Abstract
Makaluvamine J, a pyrroloiminoquinone alkaloid of marine sponge origin, and its analogs were synthesized and assessed for their potential to develop as a novel and selective growth inhibitor targeting human pancreatic cancer PANC-1 cells. Ts-damirone B, a common precursor featuring a pyrroloiminoquinone core structure, was synthesized through Bartoli indole synthesis and IBX-mediated oxidation. Late-stage diversification at N-5 and N-9 yielded makaluvamine J and several analogs. A structure–activity relationship (SAR) analysis highlighted the significance of the lipophilic side chain at N-9 for the growth inhibitory activity of PANC-1 cells. The modest alkyl group at N-5 was found to improve selectivity against other cancer cells. Among the prepared analogs, the tryptamine analog 24 showed potent and selective cytotoxicity (IC50 = 0.029 µM, selective index = 13.1), exceeding those of natural products.
1. Introduction
Marine natural products are considered rich and promising sources of drug candidates in the field of anticancer drug discovery [1,2]. Our team has concentrated on discovering novel antitumor compounds and previously developed a screening system to identify selective growth inhibitors of cancer cells under glucose-deprived conditions from marine organisms and marine-derived microorganisms [3]. For instance, novel chlorinated polyketides designated as biakamides A–D were isolated as potent and selective growth inhibitors of human pancreatic cancer PANC-1 cells under glucose-deprived conditions from the extract of marine sponge Petrosaspongia sp. Mechanistic studies have uncovered that biakamides inhibit the mitochondrial respiratory chain complex I, leading to reduced ATP synthesis in PANC-1 cells [4,5]. Many other compounds exhibiting similar bioactivity, so-called “anti-austerity agents”, have been isolated through similar bioassay-guided separation [6,7,8,9,10]. Of these, GBS-01, an extract of A. lappa containing high levels of arctigenin, has undergone testing in a phase I clinical trial for patients with advanced pancreatic cancer resistant to gemcitabine [11]. This example indicates the efficacy of natural products in the development of new drug candidates against pancreatic cancer.
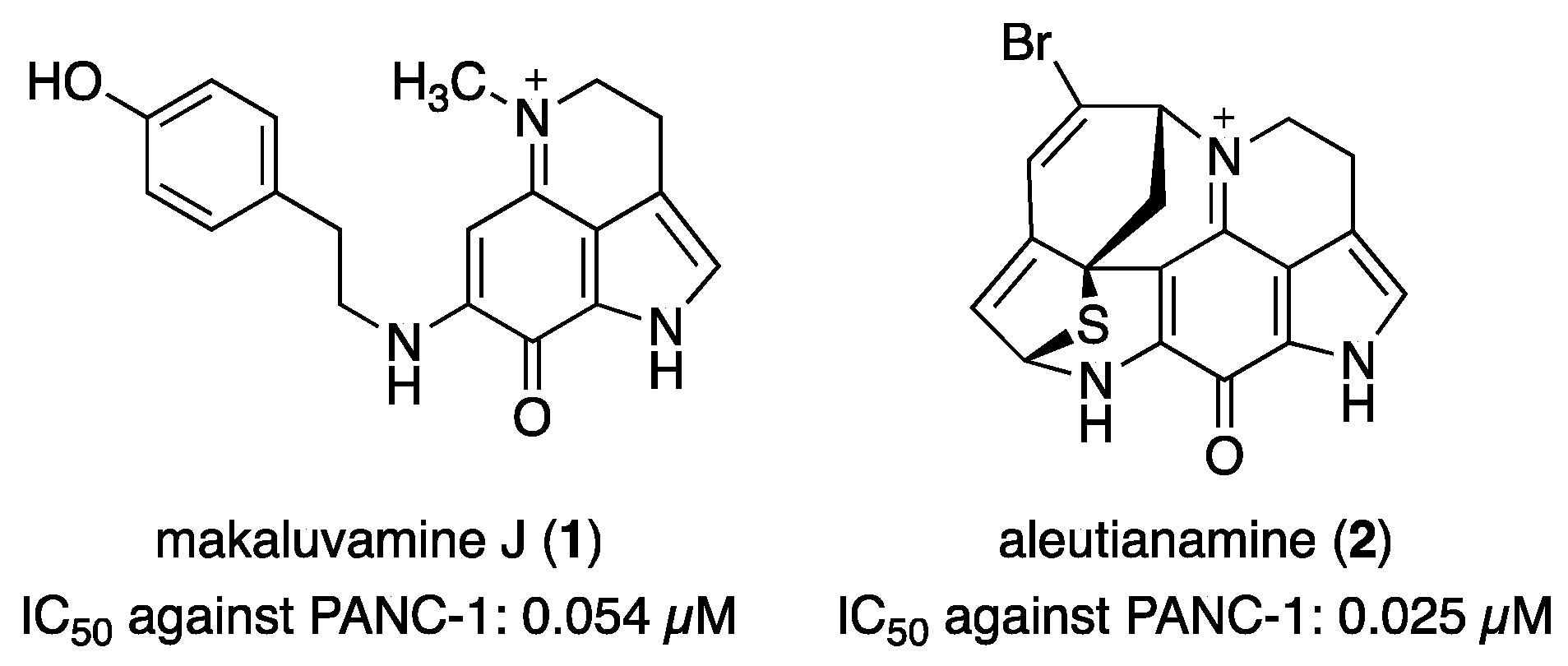
Clinically, pancreatic cancer is one of the most difficult cancers to treat, and no effective chemotherapeutic agents exist [12]. Therefore, the discovery of potent and selective growth inhibitors for pancreatic cancer cells is ongoing. Some pyrroloiminoquinone alkaloids are known to exhibit potent cytotoxicity against PANC-1 cells (Figure 1). Makaluvamine J (1) is one of the most potent active substances, with an IC50 value of 0.054 µM against PANC-1 cells [13]. Furthermore, aleutianamine (2), a novel heptacyclic pyrroloiminoquinone alkaloid obtained from an Antarctic marine sponge, demonstrated selective cytotoxicity against PANC-1 cells (IC50 = 0.025 µM) [14]. The significance of the compounds is well demonstrated by comparing them with the potency of gemcitabine (IC50 = 4 µM), a first-line chemotherapeutic agent approved by the FDA [15].
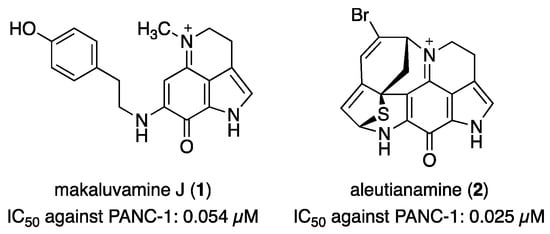
Figure 1.
The chemical structures of makaluvamine J (1) and aleutianamine (2).
These findings suggest that pyrroloiminoquinone with cationic nitrogen at N-5 could be a crucial scaffold for demonstrating potent cytotoxicity against pancreatic cancer cells. To investigate this hypothesis and develop more potent compounds as potential anti-pancreatic cancer drug candidates, we synthesized makaluvamines and their analogs. This report details the synthesis and evaluation of makaluvamine J (1) and its analogs at N-5 and C-7.
2. Results and Discussions
2.1. Reported Bioactivity and SAR of Makaluvamines against Pancreatic Cancer Cells
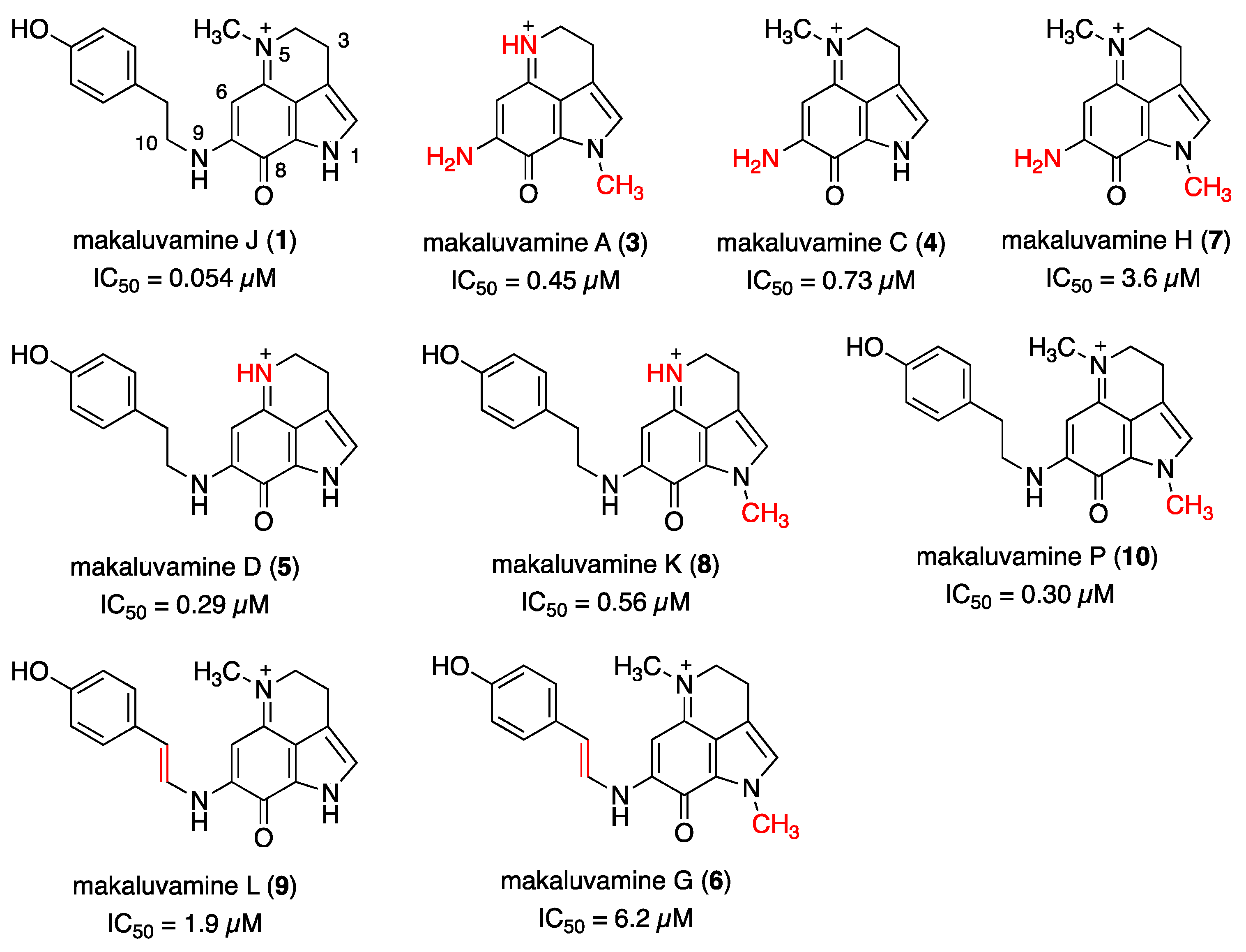
The reported structure–cytotoxicity relationship of makaluvamines in PANC-1 cells is outlined in Figure 2 [13]. The key structural feature is the substituent at N-9: makaluvamines A (3), C (4), and H (7), which lack a substituent, displayed less than one-eighth the cytotoxicity of 1 against PANC-1; meanwhile, makaluvamines G (6) and L (9) with E-alkenyl side chains exhibited relatively lower potency. Given these results and the potent cytotoxicity of aleutianamine (2), an appropriately oriented substituent at N-9 appears crucial. Conversely, demethylation of N-5 (makaluvamine D (5)) or methylation of N-1 (makaluvamine P (10)) led to approximately one-fifth less potency than compound 1, underscoring the significance of the substitution pattern at these positions.
