
Biological Evaluations, NMR Analyses, Molecular Modeling Studies, and Overview of the Synthesis of the Marine Natural Product (−)-Mucosin †

 , , , , ,
, , , , ,  and
and
Abstract
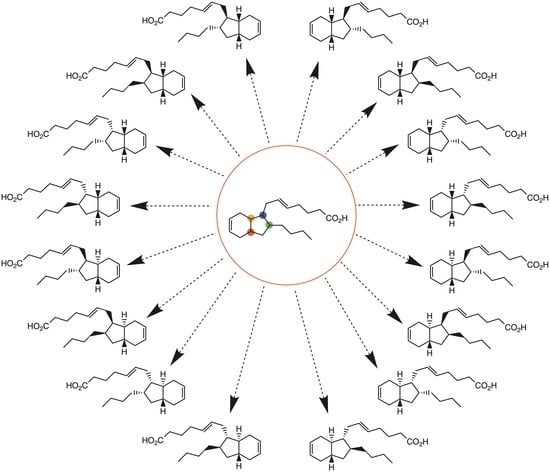
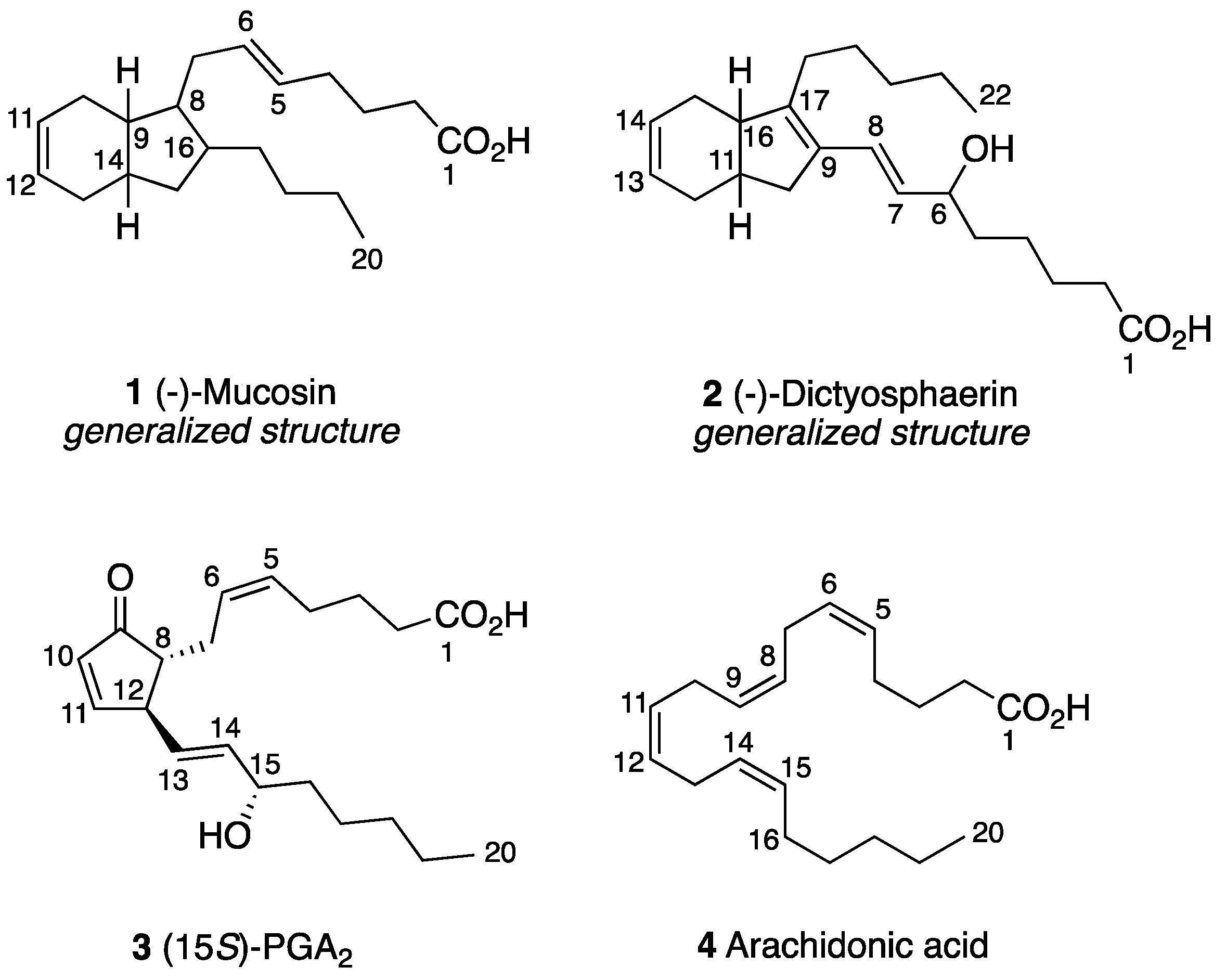
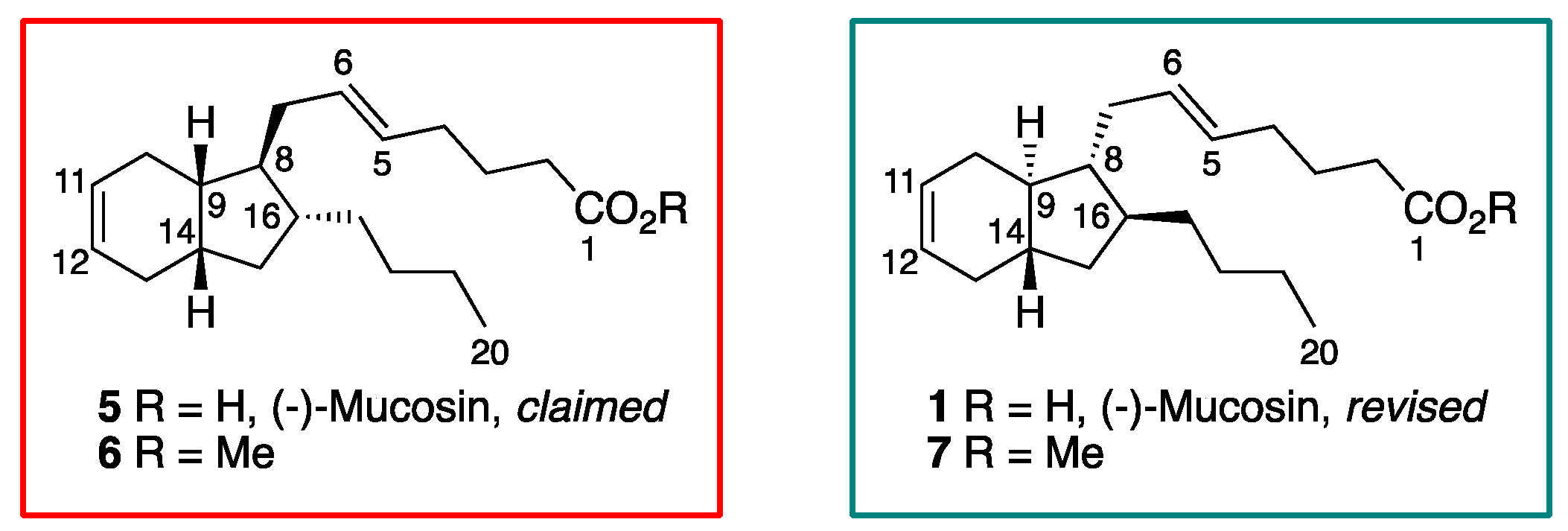
1. Introduction
2. Results and Discussion
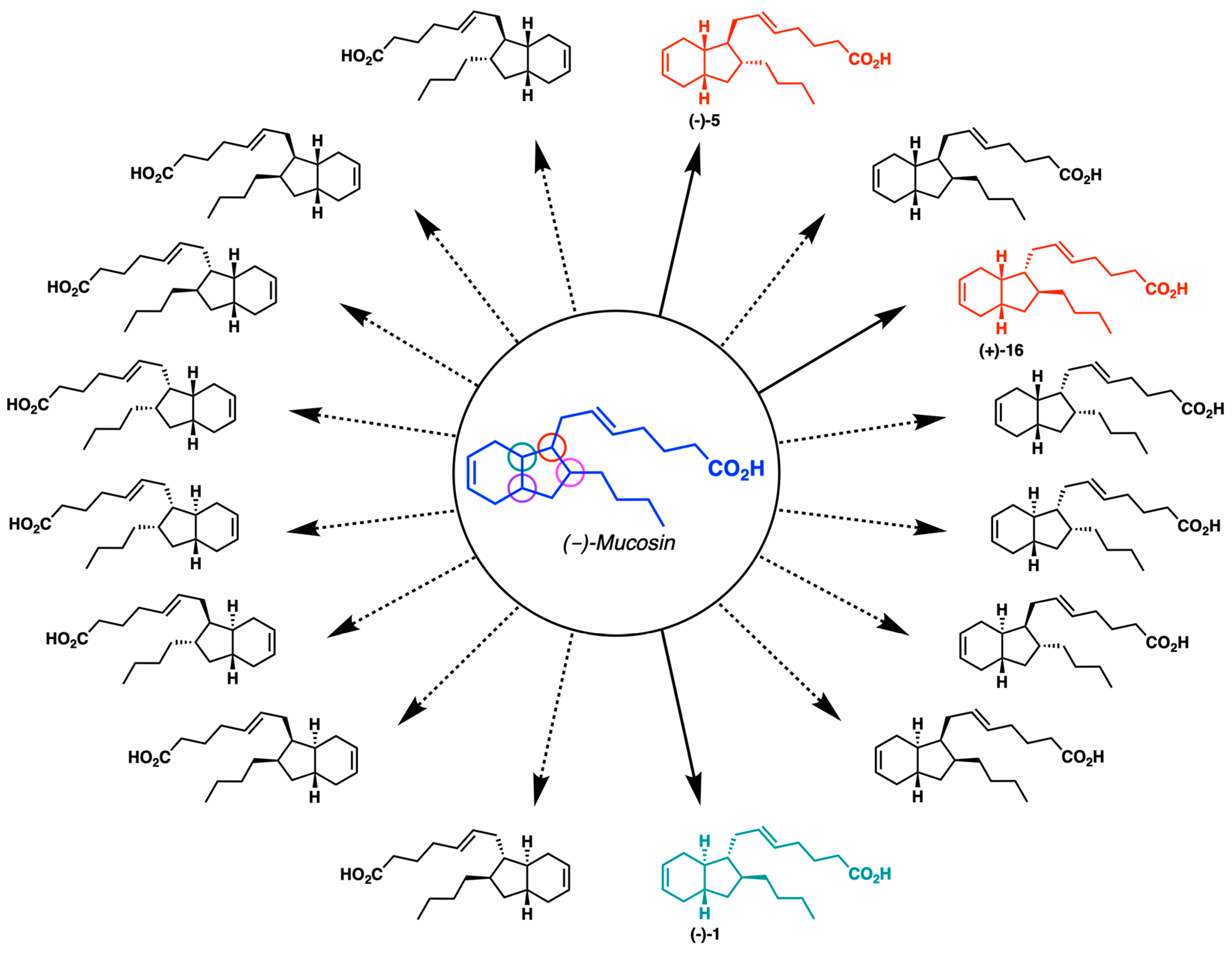
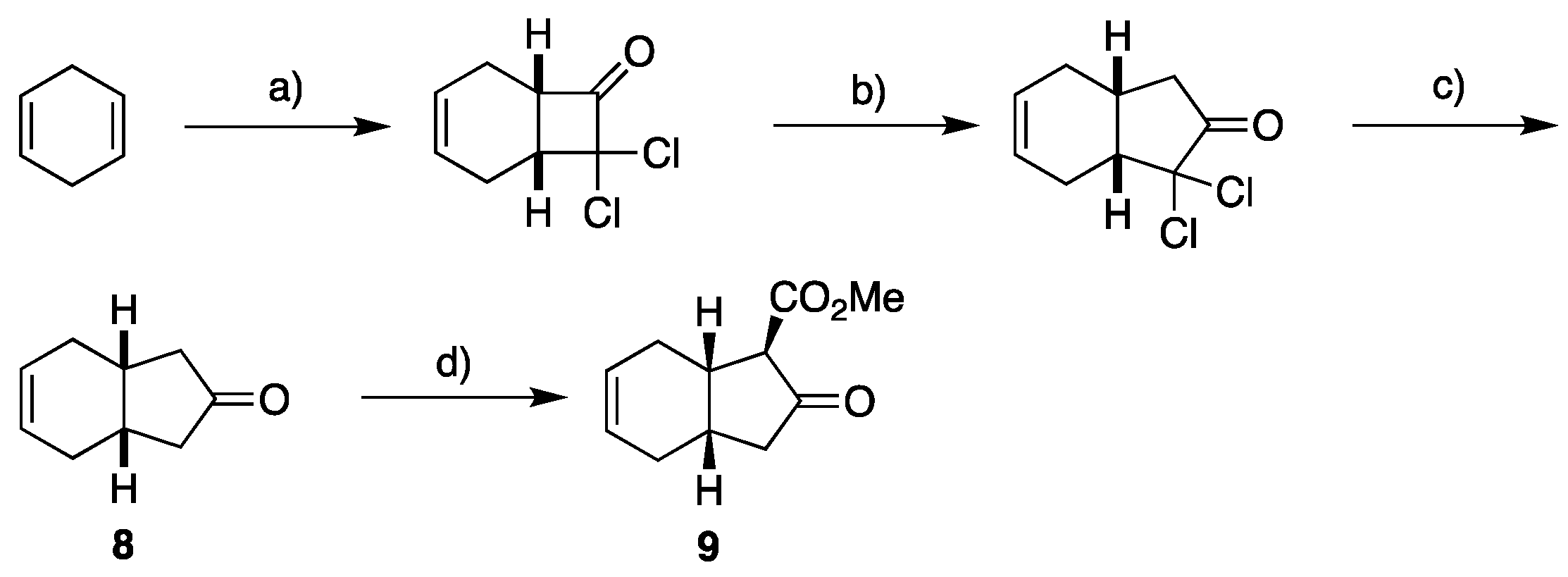
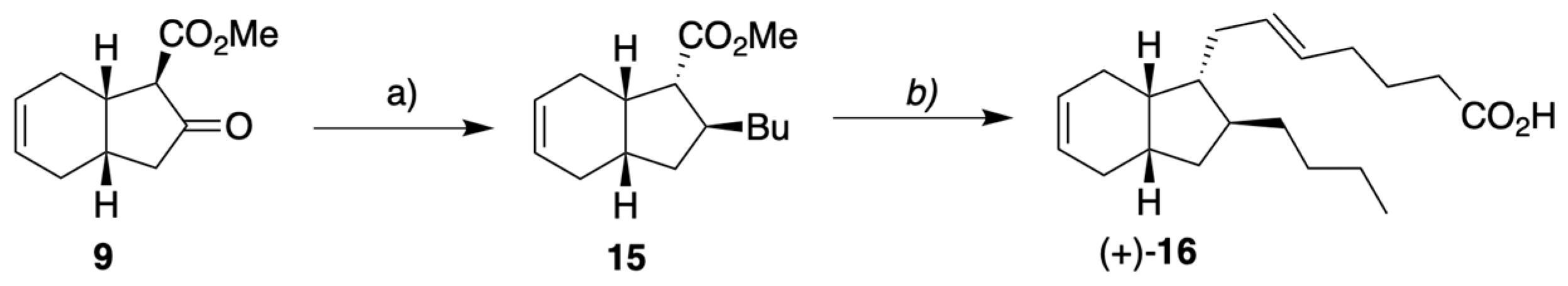
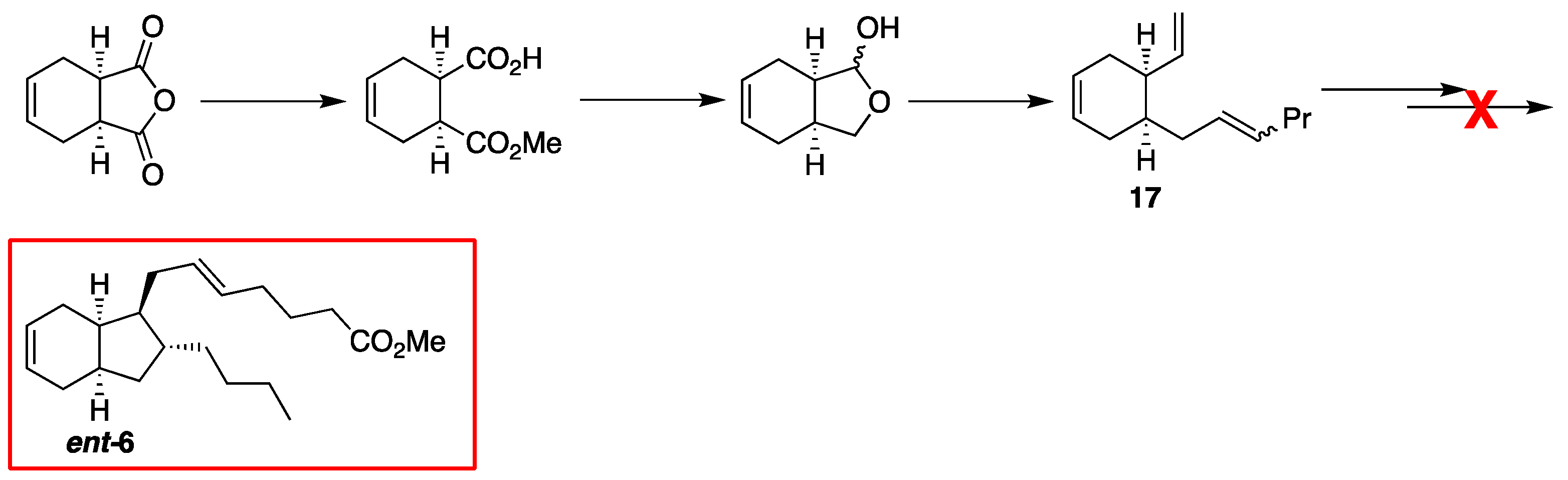
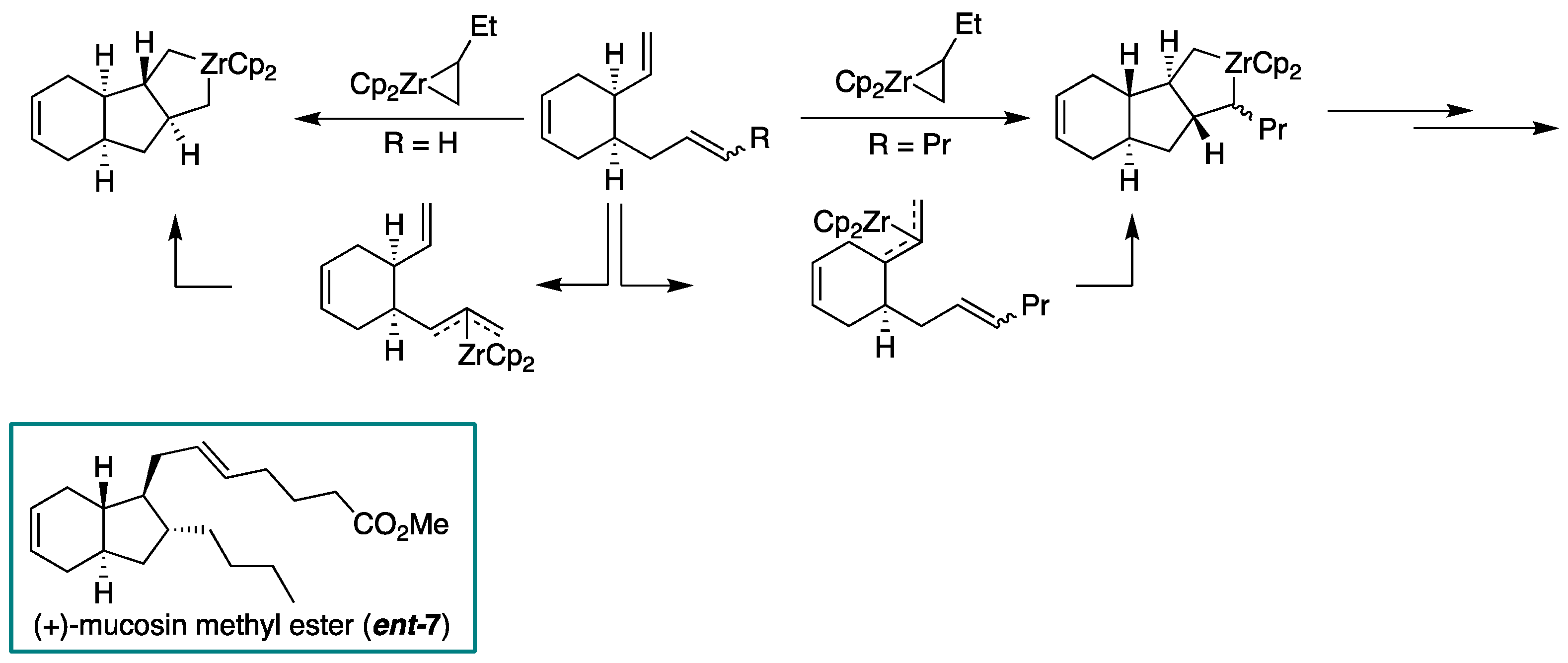
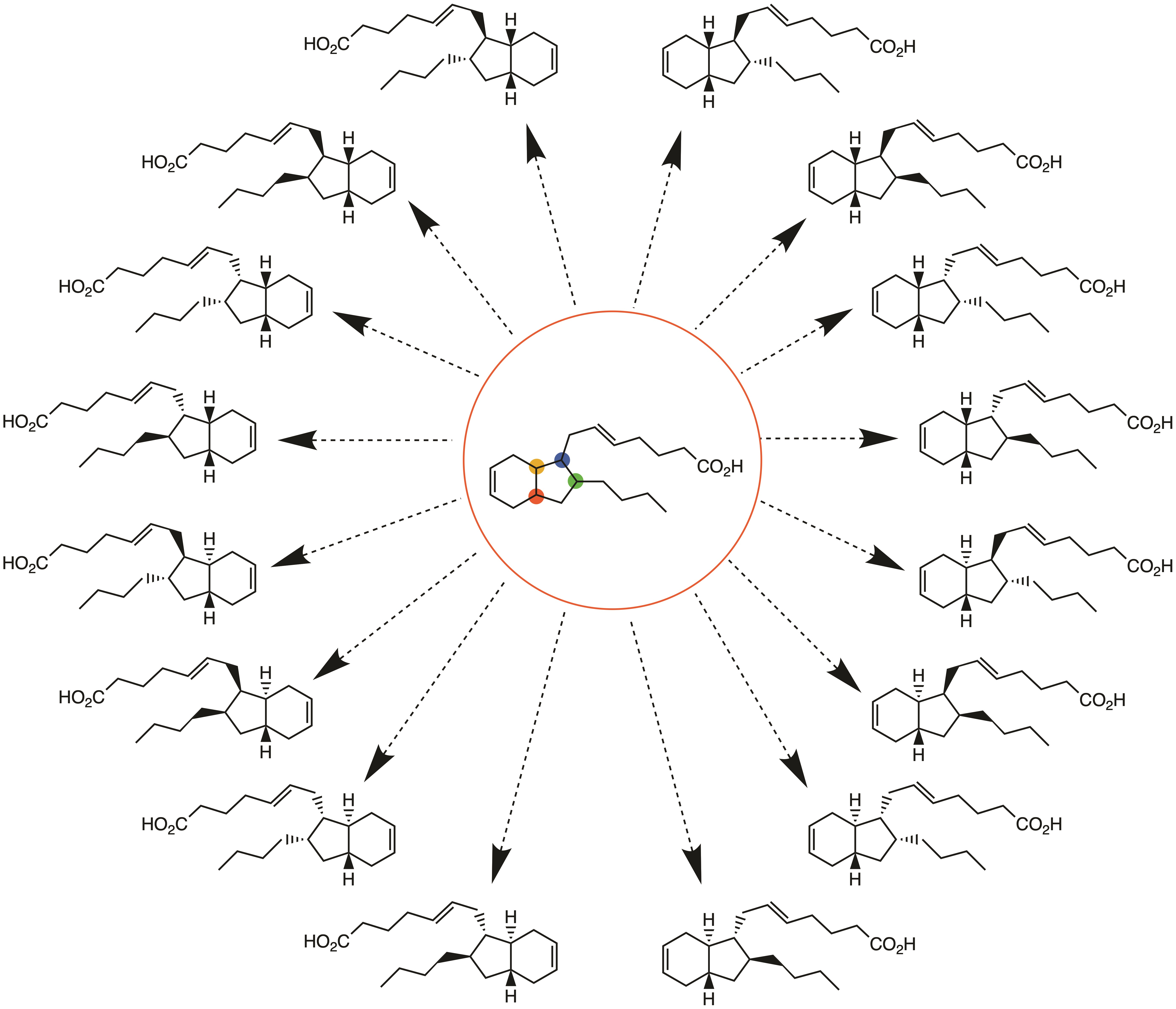
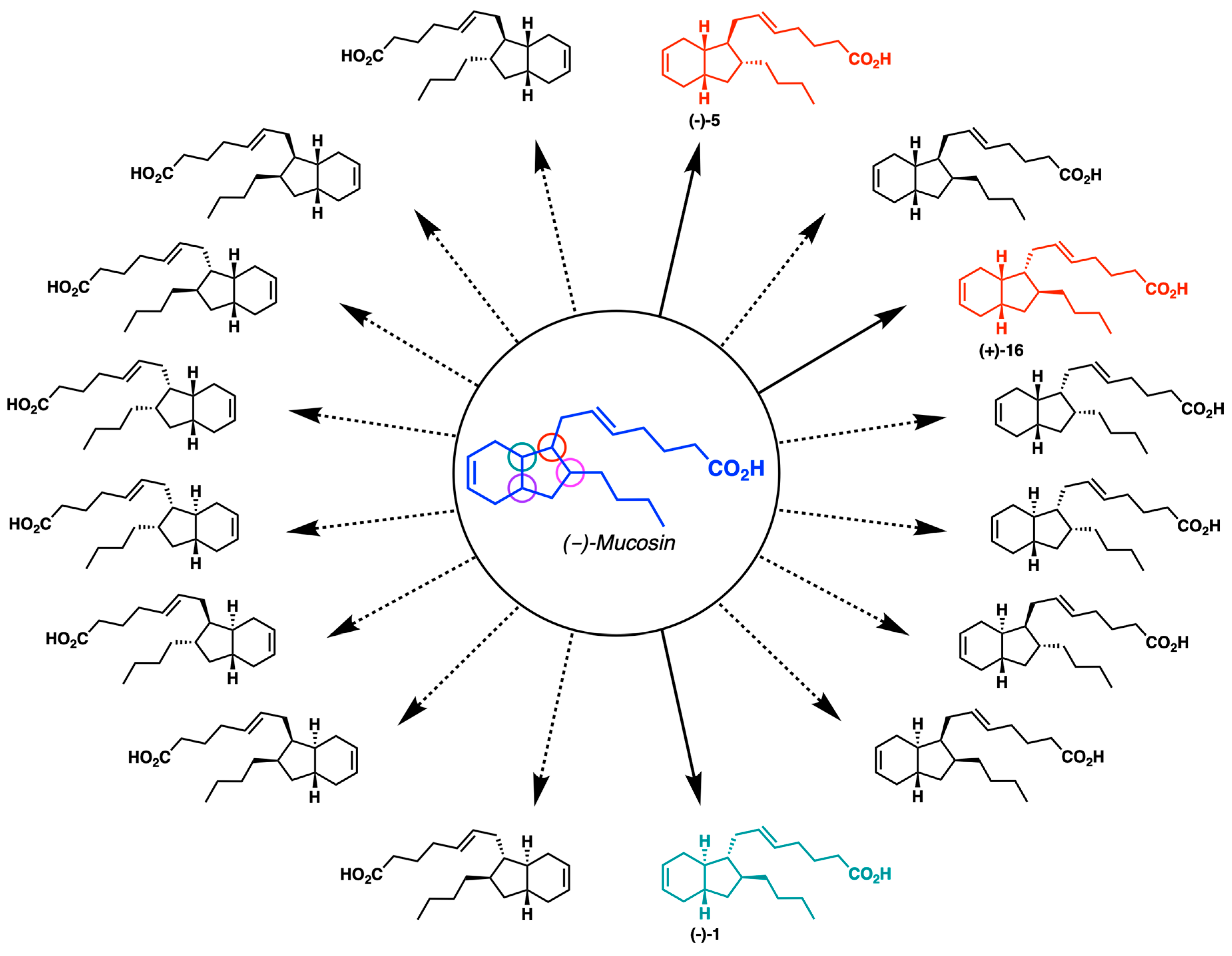
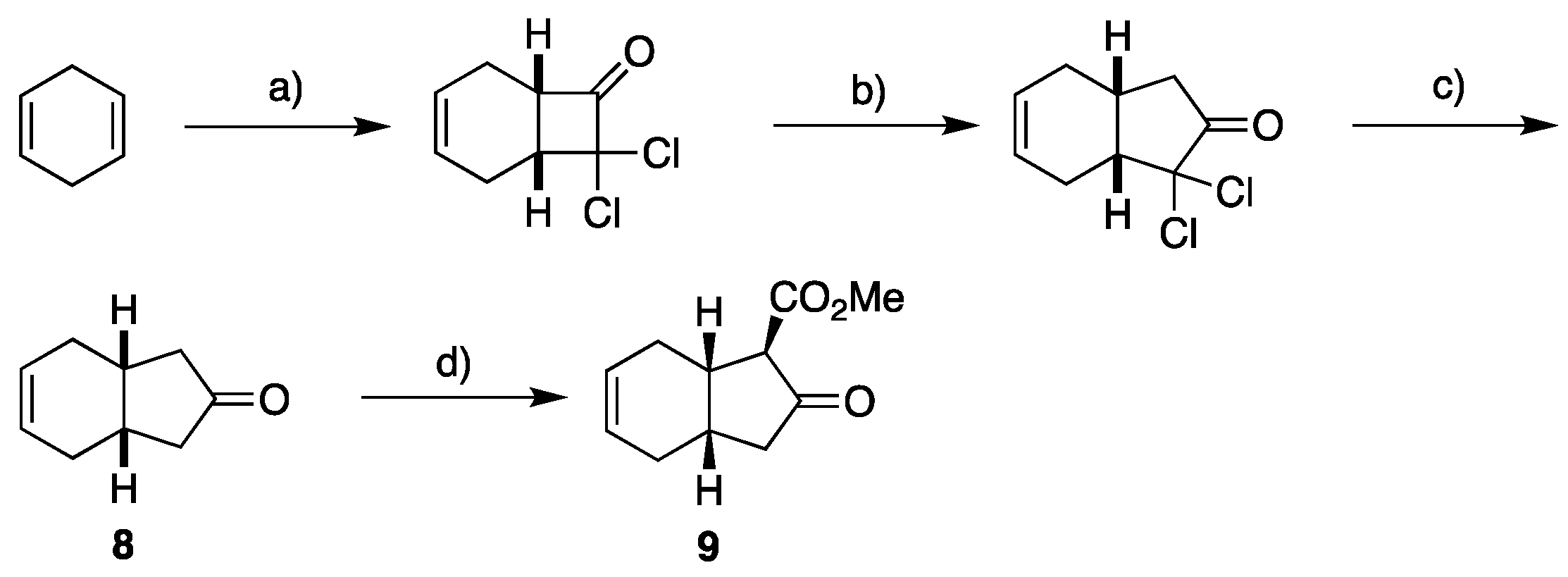
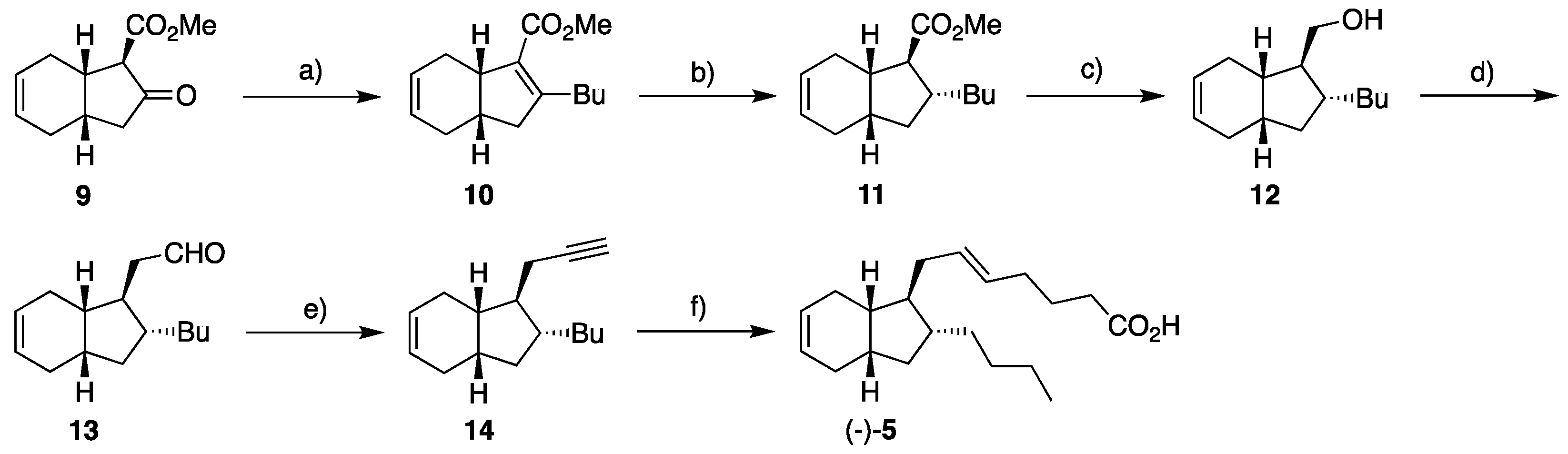
