.63495 | Halogen | Halogen (Fluorine) |
| ALA146 | 3.9193 | Hydrophobic | Pi–Sigma |
| MET163 | 3.94403 | Hydrophobic | Pi–Sigma |
| LEU193 | 3.93009 | Hydrophobic | Pi–Sigma |
| MET163 | 3.77279 | Other | Pi–Sulfur |
| TRP151 | 4.06681 | Hydrophobic | Pi–Pi Stacked |
| TRP151 | 4.54522 | Hydrophobic | Pi–Pi Stacked |
| LEU156 | 5.35624 | Hydrophobic | Alkyl |
| LEU156 | 5.08272 | Hydrophobic | Pi–Alkyl |
| LEU156 | 5.06585 | Hydrophobic | Pi–Alkyl |
| VAL188 | 4.92692 | Hydrophobic | Pi–Alkyl |
| VAL188 | 4.81405 | Hydrophobic | Pi–Alkyl |
| Substance 3 in cavity 127 Å3 | |||
| TRP151 | 3.05971 | Hydrogen Bond | Conventional Hydrogen Bond |
| LEU193 | 3.8508 | Hydrophobic | Pi–Sigma |
| TRP151 | 3.94194 | Hydrophobic | Pi–Pi Stacked |
| TRP151 | 5.00543 | Hydrophobic | Pi–Pi Stacked |
| TRP151 | 5.31489 | Hydrophobic | Pi–Pi Stacked |
| TRP151 | 4.69541 | Hydrophobic | Pi–Alkyl |
| LEU156 | 5.44551 | Hydrophobic | Pi–Alkyl |
| LEU156 | 5.2451 | Hydrophobic | Pi–Alkyl |
| ALA146 | 4.39575 | Hydrophobic | Pi–Alkyl |
| LEU156 | 5.32117 | Hydrophobic | Pi–Alkyl |
| ALA146 | 4.32019 | Hydrophobic | Pi–Alkyl |
| R59949 in cavity 179 Å3 | |||
| GLU162 | 4.64611 | Electrostatic | Attractive Charge |
| ASP136 | 3.18667 | Halogen | Halogen (Fluorine) |
| ASP168 | 3.5015 | Halogen | Halogen (Fluorine) |
| GLY173 | 3.68482 | Halogen | Halogen (Fluorine) |
| SER174 | 3.40189 | Halogen | Halogen (Fluorine) |
| LYS165 | 4.83406 | Hydrophobic | Alkyl |
| VAL135 | 4.64342 | Hydrophobic | Pi–Alkyl |
| Substance 18 in cavity 179 Å3 | |||
| SER132 | 2.12242 | Hydrogen Bond | Conventional Hydrogen Bond |
| ASP136 | 3.67142 | Electrostatic | Pi–Anion |
| ASP136 | 3.69271 | Electrostatic | Pi–Anion |
| LYS165 | 4.12278 | Hydrophobic | Alkyl |
| VAL135 | 4.17437 | Hydrophobic | Pi–Alkyl |
| VAL135 | 4.98862 | Hydrophobic | Pi–Alkyl |
| R59949 in cavity 97 Å3 | |||
| GLU166 | 4.80272 | Electrostatic | Attractive Charge |
| ARG182 | 3.12515 | Hydrogen Bond; Halogen | Conventional Hydrogen Bond; Halogen (Fluorine) |
| GLU166 | 3.7357 | Hydrogen Bond | Conventional Hydrogen Bond |
| GLU166 | 3.22464 | Hydrogen Bond | Carbon Hydrogen Bond |
| ILE167 | 3.31873 | Halogen | Halogen (Fluorine) |
| GLU179 | 3.25959 | Halogen | Halogen (Fluorine) |
| MET163 | 3.82172 | Other | Pi–Sulfur |
| MET163 | 3.91728 | Other | Pi–Sulfur |
| LEU193 | 5.31213 | Hydrophobic | Pi–Alkyl |
| ARG182 | 5.20158 | Hydrophobic | Pi–Alkyl |
| ALA183 | 4.29659 | Hydrophobic | Pi–Alkyl |
| Substance 40 in cavity 97 Å3 | |||
| GLU166 | 1.85385 | Hydrogen Bond | Conventional Hydrogen Bond |