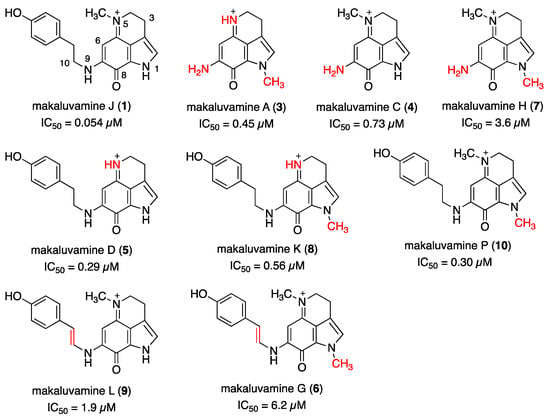
Figure 2.
The structures and bioactivities of makaluvamines (1, 3–10). Reported IC50 values of the respective compounds against PANC-1 cells are indicated. The structural differences from makaluvamine J (1) are highlighted in red.
2.2. Unified Divergent Synthesis of Makaluvamines and Analogs
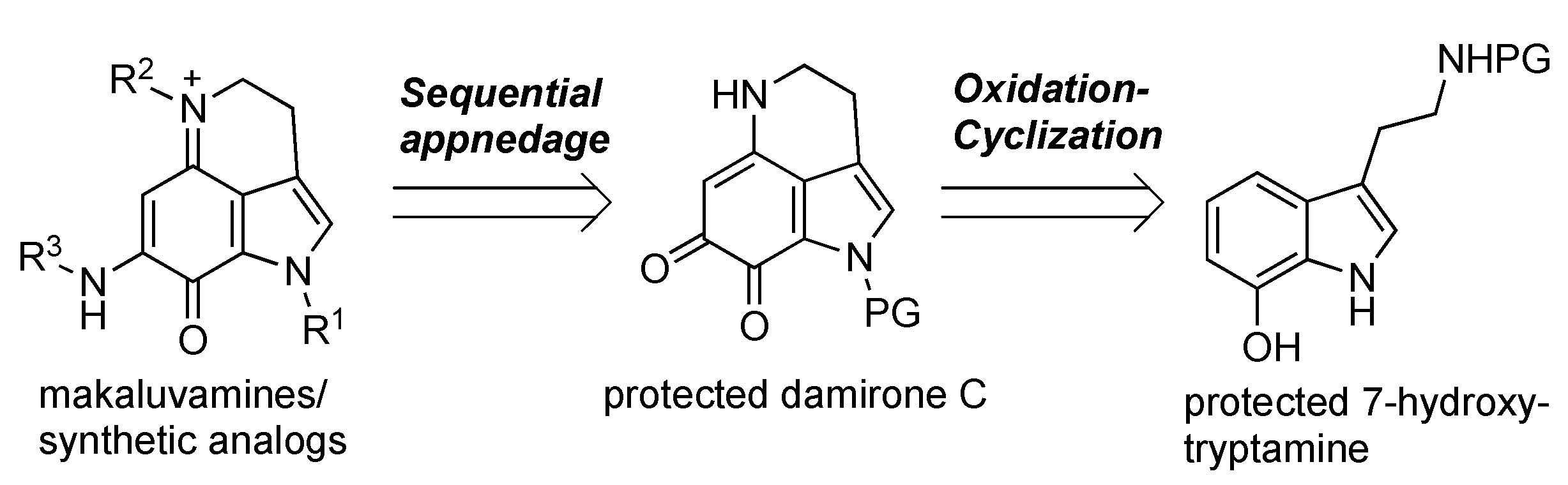
There have been a number of reports on the total synthesis of makaluvamines [16,17,18,19,20,21,22], although most of them have focused on the development of synthetic methods for the characteristic tricyclic skeleton. To gather more detailed SAR information, a unified, divergent synthesis of natural makaluvamines or various analogs and their biological evaluations is required. Scheme 1 shows an outline of our approach toward these molecules. Protected damirone C served as a common precursor, with various substituents at N-1, N-5, and N-9 appended at the final stage of the synthesis. The damirone skeleton was prepared from a 7-hydroxytryptamine derivative via oxidation/cyclization.
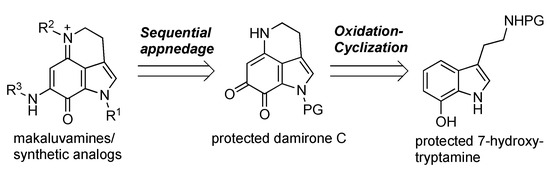
Scheme 1.
Outline of our synthetic approach toward makaluvamine derivatives.
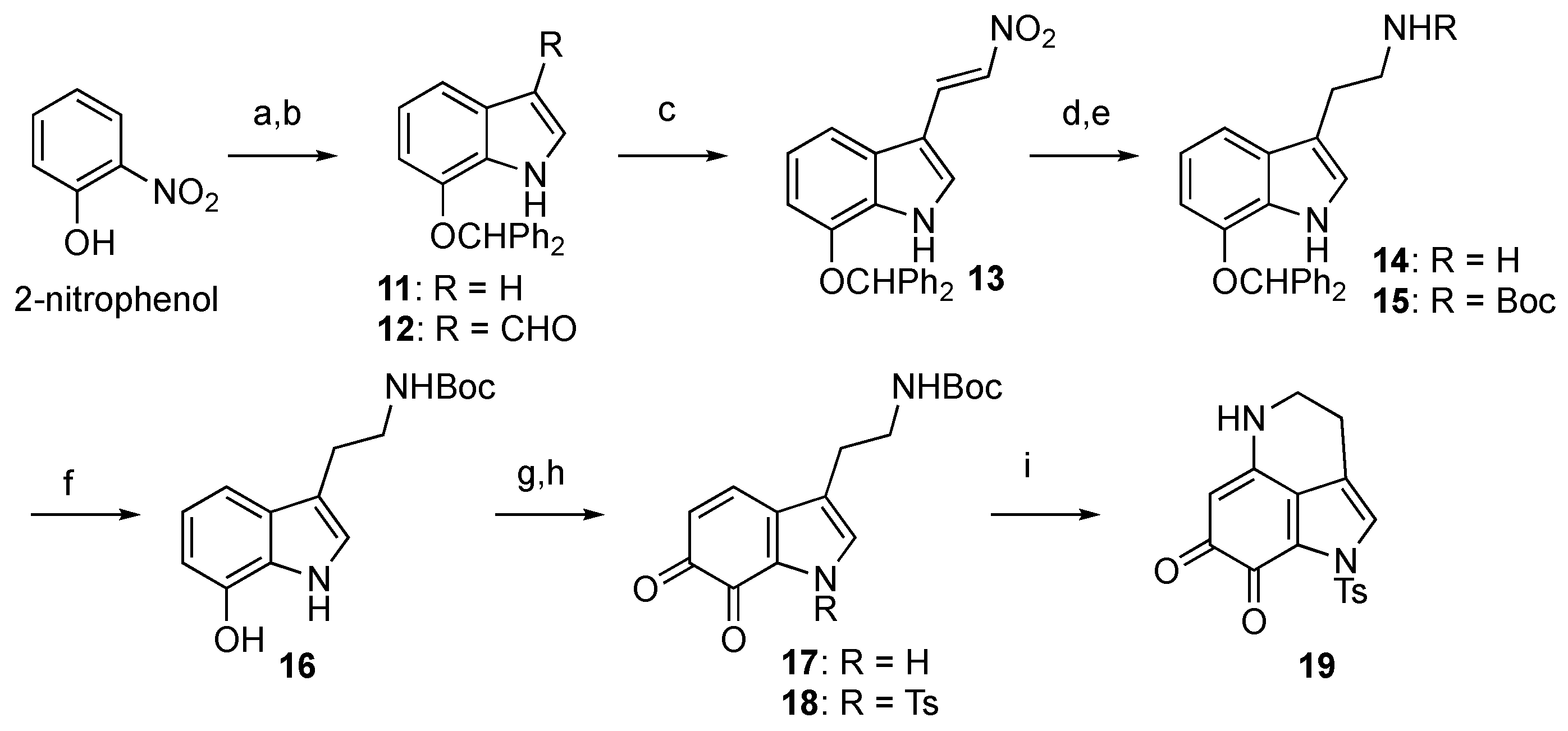
First, protected damirone C was synthesized as depicted in Scheme 2. As reported in the literature, 7-(benzhydryloxy)indole (11) was synthesized using Bartoli indole synthesis, commencing with 2-nitrophenol [23]. Then, 11 was converted into tryptamine derivative 16 as follows: regioselective introduction of the side chain was achieved through Vilsmeier–Haack formylation and a subsequent Henry reaction to give nitroalkene 13 in good yield. The reduction of nitroalkene 13 with LiAlH4 yielded amine 14, which was isolated as Boc carbamate 15 in moderate yield. Hydrogenolysis of the benzhydryl ether of 15 gave 7-hydroxytryptamine derivative 16, and subsequent oxidation to the corresponding ortho-quinone 17 was attempted. The use of 2-iodoxybenzoic acid (IBX) in N,N-dimethylformamide (DMF) [24,25] was found to be effective, although the reaction yield was moderate (<50%) with commercially available IBX (purchased from TCI Chemicals, >39% purity). However, when lab-made pure IBX was used, better results were obtained, with an isolated yield of 63% after the protection of the pyrrole nitrogen as p-toluenesulfonamide 18. The final steps involved intramolecular cyclization by removing the Boc group with TFA and subsequent Et3N treatment to yield the desired Ts-damirone C (19) in moderate yield.