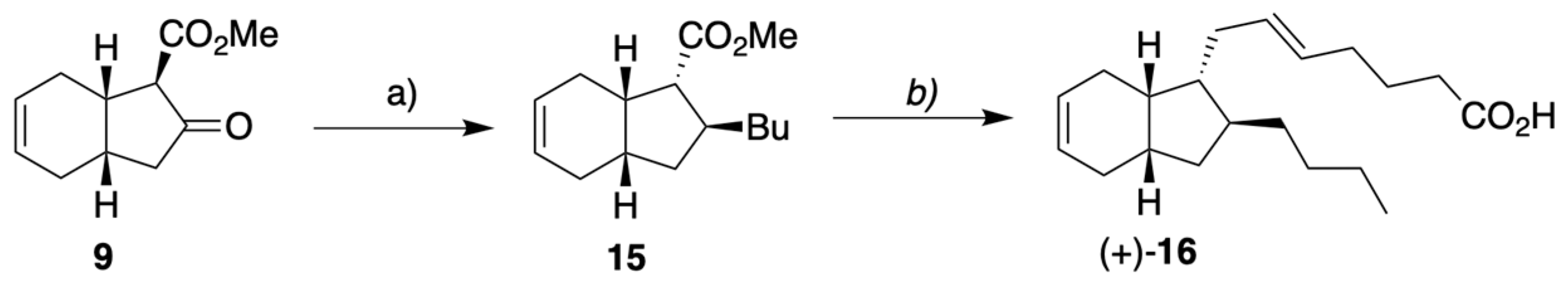
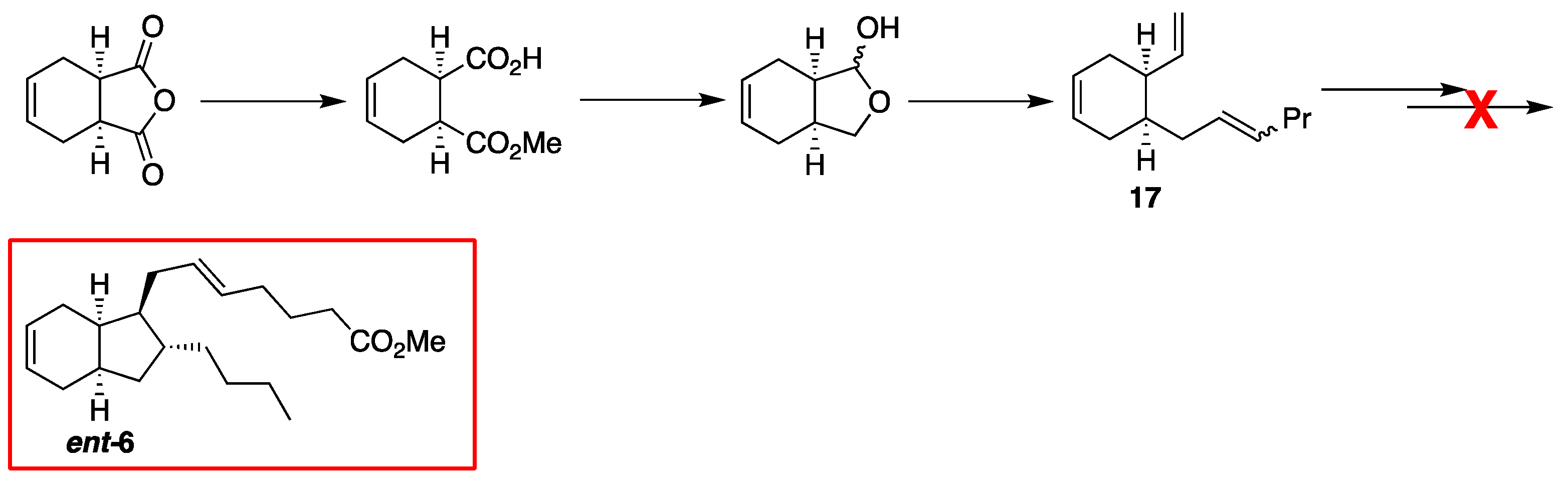
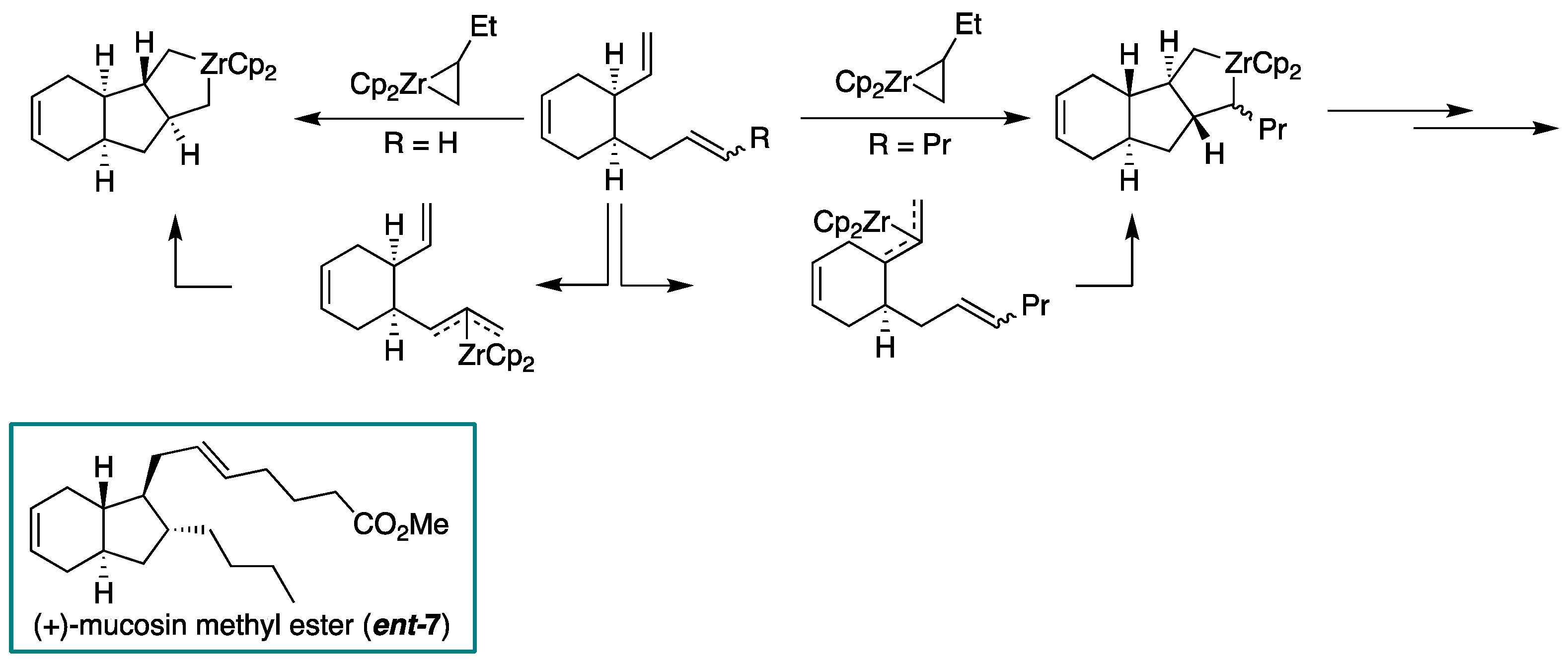
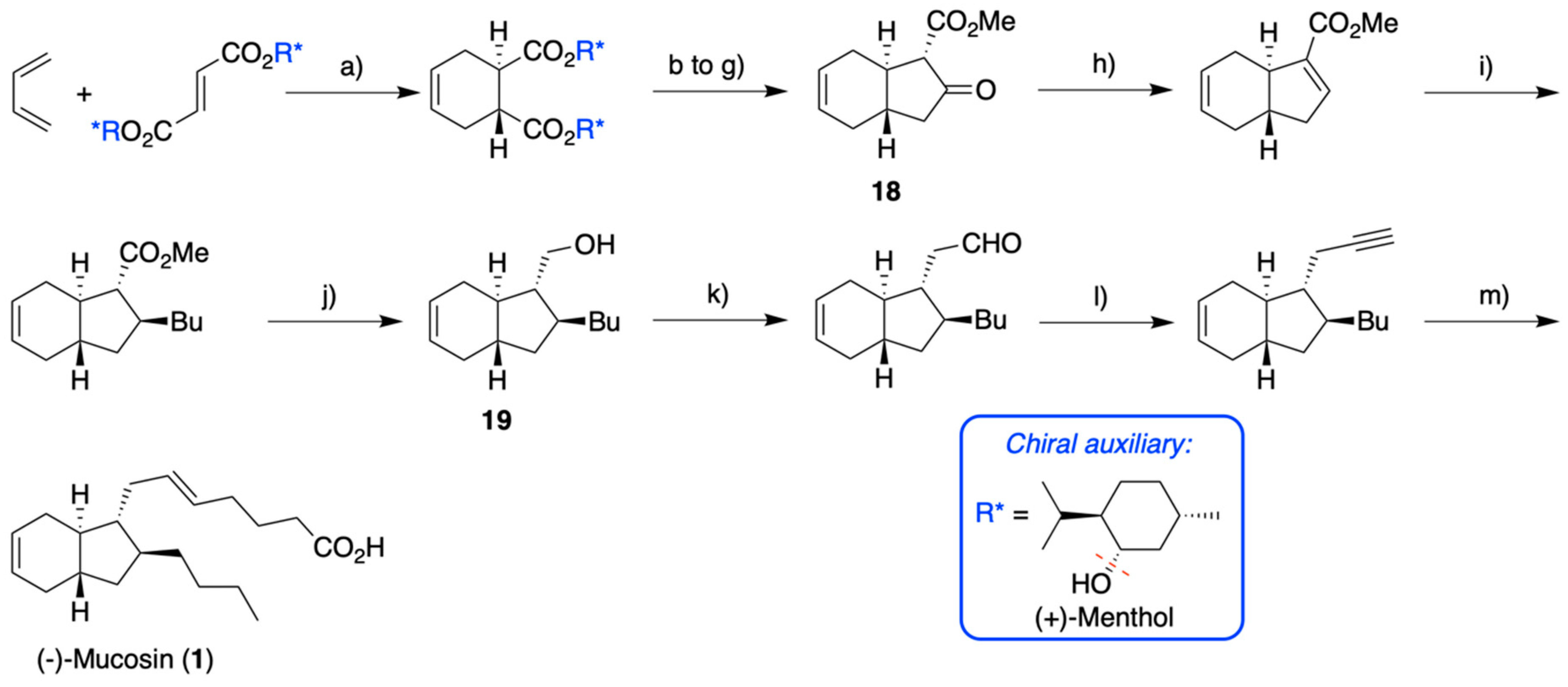
2.1. Overview of Stereoselective Synthesis of (−)-Mucosin (1) and Stereoisomers
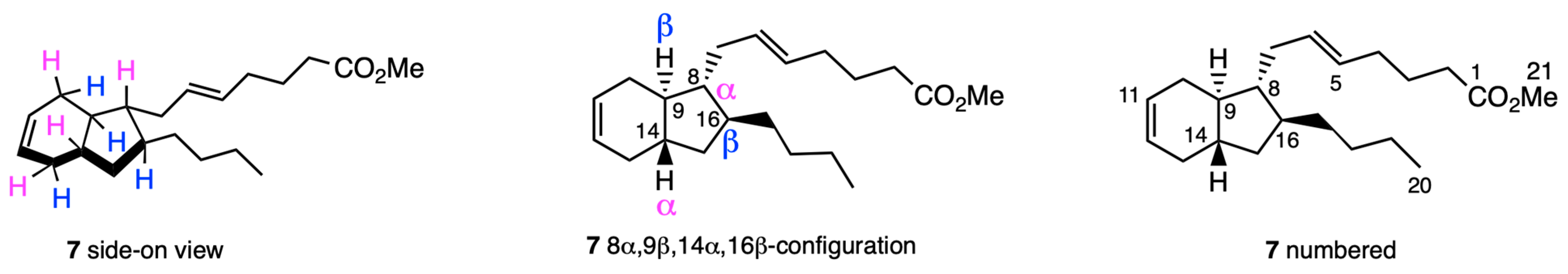
2.2. NMR Studies
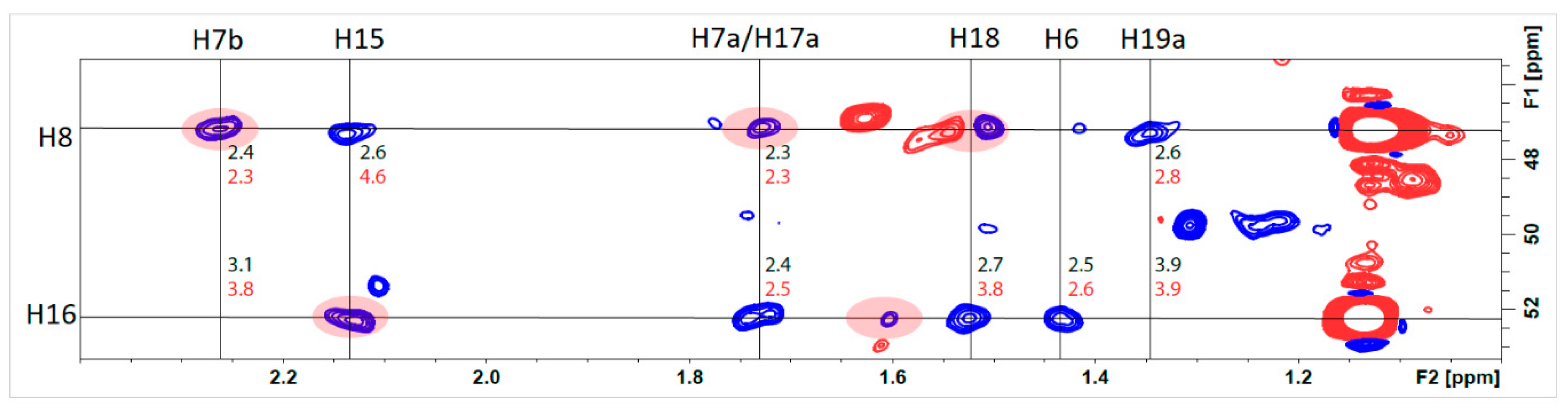
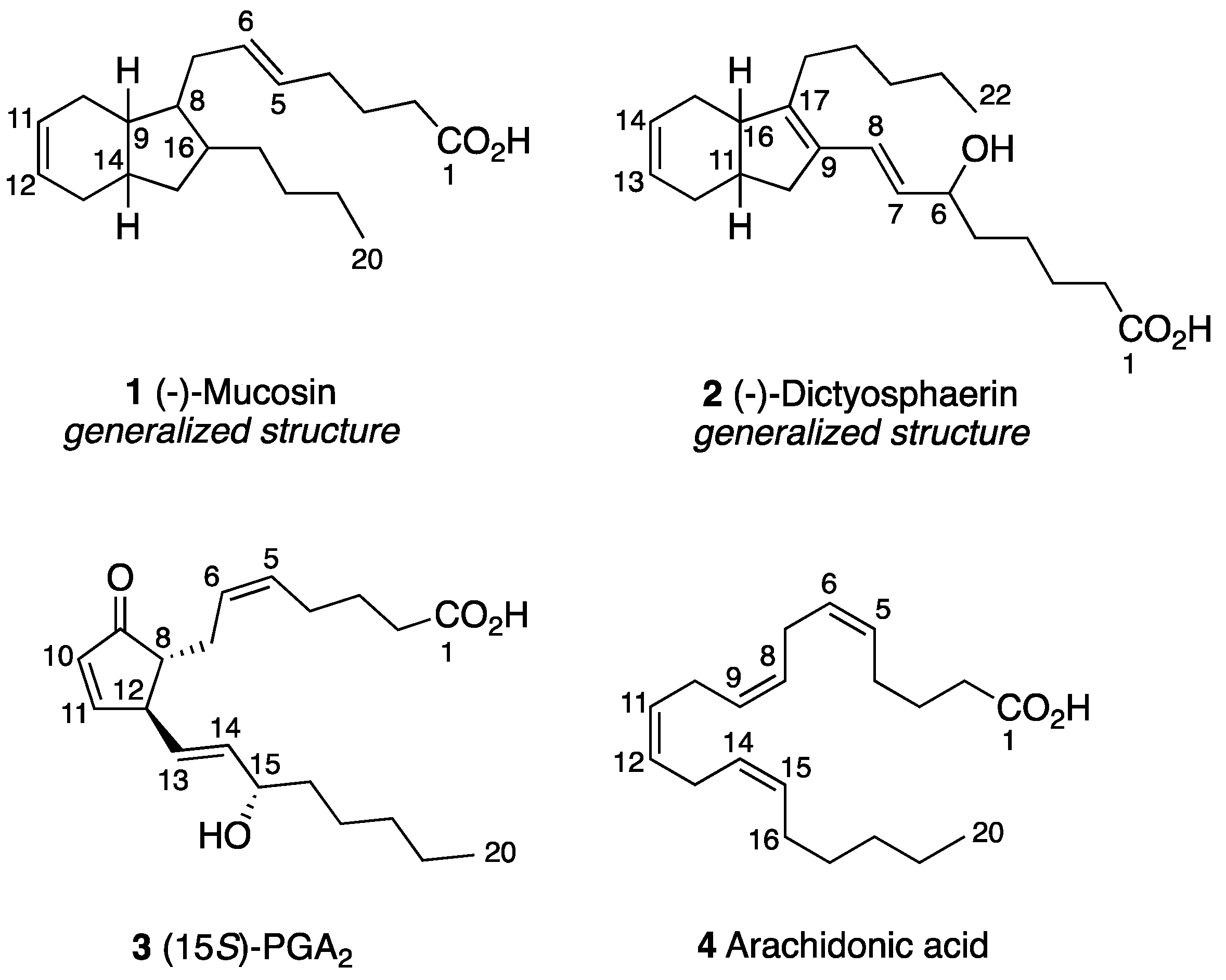
2.2.1. Preliminary Considerations
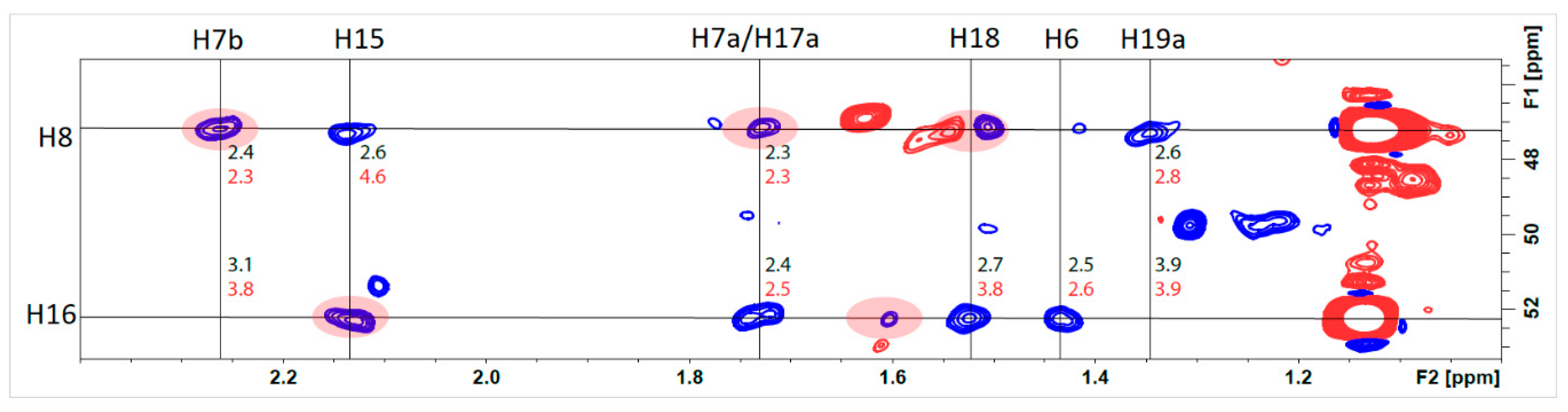
2.2.2. Structural Assignment and Discussion of NMR Data
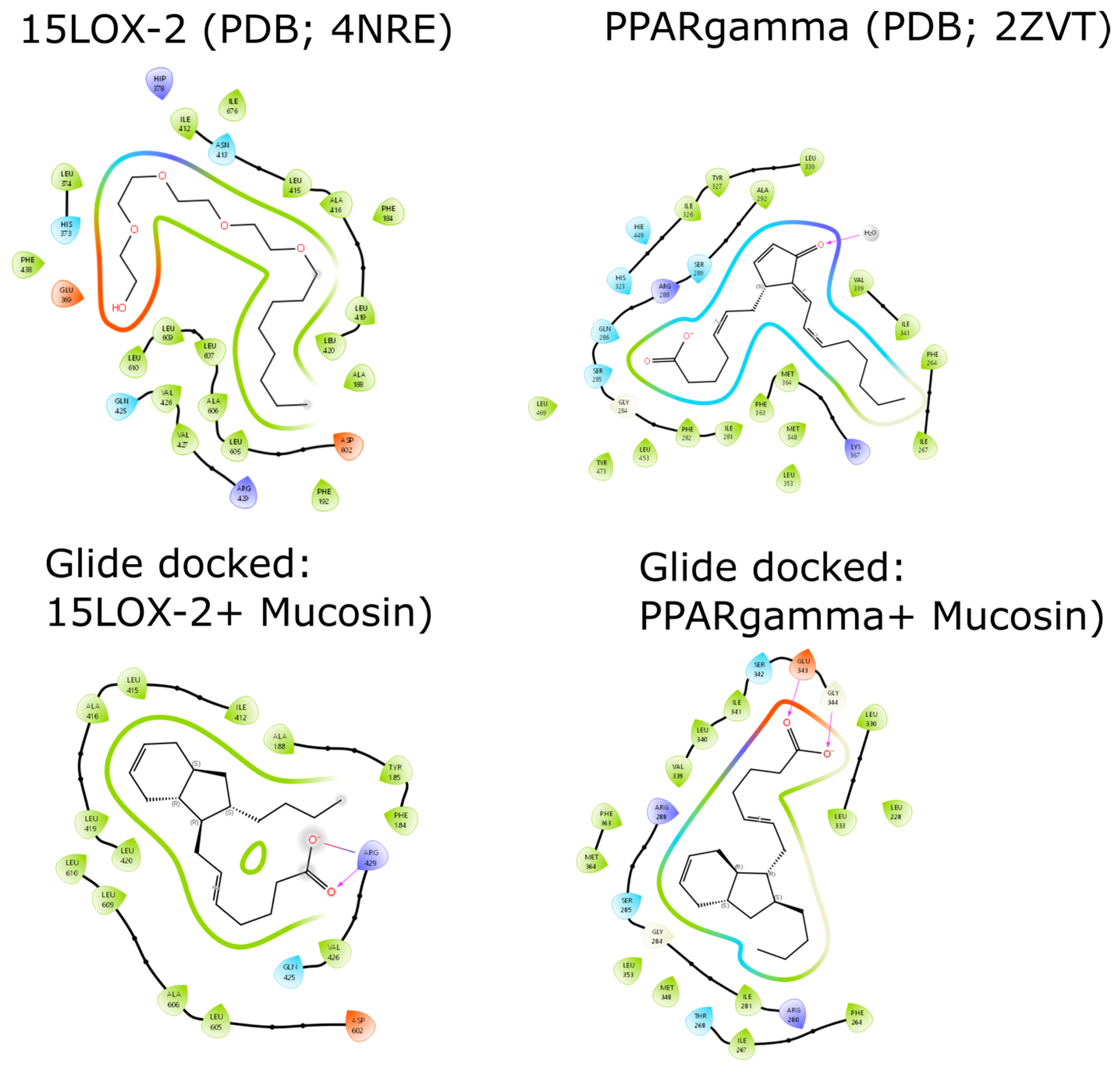
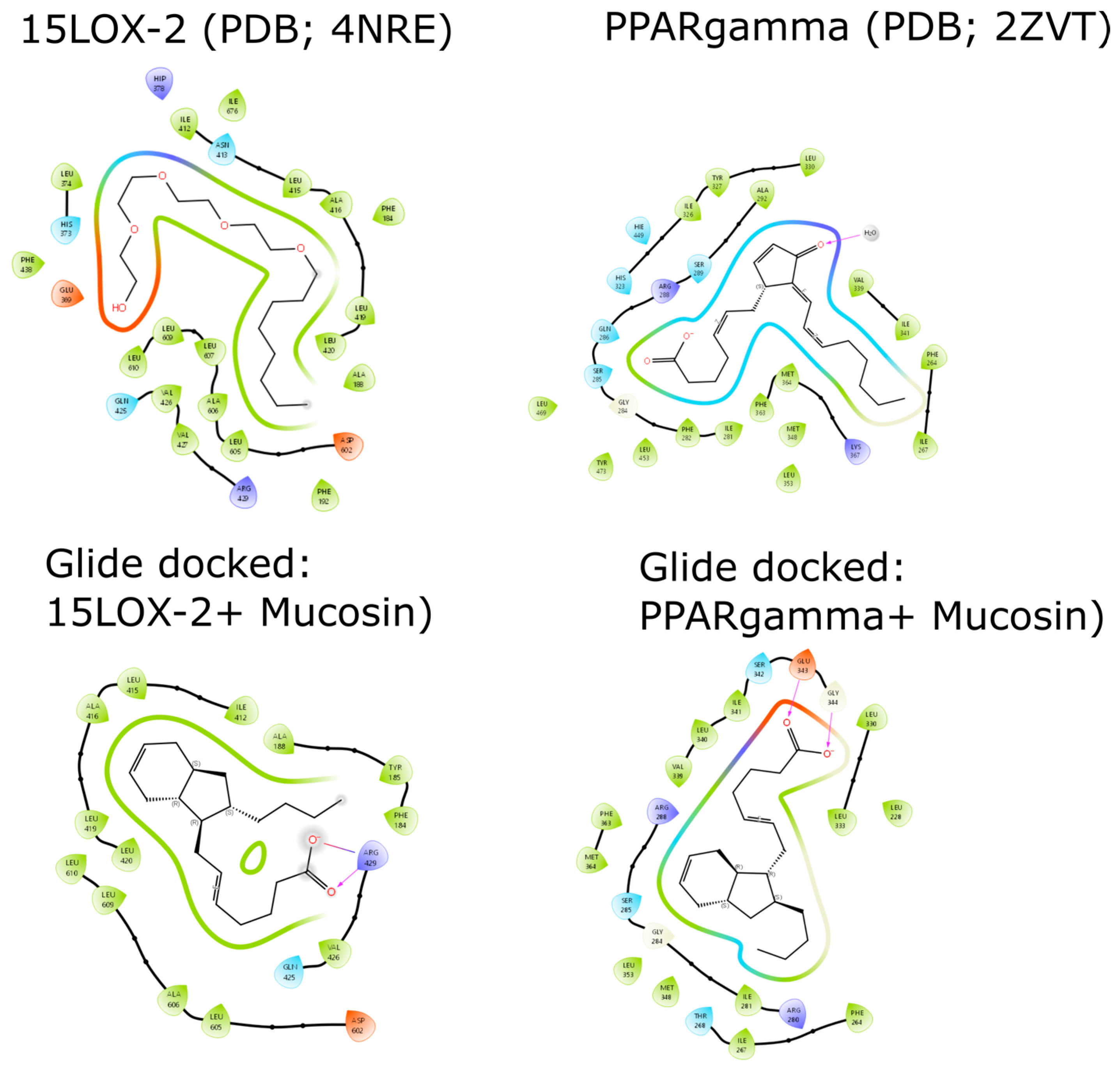
2.3. Docking Studies of (−)-Mucosin (1) with 15-LOX-2 and PPARγ
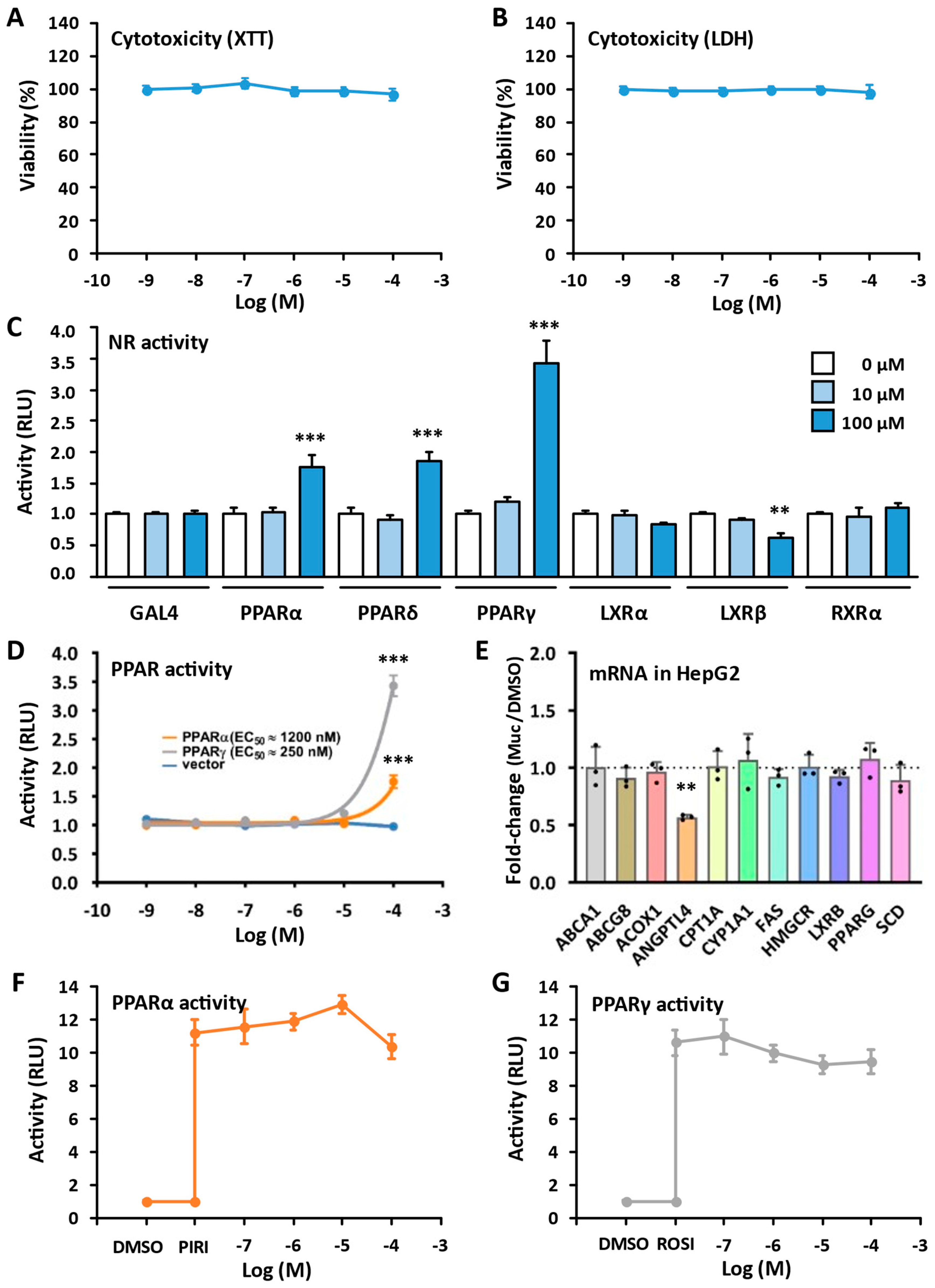
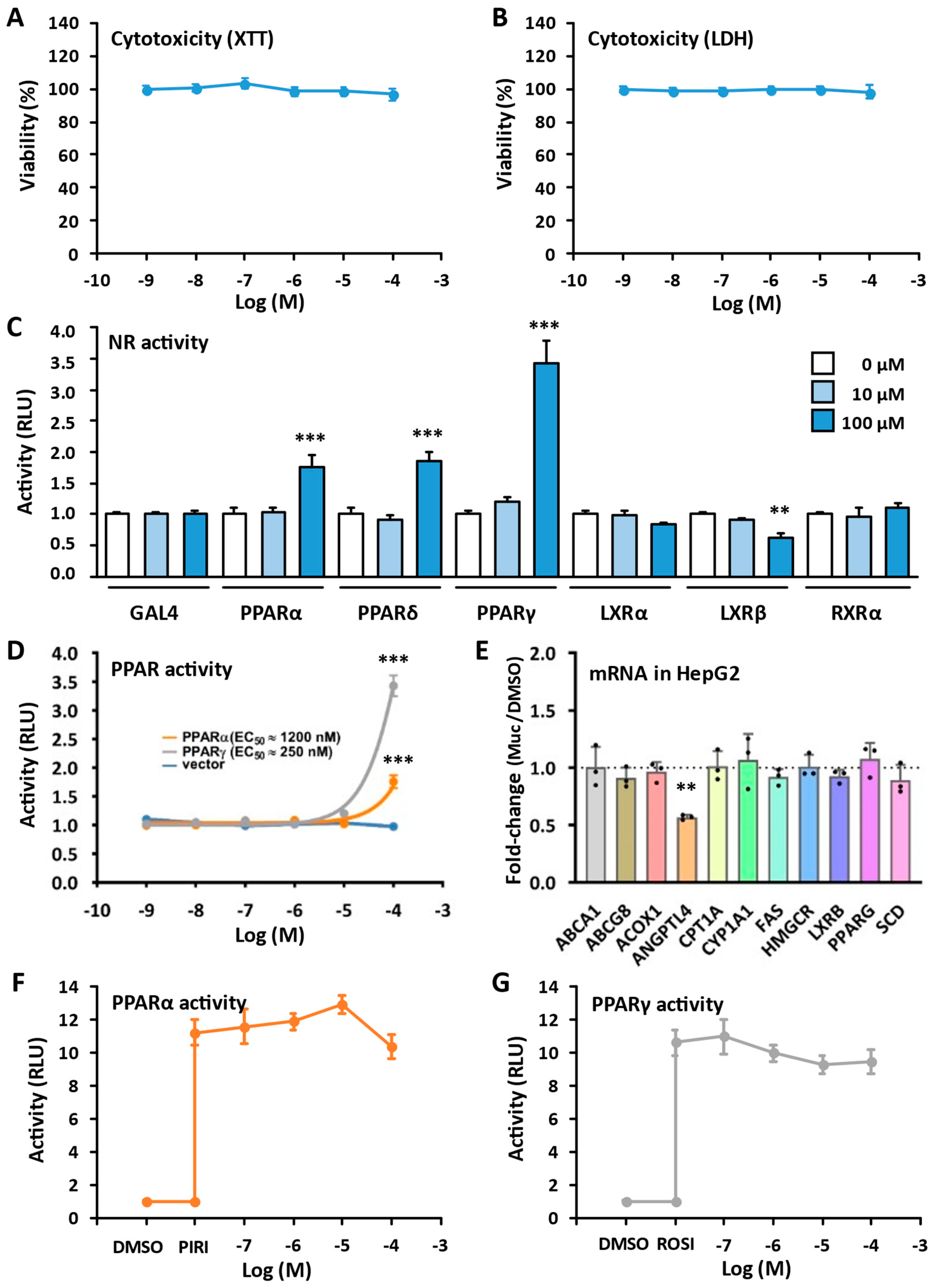