| GLU166 | 3.92296 | Electrostatic | Pi–Anion |
| THR187 | 3.27552 | Hydrogen Bond | Pi–Donor Hydrogen Bond |
| MET163 | 3.86247 | Hydrophobic | Pi–Sigma |
| ALA183 | 3.7804 | Hydrophobic | Pi–Sigma |
| MET163 | 4.04032 | Other | Pi–Sulfur |
| ARG182, ALA183 | 4.76159 | Hydrophobic | Amide–Pi Stacked |
| ILE167 | 5.38614 | Hydrophobic | Pi–Alkyl |
| ARG182 | 4.85254 | Hydrophobic | Pi–Alkyl |
| VAL188 | 4.54636 | Hydrophobic | Pi–Alkyl |
| VAL188 | 5.13921 | Hydrophobic | Pi–Alkyl |
| R59949 in cavity 121 Å3 | |||
| TYR122 | 4.03824 | Hydrogen Bond | Pi–Donor Hydrogen Bond |
| TYR122 | 3.51351 | Hydrogen Bond | Pi–Donor Hydrogen Bond |
| TYR122 | 4.19688 | Hydrophobic | Pi–Pi Stacked |
| TYR148 | 3.86497 | Hydrophobic | Pi–Pi Stacked |
| LEU121 | 5.26796 | Hydrophobic | Pi–Alkyl |
| VAL145 | 4.71276 | Hydrophobic | Pi–Alkyl |
| Substance 28 in cavity 121 Å3 | |||
| GLN141 | 3.15959 | Hydrogen Bond | Conventional Hydrogen Bond |
| VAL145 | 3.68904 | Hydrogen Bond | Carbon Hydrogen Bond |
| ARG144 | 4.65278 | Electrostatic | Pi–Cation |
| GLN141 | 3.87404 | Hydrogen Bond | Pi–Donor Hydrogen Bond |
| VAL145 | 3.69747 | Hydrophobic | Pi–Sigma |
| VAL145 | 5.03024 | Hydrophobic | Alkyl |
| LEU149 | 5.09337 | Hydrophobic | Alkyl |
| LEU149 | 5.04472 | Hydrophobic | Alkyl |
| ARG144 | 3.97846 | Hydrophobic | Pi–Alkyl |
| ARG144 | 4.07433 | Hydrophobic | Pi–Alkyl |
| R59022 in cavity 146 Å3 | |||
| THR124 | 2.99368 | Hydrogen Bond; Halogen | Conventional Hydrogen Bond; Halogen (Fluorine) |
| LEU121 | 3.16241 | Hydrogen Bond | Carbon Hydrogen Bond |
| intermolecular | 3.25258 | Hydrogen Bond | Carbon Hydrogen Bond |
| TYR122 | 3.48925 | Hydrogen Bond | Carbon Hydrogen Bond |
| LYS137 | 4.93829 | Electrostatic | Pi–Cation |
| THR124 | 4.06017 | Hydrogen Bond | Pi–Donor Hydrogen Bond |
| LEU121 | 3.60325 | Hydrophobic | Pi–Sigma |
| LEU121 | 3.94699 | Hydrophobic | Pi–Sigma |
| THR124 | 3.40756 | Hydrophobic | Pi–Sigma |
| LYS137 | 3.22303 | Hydrophobic | Pi–Sigma |
| LYS137 | 4.00354 | Hydrophobic | Pi–Alkyl |
| Substance 33 in cavity 146 Å3 | |||
| ARG126 | 3.19588 | Hydrogen Bond | Conventional Hydrogen Bond |
| LYS120 | 3.8771 | Hydrophobic | Alkyl |
| ARG126 | 4.81701 | Hydrophobic | Alkyl |
| ARG126 | 4.62315 | Hydrophobic | Alkyl |
| LYS120 | 4.5638 | Hydrophobic | Pi–Alkyl |
| ARG126 | 4.95772 | Hydrophobic | Pi–Alkyl |
| Sub | Lipinski | Ghose | Veber | Egan | Muegge |
|---|---|---|---|---|---|
| 1–10, 12–19, 21, 23, 24, 31–40 | Yes; 0 violations | Yes | Yes | Yes | Yes |
| 11, 25, 29 | Yes; 1 violation: MLOGP > 4.15 | No; 1 violation: XLOGP3 > 5 | |||