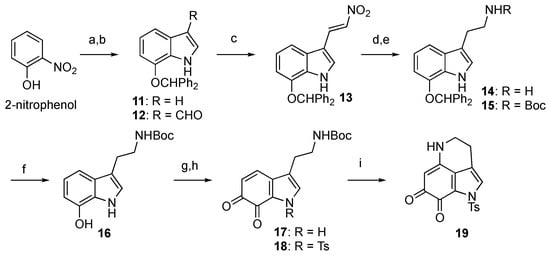
Scheme 2.
Synthesis of Ts-damirone C (19). Reagents and conditions: (a) Benzhydryl bromide, K2CO3, acetone, reflux; vinylmagnesium chloride, THF, –40 °C; (b) POCl3, DMF, 0 °C to rt, then KOH, H2O, 50 °C; (c) NH4OAc, CH3NO2, reflux, 86% (2 steps); (d) LiAlH4, THF, reflux; (e) (Boc)2O, Et3N, 1,4-dioxane, 55% (2 steps); (f) H2, Pd(OH)2-C, MeOH, 97%; (g) IBX, DMF, 0 °C; (h) TsCl, DMAP, Et3N, THF, 63% (2 steps); (i) TFA, CH2Cl2, 0 °C; Et3N, MeOH, 0 °C, 55%.
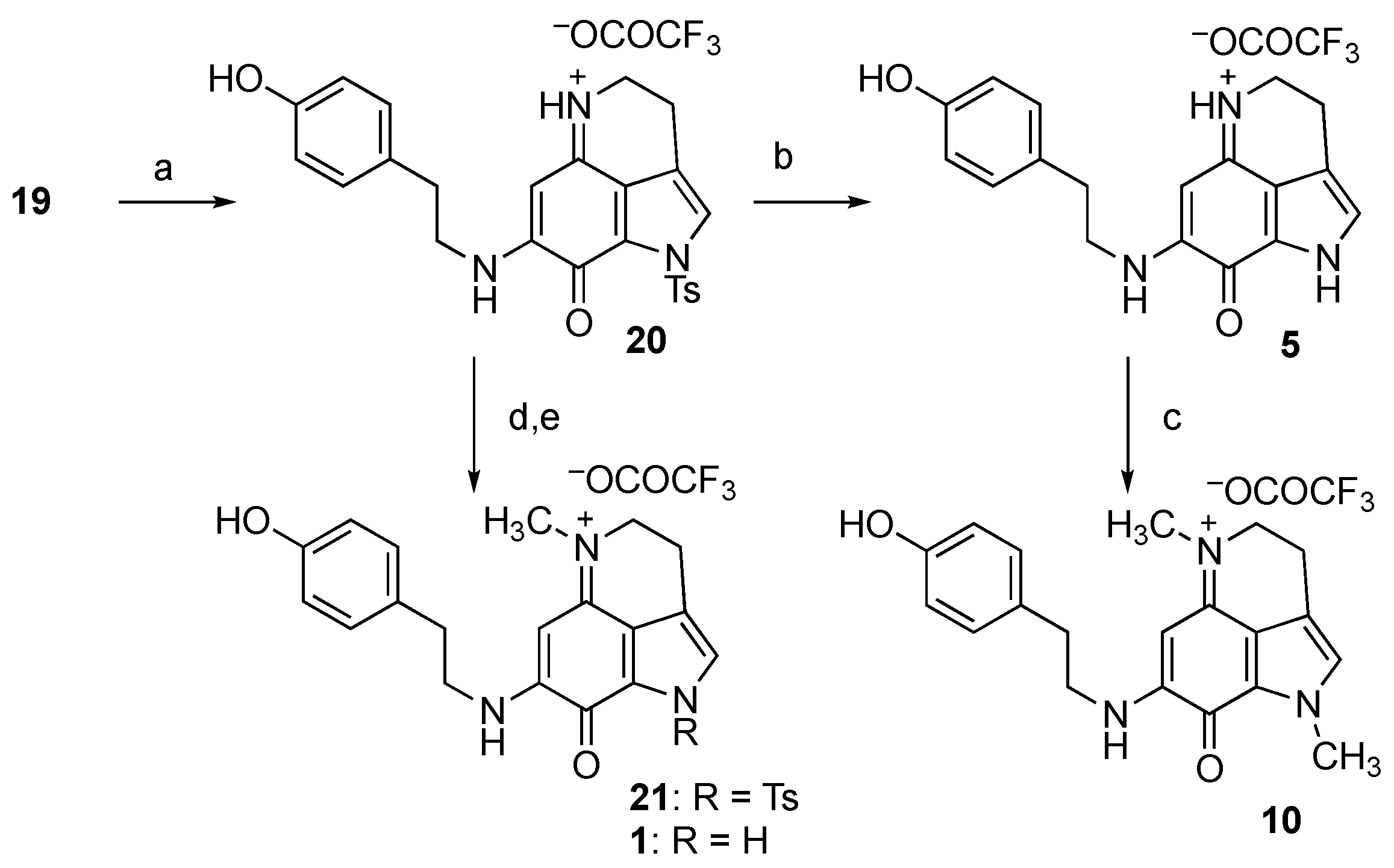
With the common precursor 19 in hand, the next step involved attempting to append the substituents at N-1, N-5, and N-9 for the total synthesis of makaluvamines, as shown in Scheme 3. The condensation with tyramine under basic conditions afforded Ts-makaluvamine D (20) in 49% yield. A total of 38% of the unreacted 19 was also isolated in this case, indicating that the yield based on the recovered starting material (brsm) was 87%. The various orders of transformation of N-1 and N-5 were then examined. In the first attempt, the Ts-protecting group was removed, followed by N-methylation to prepare makaluvamine P (10). Pyridine hydrochloride in DMF [26] was identified as the optimal reagent for removing the Ts group, yielding makaluvamine D (5) in a good yield. Other attempted detosylation conditions (Cs2CO3 in THF/MeOH [27], NaH in DMF [28], and TFA/Me2S [29]) were not effective. Double N-methylation at N-1 and N-5 occurred following treatment with iodomethane in the presence of NaH, resulting in the synthesis of makaluvamine P (10) in excellent yield.
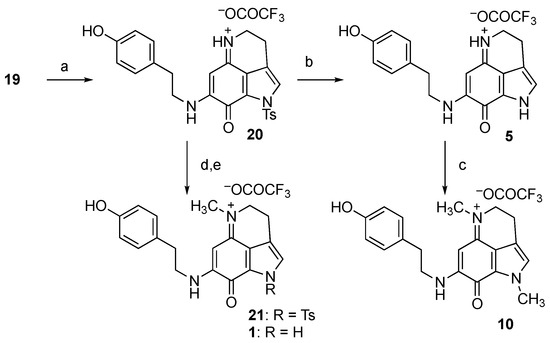
Scheme 3.
Attempted synthesis of makaluvamines. Reagents and conditions: (a) Tyramine, Et3N, MeOH, then TFA, 49%, 87% brsm; (b) pyridine hydrochloride, DMF, 80 °C, 70%; (c) MeI, NaH, DMF, 92%; (d) MeI, NaH, DMF; (e) pyridine hydrochloride, DMF, 80 °C.
The preparation of makaluvamine J (1) was attempted through N-methylation at N-5 and the subsequent removal of the Ts group, in reverse order under similar conditions as described earlier. However, the MS and 1H NMR spectra of the crude product indicated that the starting material was decomposed, and a mixture of unidentified compounds was obtained instead of 1.