2.4. Biological Evaluations
3. Experimental Section
3.1. 1D NMR Data of Methyl Ester of (−)-Mucosin (7)
3.2. Molecular Modeling Experiments
3.3. Cytotoxicity Assays
3.4. Luciferase Assays
3.5. Gene Expression in HepG2 Cells: cDNA Synthesis and Real-Time Quantitative PCR
3.6. 15-LOX Inhibition Experiment
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wiktorowska-Owczarek, A.; Berezinska, M.; Nowak, J.Z. PUFAs: Structures, metabolism and functions. Adv. Clin. Exp. Med. 2015, 24, 931–941. [Google Scholar] [CrossRef] [PubMed]
- An, J.-U.; Song, Y.-S.; Kim, K.-R.; Ko, Y.-J.; Yoon, D.-Y.; Oh, D.-K. Biotransformation of polyunsaturated fatty acids to bioactive hepoxilins and trioxilins by microbial enzymes. Nat. Commun. 2018, 9, 128. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Singh, I.P. Structural diversity of marine oxylipins. In Lipid Biotechnology; Tsung Min Kuo, H.G., Ed.; CRC Press: New York, NY, USA, 2002; pp. 249–275. [Google Scholar]
- Gerwick, W.H. Carbocyclic oxylipins of marine origin. Chem. Rev. 1993, 93, 1807–1823. [Google Scholar] [CrossRef]
- Montaser, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef] [PubMed]
- Gerhart, D.J. Prostaglandin A-2 an agent of chemical defense in the Caribbean gorgonian Plexaura homomalla. Mar. Ecol. Prog. Ser. 1984, 19, 181–187. [Google Scholar] [CrossRef]
- Flower, R.J. Prostaglandins, bioassay and inflammation. Br. J. Pharmacol. 