| 20, 22, 27, 30 | Yes; 0 violations | No; 1 violation: XLOGP3 > 5 | |||
| 26 | Yes; 1 violation: MLOGP > 4.15 | No; 1 violation: WLOGP > 5.6 | No; 1 violation: XLOGP3 > 5 | ||
| 28 | Yes; 1 violation: MLOGP > 4.15 | No; 1 violation: WLOGP > 5.6 | No; 1 violation: WLOGP > 5.88 | No; 1 violation: XLOGP3 > 5 | |
| BMS502 | Yes; 1 violation: MW > 500 | No; 2 violations: MW > 480, MR > 130 | Yes | ||
| R59022 | Yes; 1 violation: MLOGP > 4.15 | No; 1 violation: MR > 130 | Yes | No; 1 violation: XLOGP3 > 5 | |
| R59949 | Yes; 1 violation: MLOGP > 4.1 | No; 3 violations: MW > 480, WLOGP > 5.6, MR > 130 | Yes | No; 1 violation: WLOGP > 5.88 | No; 1 violation: XLOGP3 > 5 |
| Ritanserin | Yes; 1 violation: MLOGP > 4.15 | No; 2 violations: WLOGP > 5.6, MR > 130 | Yes | No; 1 violation: WLOGP > 5.88 | No; 1 violation: XLOGP3 > 5 |
| Cavity | Main Pat. Scaffold | p453 | 13 | 18 | 33 | 40 |
|---|---|---|---|---|---|---|
| Vina scores, kcal/mol | ||||||
| 179 Å3 | −6.2 | −7.1 | −8.2 | −8.5 | −8.2 | −7.9 |
| 146 Å3 | −5.7 | −7.5 | −8.4 | −7.9 | −8.3 | −7.8 |
| 127 Å3 | −5.3 | −7.0 | −7.5 | −7.9 | −7.2 | −8.2 |
| 121 Å3 | −4.8 | −6.6 | −6.8 | −6.9 | −7.5 | −6.8 |
| 97 Å3 | −4.6 | −6.6 | −6.9 | −6.6 | −7.2 | −7.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antypenko, L.; Shabelnyk, K.; Antypenko, O.; Arisawa, M.; Kamyshnyi, O.; Oksenych, V.; Kovalenko, S. In Silico Identification and Characterization of Spiro[1,2,4]triazolo[1,5-c]quinazolines as Diacylglycerol Kinase α Modulators. Molecules 2025, 30, 2324. https://doi.org/10.3390/molecules30112324
Antypenko L, Shabelnyk K, Antypenko O, Arisawa M, Kamyshnyi O, Oksenych V, Kovalenko S. In Silico Identification and Characterization of Spiro[1,2,4]triazolo[1,5-c]quinazolines as Diacylglycerol Kinase α Modulators. Molecules. 2025; 30(11):2324. https://doi.org/10.3390/molecules30112324
Chicago/Turabian StyleAntypenko, Lyudmyla, Kostiantyn Shabelnyk, Oleksii Antypenko, Mieko Arisawa, Oleksandr Kamyshnyi, Valentyn Oksenych, and Serhii Kovalenko. 2025. "In Silico Identification and Characterization of Spiro[1,2,4]triazolo[1,5-c]quinazolines as Diacylglycerol Kinase α Modulators" Molecules 30, no. 11: 2324. https://doi.org/10.3390/molecules30112324
APA StyleAntypenko, L., Shabelnyk, K., Antypenko, O., Arisawa, M., Kamyshnyi, O., Oksenych, V., & Kovalenko, S. (2025). In Silico Identification and Characterization of Spiro[1,2,4]triazolo[1,5-c]quinazolines as Diacylglycerol Kinase α Modulators. Molecules, 30(11), 2324. https://doi.org/10.3390/molecules30112324