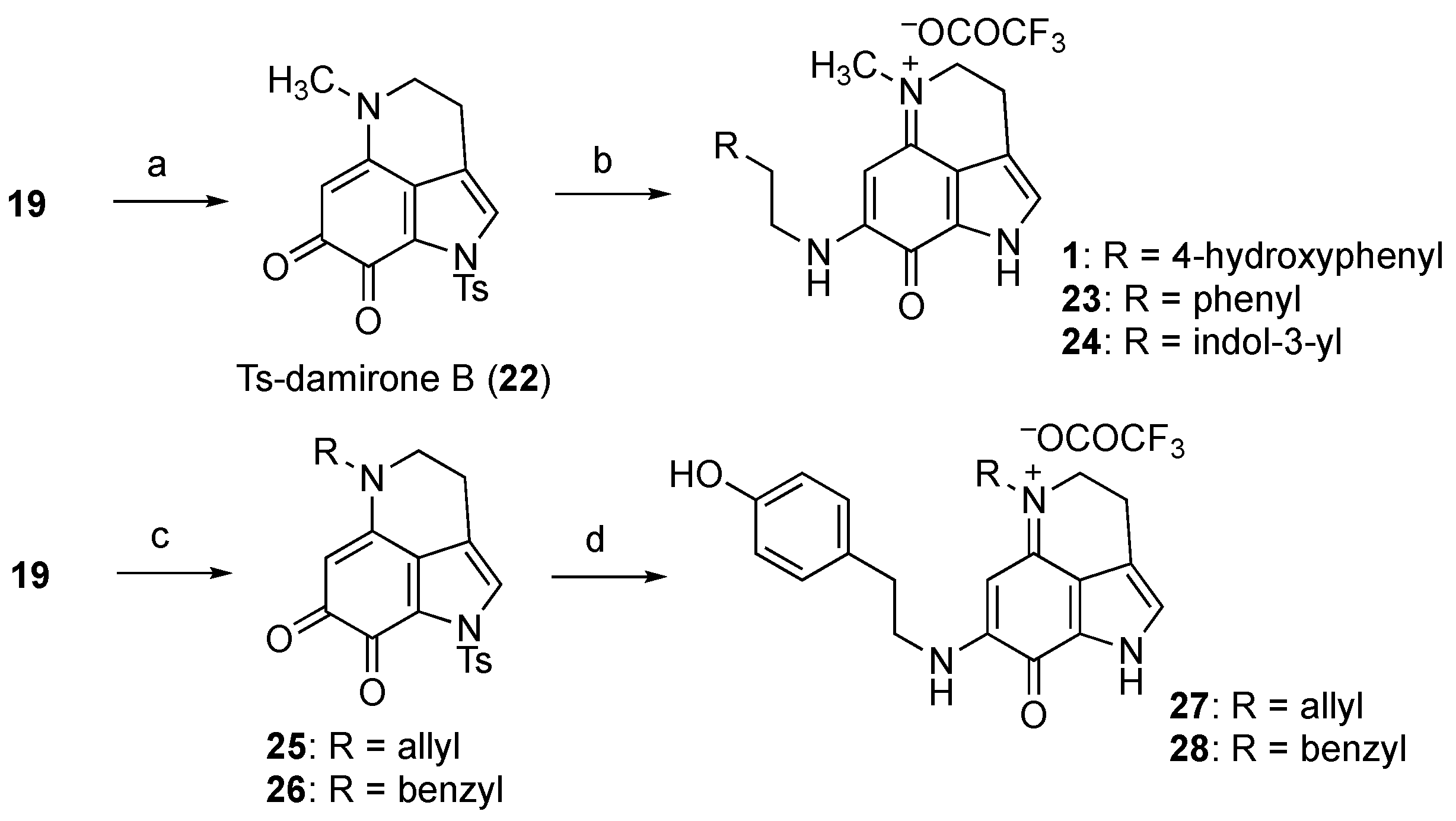
Considering these findings, we examined another synthetic route, as depicted in Scheme 4. Thus, the methylation of N-5 of 19 gave Ts-damirone B (22) in quantitative yield. Unexpectedly, the subsequent treatment of 22 with tyramine (three equiv.) yielded makaluvamine J (1) in its pure form. It appears that condensation with ketone at C-7 and deprotection of the Ts group at N-1 occurred simultaneously, suggesting that this order of the appendage might be optimal for synthesis. Indeed, the use of phenethylamine or tryptamine instead of tyramine provided the corresponding unnatural makaluvamine analogs 23 and 24, respectively.
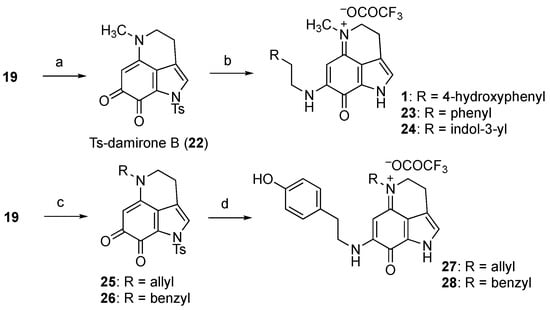
Scheme 4.
Synthesis of makaluvamine J (1) and analogs 23–28. Reagents and conditions: (a) MeI, K2CO3, DMF, quant.; (b) RCH2CH2NH2, Et3N, MeOH, 37%, 87% brsm for 1; 36%, 83% brsm for 23; 22%, 74% brsm for 24; (c) R-Br, K2CO3, DMF, 93% for 25; 77% for 26; (d) Tyramine, Et3N, MeOH, 22%, 89% brsm for 27; 17%, 76% brsm for 28.
Furthermore, we similarly obtained the N-5 analogs. Thus, 19 was treated with allyl bromide or benzyl bromide to give the corresponding damirone analogs 25 and 26, respectively. Subsequent condensation with tyramine and removal of the Ts group provided 27 and 28, which could be used to analyze the participation of the N-5 substituent of makaluvamines in cytotoxicity against pancreatic cancer cells.
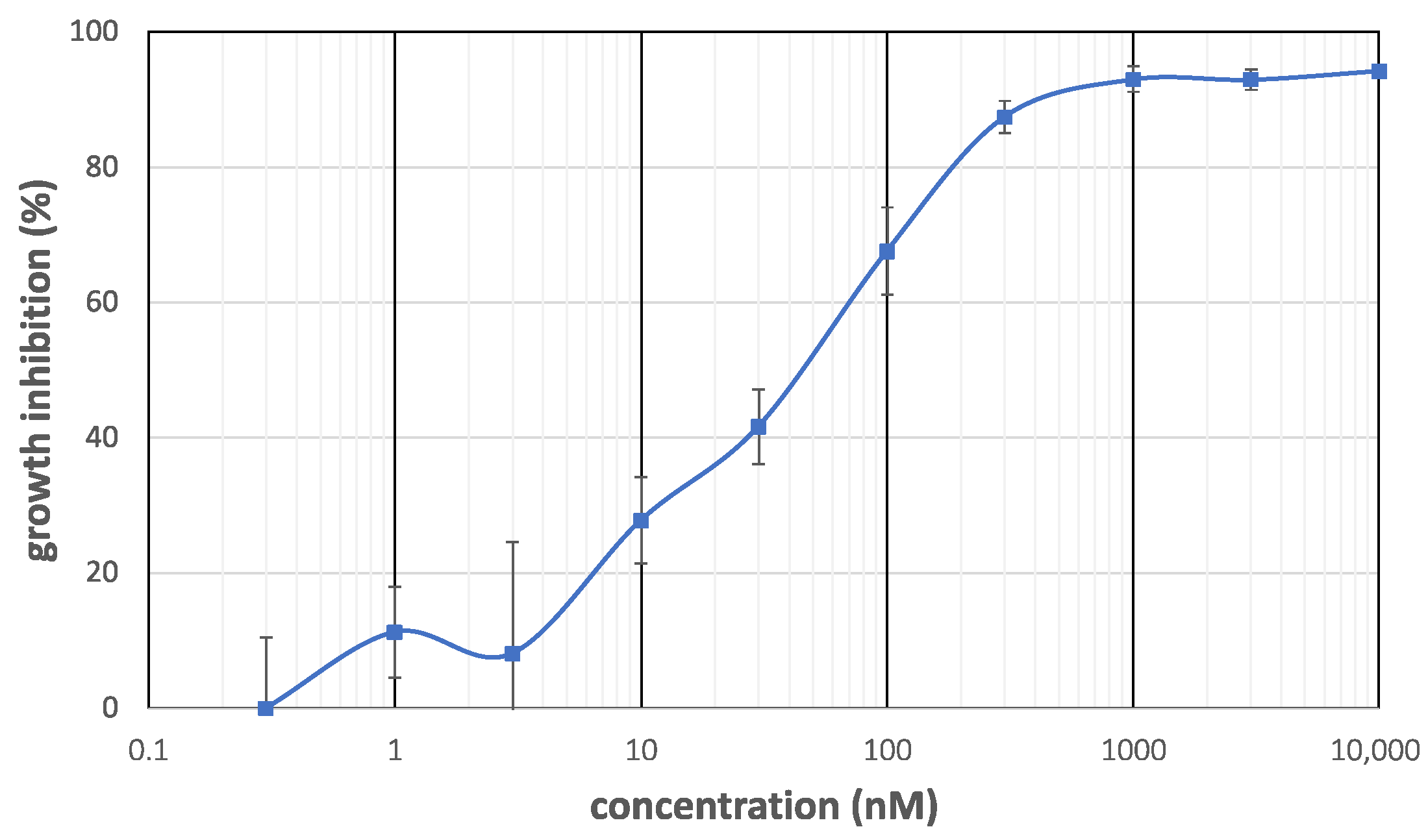
The cytotoxicity of makaluvamine J (1) and analogs 23–28 against PANC-1 cells and human epidermoid carcinoma KB3-1 cells was evaluated as summarized in Table 1. The selective index, the ratio of potency between these two cell lines, is also shown in Table 1 to clearly depict the selectivity. The growth inhibition curve of 1 against PANC-1 cells (Figure 3) shows that the cytotoxicity was raised in an almost linear and dose-dependent manner in the range between 3 and 300 nM, and the concentration of 50% inhibition (IC50) was determined to be 0.046 µM. It was almost comparable to the reported one [13] (IC50 = 0.054 µM), which ensured the validity of our experiment. It was also found that 1 exhibited relatively weak cytotoxicity against KB3-1 cells (IC50 = 0.2 µM), with a selective index of 4.3. Weaker cytotoxicity and lower cell selectivity of makaluvamines D (5) and P (10) also supported the reported SAR, the necessity of quaternary nitrogen at N-5, and the negative effect of N-1 alkylation. The makaluvamine analogs prepared in this work (23, 24, 27, and 28) also showed similar cytotoxicity against PANC-1 cells (0.027 to 0.042 µM), whereas significantly weakened cytotoxicity was observed with two damirone analogs (25 and 26). In addition, cytotoxicity testing against KB3-1 cells revealed an intriguing relationship between structure and cytotoxicity. Thus, N-9 analogs 23 and 24 showed 10-fold lower cytotoxicity in KB3-1 cells, whereas N-5 analogs 27 and 28 exhibited comparable activity. This indicates that the N-5 substituent might contribute to cancer cell selectivity, and the N-9 substituent could enhance nonselective cytotoxicity.

Table 1.
Cytotoxicity of makaluvamine J (1) and analogs against cancer cells.