2006, 147, S182–S192. [Google Scholar] [CrossRef]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Hansen, T.V.; Serhan, C.N. Protectins: Their biosynthesis, metabolism and structure-functions. Biochem. Pharmacol. 2022, 206, 115330. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef]
- Leal, M.C.; Puga, J.; Serodio, J.; Gomes, N.C.; Calado, R. Trends in the discovery of new marine natural products from invertebrates over the last two decades–where and what are we bioprospecting? PLoS ONE 2012, 7, e30580. [Google Scholar] [CrossRef]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2019, 36, 122–173. [Google Scholar] [CrossRef] [PubMed]
- Casapullo, A.; Scognamiglio, G.; Cimino, G. Mucosin: A new bicyclic eicosanoid from the Mediterranean sponge Reniera mucosa. Tetrahedron Lett. 1997, 38, 3643–3646. [Google Scholar] [CrossRef]
- Rochfort, S.J.; Watson, R.; Capon, R.J. Dictyosphaerin: A novel bicyclic lipid from a southern Australian marine green algae, Dictyosphaeria sericea. J. Nat. Prod. 1996, 59, 1154–1156. [Google Scholar] [CrossRef]
- Gallantree-Smith, H.C.; Antonsen, S.G.; Görbitz, C.H.; Hansen, T.V.; Nolsøe, J.M.; Stenstrøm, Y.H. Total synthesis based on the originally claimed structure of mucosin. Org. Biomol. Chem. 2016, 14, 8433–8437. [Google Scholar] [CrossRef] [PubMed]
- Antonsen, S.G.; Gallantree-Smith, H.; Görbitz, C.H.; Hansen, T.V.; Stenstrøm, Y.H.; Nolsøe, J.M. Stereopermutation on the Putative Structure of the Marine Natural Product Mucosin. Molecules 2017, 22, 1720. [Google Scholar] [CrossRef] [PubMed]