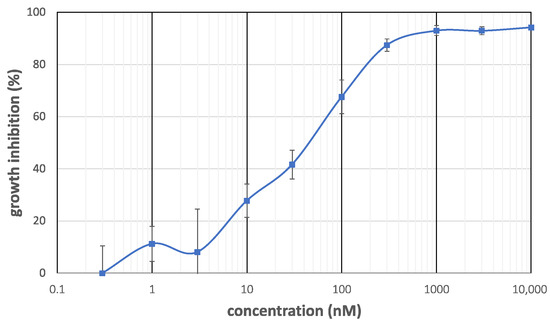
Figure 3.
Growth inhibition curve of makaluvamine J (1) against PANC-1 cells.
What surprised us the most was the potency of the analogs 23 and 24 against PANC-1 cells. The reported SAR showed that the 4-ethylphenol substituent is important for exhibiting potency against PANC-1 cells. On the other hand, analogs 23 and 24 possess 2-phenethyl and 2-(1H-indol-3-yl)ethyl moieties, respectively, instead of the 4-ethylphenol group. This indicates that the hydroxyl group at the side chain terminal is not necessary for exhibiting potent cytotoxicity. Although further studies are needed, the findings in this work are significant for optimizing the structural motif in this part.
In summary, the synthesis and biological evaluation of makaluvamine J (1) and its analogs revealed the importance of substituents N-5 and N-9 for cytotoxicity and pancreatic cancer cell selectivity. The unified synthetic method used in this study enabled easy access to makaluvamines and various analogs, yielding tryptamine analog 24 with more potent cytotoxicity against PANC-1 cells and enhanced selectivity over KB3-1 cells. Further synthesis and evaluation of various analogs will lead to the development of optimized anti-pancreatic cancer drug candidates. In addition, a mechanistic study of the compound, which will be undertaken in due course, will allow us to identify key molecules for overcoming pancreatic cancer.
3. Materials and Methods
3.1. General
The following instruments were used to obtain physical data: a JEOL JNM-ECZ500R/S1 (1H-NMR: 500 MHz, 13C-NMR: 125 MHz) spectrometer for 1H and 13C NMR data using tetramethylsilane as an internal standard; a Waters Xevo G2-XS Q-Tof mass spectrometer for ESI-Q-TOF MS. Silica gel (Kanto 63–210 μm) and pre-coated thin-layer chromatography (TLC) plates (Merck 60F254) were used for column chromatography and TLC. The spots on the TLC plates were detected by spraying with an acidic p-anisaldehyde solution (p-anisaldehyde: 25 mL, c-H2SO4: 25 mL, AcOH: 5 mL, EtOH: 425 mL) with subsequent heating. Unless otherwise noted, all reactions were performed under a N2 atmosphere. Hard copies of NMR spectra can be found as Supplementary Materials.
3.2. Antiproliferative Activity of the Compounds against Cancer Cells
PANC-1 and KB3-1 cells were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum and kanamycin (50 μg/mL). Cells were plated into 96-well microplates at 5 × 103 cells/100 μL assay medium/well, and various concentrations of test compounds were added to each well as a 10% DMSO/EtOH solution (1 µL). The plates were incubated at 37 °C in a humidified atmosphere of 5% CO2 for 72 h, and cell proliferation was determined by MTT colorimetric assay. IC50 values, drug concentrations that produced 50% cell death, were determined by interpolation of the plotted data (the averaged values of the triplicate experiment).
3.3. Synthesis
3.3.1. 7-(Benzhydryloxy)-1H-indole (11)
K2CO3 (30.3 g, 219 mmol, 1.6 equiv.) and benzhydryl bromide (33.9 g, 137 mmol, 1.0 equiv.) were added to a solution of 2-nitrophenol (19.0 g, 137 mmol) in acetone (350 mL), and the red mixture was stirred with reflux for 6 h. After cooling to rt, the reaction mixture was filtered and concentrated in vacuo. The residue was extracted with Et2O, and the precipitates were removed by filtration. The filtrate was again concentrated in vacuo, and the residue was triturated with hexane (80 °C). The resulting brown solid was isolated by filtration, washed with hexane, and dried in vacuo to afford 2-benzhydryloxynitrobenzene (29.1 g, 70%) as a brown solid.
Vinylmagnesium chloride solution (1.4 mol/L in THF, 100 mL, 140 mmol, 3.5 equiv.) was added dropwise over a few minutes to a solution of an aliquot of the above product (12.2 g, 40.0 mmol) in dry THF (500 mL) at −40 °C, and the mixture was stirred for 45 min. Then, the reaction mixture was quenched with sat. NH4Cl aq. and the whole mixture was extracted with Et2O three times. The combined organic layer was dried over Na2SO4 and was concentrated in vacuo. The crude brown oil was purified by silica gel column chromatography (hexane/EtOAc = 7:1) to give 11 (6.0 g, 50%) as a pale yellow solid.
All the spectral data were identical to the reported ones [23].
3.3.2. 7-(Benzhydryloxy)-1H-indole-3-carbaldehyde (12)
POCl3 (2.00 mL, 22.0 mmol, 1.1 equiv.) was added dropwise to dry DMF (20 mL) at 0 °C, and a solution of 11 (6.01 g, 20.0 mmol) in dry DMF (10 mL) was added dropwise to the reaction mixture. The resulting yellow mixture was allowed to warm up to rt with stirring for 3 h. After cooling to 0 °C, a solution of KOH (6.80 g) in H2O (30 mL) was added to the reaction mixture, and the whole mixture was stirred at 50 °C for 90 min. After cooling to rt, the resulting mixture was diluted with EtOAc, and the whole mixture was washed with H2O and brine. The organic layer was dried over Na2SO4 and was concentrated in vacuo to afford a crude 12 (5.98 g) as a brown oil. The residue was subjected to the next reaction without further purification.
A characterization sample could be obtained by silica gel column chromatography (hexane/EtOAc = 4:1) as a pale-yellow powder.
1H NMR (500 MHz, CDCl3): δ 9.97 (s, 1H), 9.56 (br s, 1H), 7.86 (d, J = 7.9 Hz, 1H), 7.73 (d, J = 2.9 Hz, 1H), 7.43 (d, J = 7.3 Hz, 4H), 7.34 (t, J = 7.4 Hz, 4H), 7.30 (t, J = 7.3 Hz, 2H), 7.08 (t, J = 8.0 Hz, 1H), 6.70 (d, J = 7.9 Hz, 1H), 6.40 (s, 1H). 13C NMR (125 MHz, CDCl3): δ 185.6, 144.6, 140.7, 135.1, 128.8, 128.2, 127.8, 127.1, 126.1, 123.7, 120.0, 114.7, 107.7, 82.5. MS (ESI-Q-TOF) m/z: 328 [M + H]+. HRMS (ESI-Q-TOF) m/z: 328.1338, calcd for C22H18NO2; found: 328.1354.
3.3.3. (E)-7-(Benzhydryloxy)-3-(2-nitrovinyl)-1H-indole (13)
Ammonium acetate (1.23 g, 22.0 mmol, 1.1 equiv.) was added to a solution of crude 12 (5.98 g, 18.3 mmol) in nitromethane (50 mL), and the red mixture was stirred with reflux for 2 h. After cooling to rt, the reaction mixture was concentrated in vacuo. The residue was diluted with CH2Cl2, and the whole mixture was washed with H2O and brine. The organic layer was dried over Na2SO4 and was concentrated in vacuo to a red solid. Recrystallization from CH2Cl2 was performed to afford pure 13 (6.43 g, 86% over 2 steps) as an orange solid.
1H NMR (500 MHz, (CD3)2CO): δ 11.71 (br s, 1H), 8.37 (d, J = 13.4 Hz, 1H), 8.14 (d, J = 3.0 Hz, 1H), 7.87 (d, J = 13.4 Hz, 1H), 7.62 (d, J = 7.4 Hz, 4H), 7.48 (d, J = 8.0 Hz, 1H), 7.36 (t, J = 7.5 Hz, 4H), 7.27 (t, J = 7.4 Hz, 2H), 7.09 (t, J = 8.0 Hz, 1H), 6.91 (d, J = 8.0 Hz, 1H), 6.73 (1H, s). 13C NMR (125 MHz, (CD3)2CO): δ 145.7, 142.5, 135.1, 134.8, 132.9, 129.6, 129.4, 128.6, 127.6, 123.5, 114.0, 110.2, 108.0, 81.9. MS (ESI-Q-TOF) m/z: 371 [M + H]+. HRMS (ESI-Q-TOF) m/z: 371.1396, calcd for C23H19N2O3; found: 371.1391.
3.3.4. 2-(7-(Benzhydryloxy)-1H-indol-3-yl)ethan-1-amine (14)
LiAlH4 (3.98 g, 104 mmol, 6.0 equiv.) was carefully added to a solution of 13 (6.43 g, 17.4 mmol) in dry THF (90 mL) at 0 °C, and the green mixture was stirred with reflux for 3 h. After cooling to 0 °C, the reaction mixture was quenched by slow addition of sat. Na2SO4 aq. (90 mL). The reaction mixture was passed through a pad of Celite and was washed with EtOAc. Brine was added to the filtrate and extracted with EtOAc twice. The organic layer was dried over Na2SO4 and was concentrated in vacuo to afford a crude 14 (5.43 g) as a pale yellow solid. The residue was subjected to the next reaction without further purification.
A characterization sample could be obtained by silica gel column chromatography (hexane/EtOAc = 4:1) as a white solid.
1H NMR (500 MHz, CDCl3): δ 9.04 (br s, 1H), 7.46 (d, J = 7.3 Hz, 4H), 7.37 (t, J = 7.3 Hz, 4H), 7.32 (t, J = 7.3 Hz, 2H), 7.21 (d, J = 7.9 Hz, 1H), 6.96 (s, 1H), 6.92 (t, J = 7.9 Hz, 1H), 6.59 (d, J = 7.7 Hz, 1H), 6.41 (s, 1H), 2.99 (t, J = 6.6 Hz, 2H), 2.88 (t, J = 6.6 Hz, 2H), 1.59 (br s, 2H). 13C NMR (125 MHz, CDCl3): δ 144.8, 141.2, 129.1, 128.7, 127.9, 127.5, 127.1, 121.9, 119.5, 113.9, 112.0, 105.0, 82.1, 42.3, 29.5. MS (ESI-Q-TOF) m/z: 343 [M + H]+. HRMS (ESI-Q-TOF) m/z: 343.1810, calcd for C23H23N2O; found: 343.1815.
3.3.5. tert-Butyl (2-(7-(benzhydryloxy)-1H-indol-3-yl)ethyl)carbamate (15)
Et3N (5.51 mL, 39.5 mmol) and (Boc)2O (3.75 mL, 17.4 mmol) were added dropwise to a suspended solution of crude 14 (5.43 g) in 1,4-dioxane (30 mL), and the whole mixture was stirred for 30 min. The reaction mixture was concentrated in vacuo, and the crude product was purified by silica gel column chromatography (hexane/EtOAc = 6:1) to give 15 (4.23 g, 55% over 2 steps) as a pale yellow powder.
1H NMR (500 MHz, CDCl3): δ 8.40 (br s, 1H), 7.46 (d, J = 7.0 Hz, 4H), 7.38 (t, J = 7.5 Hz, 4H), 7.32 (t, J = 7.3 Hz, 2H), 7.21 (d, J = 7.9 Hz, 1H), 6.97 (s, 1H), 6.92 (t, J = 7.9 Hz, 1H), 6.60 (d, J = 7.9 Hz, 1H), 6.40 (s, 1H), 4.67 (br s, 1H), 3.46 (d, J = 6.6 Hz, 2H), 2.95 (t, J = 6.6 Hz, 2H), 1.48 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 156.1, 144.8, 141.3, 129.0, 128.8, 128.0, 127.5, 127.1, 121.8, 119.8, 113.7, 112.0, 105.3, 82.2, 79.2, 40.9, 28.5, 26.0. MS (ESI-Q-TOF) m/z: 443 [M + H]+. HRMS (ESI-Q-TOF) m/z: 443.2335, calcd for C28H31N2O3; found: 443.2315.
3.3.6. tert-Butyl (2-(7-hydroxy-1H-indol-3-yl)ethyl)carbamate (16)
Pd(OH)2-C (20% w/w, 0.20 g, 0.03 mmol, 0.03 equiv.) was added to a solution of 15 (4.23 g, 9.56 mmol) in MeOH (50 mL), and the mixture was stirred for 8 h under a H2 atmosphere. The reaction mixture was filtered through a pad of Celite and was concentrated in vacuo. The crude product was purified by silica gel column chromatography (hexane/EtOAc = 3:2) to give 16 (2.55 g, 97%) as a white powder.
The spectroscopic and physical data were identical to those reported [25].
3.3.7. tert-Butyl (2-(6,7-dioxo-6,7-dihydro-1H-indol-3-yl)ethyl)carbamate (17)
IBX (2.84 g, 10.2 mmol, 1.1 equiv.) was added to a solution of 16 (2.55 g, 9.23 mmol) in dry DMF (50 mL) at 0 °C, and the mixture was stirred for 2 h. The red mixture was quenched with sat. NaHCO3 aq. and the whole mixture was extracted with EtOAc. The combined organic layer was washed with H2O, dried over Na2SO4, and concentrated in vacuo to afford crude 17 (2.23 g) as a brown solid. The residue was subjected to the next reaction without further purification.
A characterization sample could be obtained by silica gel column chromatography (hexane/EtOAc = 1:2) as an orange solid.
The spectroscopic and physical data were identical to those reported [25].
3.3.8. tert-Butyl (2-(6,7-dioxo-1-tosyl-6,7-dihydro-1H-indol-3-yl)ethyl)carbamate (18)
Et3N (3.19 mL, 23.0 mmol) was added dropwise to a solution of crude 17 (2.23 g) in THF (150 mL) at 0 °C. Then, 4-dimethylaminopyridine (0.23 g, 1.92 mmol) and p-toluenesulfonyl chloride (3.66 g, 19.2 mmol) were successively added to the solution, and the whole mixture was stirred for 1 h. The reaction mixture was quenched with 0.2 mol/L HCl aq. and extracted with EtOAc three times. The organic layer was dried over Na2SO4 and was concentrated in vacuo. The crude material was purified by silica gel column chromatography (CH2Cl2/EtOAc = 15:1) to give 18 (2.58 g, 63% over 2 steps) as a red solid.
The spectroscopic and physical data were identical to those reported [25].
3.3.9. 1-Tosyl-1,3,4,5-tetrahydropyrrolo[4,3,2-de]quinoline-7,8-dione (Ts-damirone C, 19)
TFA (11.5 mL, 150 mmol, 50 equiv.) was added to a solution of 18 (1.34 g, 3.00 mmol) in CH2Cl2 (50 mL) over 10 min at 0 °C, and the mixture was stirred for 70 min. The reaction mixture was concentrated in vacuo, and the resulting red-brown oil was redissolved in MeOH (60 mL). Et3N (4.16 mL, 30 mmol, 10.0 equiv.) was added to a solution over 10 min at 0 °C, and the mixture was stirred for 20 min. The reaction mixture was concentrated in vacuo, and the residue was diluted with CH2Cl2 and H2O. The layers were separated, and the aqueous layer was reextracted with CH2Cl2 twice. The combined organic layer was washed, saturated NaHCO3 aq., and the aqueous layer was reextracted with CH2Cl2. The organic layer was dried over Na2SO4 and was concentrated in vacuo. The crude was purified by silica gel column chromatography (CH2Cl2/MeOH = 10:1) to give 19 (0.57 g, 55%) as a purple solid.
The spectroscopic and physical data were identical to those reported [25].
3.3.10. Ts-makaluvamine D (20)
Tyramine (37.9 mg, 0.28 mmol, 3.0 equiv.) was added to a solution of 19 (107.2 mg, 0.31 mmol) in dry MeOH/THF (1.6:1, 6.5 mL) at rt, and the mixture was stirred for 16 h. The reaction mixture was concentrated in vacuo, and the crude material was purified by silica gel column chromatography (10% MeOH/CH2Cl2, containing 0.1% TFA) to give 20 (88.2 mg, 49%) as a purple solid and recovered 19 (41.1 mg, 87% brsm).
The spectroscopic and physical data were identical to those reported [25].
3.3.11. Makaluvamine D (5)
Pyridine hydrochloride (74.1 mg, 0.64 mmol, 10 equiv.) was added to a solution of 20 (36.9 mg, 64 µmol) in dry DMF (3 mL) at rt, and the mixture was stirred at 80 °C for 2 h. The reaction mixture was concentrated in vacuo, and the crude material was purified by silica gel column chromatography (10% MeOH/CH2Cl2, containing 0.1% TFA) to give 5 (18.9 mg, 70%) as a red-brown solid.
The spectroscopic and physical data were identical to those reported [13].
3.3.12. Makaluvamine P (10)
NaH (50% w/w, 4.3 mg, 90 µmol, 2.0 equiv.) was added to a solution of 5 (18.9 mg, 45 µmol) in dry DMF (5 mL) at 0 °C, and the mixture was stirred for 2 h. Then, CH3I (0.81 mol/L in dry DMF, 0.17 mL, 0.13 mmol, 3.0 equiv.) was added dropwise to the reaction mixture, and the whole mixture was stirred for 2 h. The reaction mixture was quenched with MeOH and concentrated in vacuo. The crude material was purified by silica gel column chromatography (5–20% MeOH/CH2Cl2 gradient, containing 0.1% TFA) to give 10 (18.5 mg, 92%) as a red-brown solid.
The spectroscopic and physical data were identical to those reported [30].
3.3.13. 5-Methyl-1-tosyl-1,3,4,5-tetrahydropyrrolo[4,3,2-de]quinoline-7,8-dione (Ts-damirone B, 22)
K2CO3 (73.3 mg, 0.53 mmol, 2.0 equiv.) was added to a solution of 19 (90.8 mg, 0.26 mmol) in dry DMF (5 mL) at rt. Then, CH3I (1.61 M in dry DMF, 0.49 mL, 0.80 mmol, 3.0 equiv.) was added dropwise to the solution, and the whole mixture was stirred for 16 h. The reaction mixture was quenched with H2O and extracted with EtOAc four times. The organic layer was dried over Na2SO4 and was concentrated in vacuo. The crude material was purified by silica gel column chromatography (CH2Cl2/MeOH = 15:1) to give 22 (93.2 mg, quant.) as a red solid.
The spectroscopic and physical data were identical to those reported [31].
3.3.14. Makaluvamine J (1)
Tyramine (37.9 mg, 0.28 mmol, 3.0 equiv.) was added to a solution of 22 (33.0 mg, 92.1 µmol) in dry MeOH/THF (1.6:1, 6.5 mL) at rt, and the whole mixture was stirred for 16 h. The reaction mixture was concentrated in vacuo, and the crude material was purified by silica gel column chromatography (5–20% MeOH/CH2Cl2 gradient, containing 0.1% TFA) to give 1 (14.8 mg, 37%) as a green solid and recovered 22 (16.4 mg, 87% brsm).
The spectroscopic and physical data were identical to those reported [13].
3.3.15. 5-Methyl-8-oxo-7-(phenethylamino)-1,3,4,8-tetrahydropyrrolo[4,3,2-de]quinolin-5-ium trifluoroacetate (23)
Phenethylamine (0.79 M in dry MeOH/THF, 0.32 mL, 0.25 mmol, 3.0 equiv.) was added to a solution of 22 (30.0 mg, 84 µmol) in dry MeOH/THF (1.6:1, 6.5 mL) at rt, and the whole mixture was stirred for 16 h. The reaction mixture was concentrated in vacuo, and the crude material was purified by silica gel column chromatography (5–20% MeOH/CH2Cl2 gradient, containing 0.1% TFA) to give 23 (12.7 mg, 36%) as a green solid and recovered 22 (14.0 mg, 83% brsm).
1H NMR (500 MHz, CD3OD): δ 7.32–7.21 (m, 5H), 7.14 (s, 1H), 5.40 (s, 1H), 3.92 (t, J = 7.5 Hz, 2H), 3.72 (t, J = 7.0 Hz, 2H), 3.36 (s, 3H), 3.02 (t, J = 7.0 Hz, 2H), 3.00 (t, J = 7.5 Hz, 2H). 13C NMR (125 MHz, CD3OD): δ 168.1, 158.0, 155.2, 139.6, 130.1, 129.8, 127.8, 127.0, 124.9, 124.7, 120.0, 84.3, 54.4, 46.2, 39.7, 35.4, 20.3. MS (ESI-Q-TOF) m/z: 306 [M + H]+. HRMS (ESI-Q-TOF) m/z: 306.1606, calcd for C19H20N3O; found: 306.1607.
3.3.16. 7-((2-(1H-Indol-3-yl)ethyl)amino)-5-methyl-8-oxo-1,3,4,8-tetrahydropyrrolo[4,3,2-de]quinolin-5-ium trifluoroacetate (24)
Tryptamine (36.9 mg, 0.23 mmol, 3.0 equiv.) was added to a solution of 22 (27.5 mg, 77 µmol) in dry MeOH/THF (1.6:1, 6.5 mL) at rt, and the whole mixture was stirred for 16 h. The reaction mixture was concentrated in vacuo, and the crude material was purified by silica gel column chromatography (5–20% MeOH/CH2Cl2 gradient, containing 0.1% TFA) to give 24 (7.7 mg, 22%) as a green solid and recovered 22 (15.2 mg, 74% brsm).
1H NMR (500 MHz, CD3OD): δ 7.61 (d, J = 7.8 Hz, 1H), 7.32 (d, J = 8.1 Hz, 1H), 7.08–6.99 (m, 4H) 4.96 (s, 1H), 3.76–3.73 (m, 4H), 3.14 (t, J = 6.3 Hz, 2H), 2.89 (t, J = 7.1 Hz, 2H), 2.88 (s, 3H). 13C NMR (125 MHz, CD3OD): δ 168.1, 157.4, 155.5, 138.3, 128.7, 127.0, 124.7, 124.5, 122.6, 120.0, 119.3, 119.1, 112.5, 112.3, 84.0, 54.1, 45.5, 39.2 25.9, 20.3. MS (ESI-Q-TOF) m/z: 345 [M + H]+. HRMS (ESI-Q-TOF) m/z: 345.1715, calcd for C21H21N4O; found: 345.1721.
3.3.17. 1-Tosyl-5-vinyl-1,3,4,5-tetrahydropyrrolo[4,3,2-de]quinoline-7,8-dione (25)
K2CO3 (24.9 mg, 0.18 mmol, 2.0 equiv.) was added to a solution of 19 (30.8 mg, 90 µmol) in dry DMF (5 mL) at rt. Then, Allyl bromide (1.18 M in dry DMF, 0.23 mL, 0.27 mmol, 3.0 equiv.) was added dropwise to the solution, and the whole mixture was stirred for 16 h. The reaction mixture was quenched with H2O and extracted with EtOAc for four times. The organic layer was dried over Na2SO4 and was concentrated in vacuo. The crude material was purified by silica gel column chromatography (CH2Cl2/MeOH = 30:1) to give 25 (31.9 mg, 93%) as a red solid.
1H NMR (500 MHz, CDCl3): δ 8.11 (d, J = 8.5 Hz, 2H), 7.54 (s, 1H), 7.32 (d, J = 8.5 Hz, 2H), 5.82–5.75 (m, 1H), 5.38 (s, 1H), 5.30–5.23 (m, 2H), 3.94 (d, J = 5.6 Hz, 2H), 3.57 (d, J = 6.7 Hz, 2H), 2.90 (d, J = 6.7 Hz, 2H), 2.41 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 177.8, 168.5, 151.6, 146.4, 133.6, 130.1, 130.0, 129.9, 129.4, 125.7, 124.5, 119.3, 116.7, 95.3, 54.0, 49.0, 21.9, 20.2. MS (ESI-Q-TOF) m/z: 383 [M + H]+. HRMS (ESI-Q-TOF) m/z: 383.1066, calcd for C20H19N2O4S; found: 383.1063.
3.3.18. 5-Benzyl-1-tosyl-1,3,4,5-tetrahydropyrrolo[4,3,2-de]quinoline-7,8-dione (26)
K2CO3 (15.0 mg, 0.11 mmol, 2.0 equiv.) was added to a solution of 19 (18.6 mg, 54 µmol) in dry DMF (5 mL) at rt. Benzyl bromide (0.84 M in dry DMF, 0.19 mL, 0.16 mmol, 3.0 equiv.) was added dropwise to a solution, and the mixture was stirred for 16 h. The reaction mixture was quenched with H2O and extracted with EtOAc four times. The organic layer was dried over Na2SO4 and was concentrated in vacuo. The crude material was purified by silica gel column chromatography (CH2Cl2/MeOH = 30:1) to give 26 (18.2 mg, 77%) as a purple solid.
1H NMR (500 MHz, CDCl3): δ 8.12 (d, J = 8.5 Hz, 2H), 7.55 (s, 1H), 7.36–7.30 (m, 5H), 7.23 (d, J = 6.6 Hz, 2H), 5.51 (s, 1H), 4.52 (s, 2H), 3.58 (t, J = 6.7 Hz, 2H), 2.87 (t, J = 6.7 Hz, 2H), 2.41 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 177.8, 168.5, 152.2, 146.5, 134.9, 133.6, 130.0, 129.9, 129.5, 129.2, 128.3, 127.5, 125.8, 124.7, 116.8, 95.3, 54.8, 49.1, 21.9, 20.2. MS (ESI-Q-TOF) m/z: 433 [M + H]+. HRMS (ESI-Q-TOF) m/z: 433.1222, calcd for C24H21N2O4S; found: 433.1201.
3.3.19. 5-Allyl-7-((4-hydroxyphenethyl)amino)-8-oxo-1,3,4,8-tetrahydropyrrolo[4,3,2-de]quinolin-5-ium trifluoroacetate (27)
Tyramine (29.9 mg, 0.22 mmol, 3.0 equiv.) was added to a solution of 25 (27.8 mg, 72.7 µmol) in dry MeOH/THF (1.6:1, 6.5 mL) at rt, and the whole mixture was stirred for 16 h. The reaction mixture was concentrated in vacuo, and the crude material was purified by silica gel column chromatography (5–20% MeOH/CH2Cl2 gradient, containing 0.1% TFA) to give 27 (7.5 mg, 22%) as a green solid and recovered 25 (18.5 mg, 89% brsm).
1H NMR (500 MHz, CD3OD): δ 7.15 (s, 1H), 7.06 (d, J = 8.5 Hz, 2H), 6.71 (d, J = 8.5 Hz, 2H), 5.97–5.90 (m, 1H), 5.41–5.36 (m, 3H), 4.32 (d, J = 5.7 Hz, 2H), 3.91 (t, J = 7.5 Hz, 2H), 3.65 (t, J = 6.9 Hz, 2H), 2.99 (t, J = 7.5 Hz, 2H), 2.89 (t, J = 6.9 Hz, 2H). 13C NMR (125 MHz, CD3OD): δ 168.0, 158.0, 157.5, 155.6, 131.6, 131.0, 130.1, 127.1, 125.1, 124.9, 119.9, 119.4, 116.5, 84.5, 55.2, 52.5, 46.6, 34.8, 20.5. MS (ESI-Q-TOF) m/z: 348 [M + H]+. HRMS (ESI-Q-TOF) m/z: 348.1712, calcd for C21H22N3O2; found: 348.1729.
3.3.20. 5-Benzyl-7-((4-hydroxyphenethyl)amino)-8-oxo-1,3,4,8-tetrahydropyrrolo[4,3,2-de]quinolin-5-ium trifluoroacetate (28)
Tyramine (27.9 mg, 0.20 mmol, 3.0 equiv.) was added to a solution of 26 (29.3 mg, 68 µmol) in dry MeOH/THF (1.6:1, 6.5 mL) at rt, and the whole mixture was stirred for 16 h. The reaction mixture was concentrated in vacuo, and the crude material was purified by silica gel column chromatography (5–20% MeOH/CH2Cl2 gradient, containing 0.1% TFA) to give 28 (5.8 mg, 17%) as a green solid and recovered 26 (17.3 mg, 76% brsm).
1H NMR (500 MHz, CD3OD): δ 7.44 (t, J = 7.3 Hz, 2H), 7.37(m, 3H), 7.16 (s, 1H), 7.00 (d, J = 8.5 Hz, 2H), 6.69 (d, J = 8.5 Hz, 2H), 5.57 (s, 1H), 4.93 (s, 2H), 3.91 (t, J = 7.5 Hz, 2H), 3.62 (t, J = 7.0 Hz, 2H), 2.95 (t, J = 7.5 Hz, 2H), 2.83 (t, J = 7.0 Hz, 2H). 13C NMR (125 MHz, CD3OD): δ 168.0, 158.3, 157.5, 155.8, 136.0, 131.0, 130.4, 130.0, 129.6, 128.7, 127.2, 125.2, 124.9, 119.9, 116.5, 84.8, 56.1, 52.7, 46.6, 34.7, 20.5. MS (ESI-Q-TOF) m/z: 398 [M + H]+. HRMS (ESI-Q-TOF) m/z: 398.1869, calcd for C25H24N3O2; found: 398.1884.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29061389/s1, Supplementary Data S1: The NMR spectra of new compounds.
Author Contributions
Conceptualization, N.K.; methodology, N.K.; validation, N.K.; formal analysis, N.K.; investigation, Y.K. and K.F.; data curation, Y.K., K.F., A.K., and K.I.; writing—original draft preparation, Y.K.; writing—review and editing, N.K.; supervision, N.K.; project administration, N.K.; funding acquisition, N.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Research Promotion Program for Acquiring KAKENHI, grant no. B21-0051, from Ritsumeikan University, and Grant-in-Aid for Scientific Research C, grant no. 22K05339, from the Japan Society for the Promotion of Science (JSPS) to N.K.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
The human pancreatic carcinoma cell line, PANC-1 (RCB2095), was provided by RIKEN BRC through the National Bio-Resource Project of the MEXT/AMED, Japan.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; the International Natural Product Sciences Taskforce; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Kotoku, N. Marine Natural Products Targeting Tumor Microenvironment. In New Tide of Natural Product Chemistry; Ishikawa, H., Takayama, H., Eds.; Springer: Singapore, 2023; Volume 20, Chapter 3; pp. 35–58. [Google Scholar] [CrossRef]
- Kotoku, N.; Ishida, R.; Matsumoto, H.; Arai, M.; Toda, K.; Setiawan, A.; Muraoka, O.; Kobayashi, M. Biakamides A-D, Unique Polyketides from a Marine Sponge, Act as Selective Growth Inhibitors of Tumor Cells Adapted to Nutrient Starvation. J. Org. Chem. 2017, 82, 1705–1718. [Google Scholar] [CrossRef]
- Ishida, R.; Matsumoto, H.; Ichii, S.; Kobayashi, M.; Arai, M.; Kotoku, N. Structure-Activity Relationship of Biakamide, Selective Growth Inhibitors under Nutrient-Starved Condition from Marine Sponge. Chem. Pharm. Bull. 2019, 67, 210–223. [Google Scholar] [CrossRef]
- Lu, J.; Kunimoto, S.; Yamazaki, Y.; Kaminishi, M.; Esumi, H. Kigamicin D, A Novel Anticancer Agent Based on A New Anti-Austerity Strategy Targeting Cancer Cells’ Tolerance to Nutrient Starvation. Cancer Sci. 2004, 95, 547–552. [Google Scholar] [CrossRef]
- Awale, S.; Lu, J.; Kalauni, S.K.; Kurashima, Y.; Tezuka, Y.; Kadota, S.; Esumi, H. Identification of Arctigenin as an Antitumor Agent Having the Ability to Eliminate the Tolerance of Cancer Cells to Nutrient Starvation. Cancer Res. 2006, 66, 1751–1757. [Google Scholar]
- Arai, M.; Kamiya, K.; Shin, D.; Matsumoto, H.; Hisa, T.; Setiawan, A.; Kotoku, N.; Kobayashi, M. N-Methylniphatyne A, a New 3-Alkylpyridine Alkaloid as an Inhibitor of the Cancer Cells Adapted to Nutrient Starvation, from an Indonesian Marine Sponge of Xestospongia sp. Chem. Pharm. Bull. 2016, 64, 766–771. [Google Scholar] [CrossRef]
- Arai, M.; Shin, D.; Kamiya, K.; Ishida, R.; Setiawan, A.; Kotoku, N.; Kobayashi, M. Marine Spongean Polybrominated Diphenyl Ethers, Selective Growth Inhibitors against the Cancer Cells Adapted to Glucose Starvation, Inhibits Mitochondrial Complex II. J. Nat. Med. 2017, 71, 44–49. [Google Scholar] [CrossRef]
- Matsumoto, H.; Hisa, T.; Toda, K.; Ishida, R.; Setiawan, A.; Arai, M.; Kotoku, N. Fasciospyrinadinone and Fasciospyrinadinol, Novel 3-Alkylpyridine Sesquiterpenoids from an Indonesian Marine Sponge, as Selective Growth Inhibitors of the Cancer Cells under Nutrient Starvation. Heterocycles 2021, 103, 827–838. [Google Scholar] [CrossRef]
- Ikeda, M.; Sato, A.; Mochizuki, N.; Toyosaki, K.; Miyoshi, C.; Fujioka, R.; Mitsunaga, S.; Ohno, I.; Hashimoto, Y.; Takahashi, H.; et al. Phase I Trial of GBS-01 for Advanced Pancreatic Cancer Refractory to Gemcitabine. Cancer Sci. 2016, 107, 1818–1824. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Lin, S.; McCauley, E.P.; Lorig-Roach, N.; Tenney, K.; Naphen, C.N.; Yang, A.-M.; Johnson, T.A.; Hernadez, T.; Rattan, R.; Valeriote, F.A.; et al. Another Look at Pyrroloiminoquinone Alkaloids—Perspectives on Their Therapeutic Potential from Known Structures and Semisynthetic Analogues. Mar. Drugs 2017, 15, 98. [Google Scholar] [CrossRef]
- Zou, Y.; Wang, X.; Sims, J.; Wang, B.; Pandey, P.; Welsh, C.L.; Stone, R.P.; Avery, M.A.; Doerksen, R.J.; Ferreira, D.; et al. Computationally Assisted Discovery and Assignment of a Highly Strained and PANC-1 Selective Alkaloid from Alaska’s Deep Ocean. J. Am. Chem. Soc. 2019, 141, 4338–4344. [Google Scholar] [CrossRef]
- Wang, B.; Shen, C.; Li, Y.; Zhang, T.; Huang, H.; Ren, J.; Hu, Z.; Xu, J.; Xu, B. Oridonin overcomes the gemcitabine resistant PANC-1/Gem cells by regulating GST pi and LRP/1 ERK/JNK signaling. Onco Targets Ther. 2019, 12, 5751–5765. [Google Scholar] [CrossRef]
- Fujioka, H.; Kita, Y. Marine Pyrroloiminoquinone Alkaloids, Makaluvamines and Discorhabdins, and Marine Pyrrole-Imidazole Alkaloids. In Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes; Ramawat, K.G., Mérillon, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 251–283. [Google Scholar]
- Hu, J.; Fan, H.; Xiong, J.; Wu, S. Discorhabdins and Pyrroloiminoquinone-Related Alkaloids. Chem. Rev. 2011, 111, 5465–5491. [Google Scholar] [CrossRef] [PubMed]
- White, J.D.; Yager, K.M.; Yakura, T. Synthetic Studies of the Pyrroloquinoline Nucleus of the Makaluvamine Alkaloids. Synthesis of the Topoisomerase II Inhibitor Makaluvamine D. J. Am. Chem. Soc. 1994, 116, 1831–1838. [Google Scholar] [CrossRef]
- Izawa, T.; Nishiyama, S.; Yamamura, S. Total syntheses of makaluvamines A, B, C, D and E, cytotoxic pyrroloiminoquinone alkaloids isolated from marine sponge bearing inhibitory activities against topoisomerase II. Tetrahedron 1994, 50, 13593–13600. [Google Scholar] [CrossRef]
- Sadanandan, E.V.; Pillai, S.K.; Lakshmikantham, M.V.; Billimoria, A.D.; Culpepper, J.S.; Cava, M.P. Efficient Syntheses of the Marine Alkaloids Makaluvamine D and Discorhabdin C: The 4,6,7-Trimethoxyindole Approach. J. Org. Chem. 1995, 60, 1800–1805. [Google Scholar] [CrossRef]
- Iwao, M.; Motoi, O.; Fukuda, T.; Ishibashi, F. New synthetic approach to pyrroloiminoquinone marine alkaloids. Total synthesis of makaluvamines A, D, I, and K. Tetrahedron 1998, 54, 8999–9010. [Google Scholar] [CrossRef]
- An, J.; Jackson, R.K., III; Tuccinardi, J.P.; Wood, J.L. Pyrroloiminoquinone Alkaloids: Total Synthesis of Makaluvamines A and K. Org. Lett. 2023, 25, 1868–1871. [Google Scholar] [CrossRef]
- Dobson, D.; Todd, A.; Gilmore, J. The synthesis of 7-Alkoxyindoles. Synth. Commun. 1991, 21, 611–617. [Google Scholar] [CrossRef]
- Magdziak, D.; Rodriguez, A.A.; Van De Water, R.W.; Pettus, T.R.R. Regioselective Oxidation of Phenols to o-Quinones with o-Iodoxybenzoic Acid (IBX). Org. Lett. 2002, 4, 285–288. [Google Scholar] [CrossRef]
- Smith, M.W.; Falk, I.D.; Ikemoto, H.; Burns, N.Z. A convenient C–H functionalization platform for pyrroloiminoquinone alkaloid synthesis. Tetrahedron 2019, 75, 3366–3370. [Google Scholar] [CrossRef]
- Beyerman, H.C.; Hirt, J.; Kranenburg, P.; Syrier, J.L.M.; van Zon, A. Excess mixed anhydride peptide synthesis with histidine derivatives. Recl. Trav. Chim. Pays-Bas 1974, 93, 256–257. [Google Scholar] [CrossRef]
- Bajwa, J.S.; Chen, G.P.; Prasad, K.; Oljan, R.; Blacklock, T.J. Deprotection of N-tosylated indoles and related structures using cesium carbonate. Tetrahedron Lett. 2006, 47, 6425–6427. [Google Scholar] [CrossRef]
- Sun, W.; Chen, X.; Hu, Y.; Geng, H.; Jiang, Y.; Zhou, Y.; Zhu, W.; Hu, M.; Hu, H.; Wang, X.; et al. A NaH-promoted N-detosylation reaction of diverse p-toluenesulfonamides. Tetrahedron Lett. 2020, 61, 152442. [Google Scholar] [CrossRef]
- Kitagawa, K.; Kitade, K.; Kiso, Y.; Akita, T.; Funakoshi, S.; Fujii, N.; Yajima, H. Studies on Peptides. LXXXV. A New Deprotecting Procedure for p-Toluenesulfonyl and p-Methoxybenzenesulfonyl Groups from the Nim-Function of Histidine. Chem. Pharm. Bull. 1980, 28, 926–931. [Google Scholar] [CrossRef]
- Casapullo, A.; Cutignano, A.; Bruno, I.; Bifulco, G.; Debitus, C.; Gomez-Paloma, L.; Riccio, R. Makaluvamine P, a New Cytotoxic Pyrroloiminoquinone from Zyzzya cf. fuliginosa. J. Nat. Prod. 2001, 64, 1354–1356. [Google Scholar] [CrossRef]
- Inoue, K.; Ishikawa, Y.; Nishiyama, S. Synthesis of Tetrahydropyrroloiminoquinone Alkaloids Based on Electrochemically Generated Hypervalent Iodine Oxidative Cyclization. Org. Lett. 2010, 12, 436–439. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).