- Nolsøe, J.M.; Antonsen, S.; Görbitz, C.H.; Hansen, T.V.; Nesman, J.I.; Røhr, Å.K.; Stenstrøm, Y.H. Total Synthesis of (−)-Mucosin and Revision of Structure. J. Org. Chem. 2018, 83, 15066–15076. [Google Scholar] [CrossRef]
- Nolsøe, J.M.; Aursnes, M.; Stenstrøm, Y.H.; Hansen, T.V. Some Biogenetic Considerations Regarding the Marine Natural Product (−)-Mucosin. Molecules 2019, 24, 4147. [Google Scholar] [CrossRef] [PubMed]
- Antonsen, S.G. Total Synthesis as a Tool for Structural Elucidation of Some Marine Lipid Natural Products. Ph.D. Thesis, Norwegian University of Life Sciences, Ås, Norway, 2017. [Google Scholar]
- Nicolaou, K.; Snyder, S.A. Chasing molecules that were never there: Misassigned natural products and the role of chemical synthesis in modern structure elucidation. Angew. Chem. Int. Ed. 2005, 44, 1012–1044. [Google Scholar] [CrossRef]
- Henderson, A.R.; Stec, J.; Owen, D.R.; Whitby, R.J. The first total synthesis of (+)-mucosin. Chem. Commun. 2012, 48, 3409–3411. [Google Scholar] [CrossRef]
- Heathcock, C.H.; Davis, B.R.; Hadley, C.R. Synthesis and biological evaluation of a monocyclic, fully functional analog of compactin. J. Med. Chem. 1989, 32, 197–202. [Google Scholar] [CrossRef]
- Friebolin, H. Basic One-and Two-Dimensional NMR Spectroscopy, 5th ed.; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Dodziuk, H.; Jaszuński, M.; Schilf, W. 1H and 13C NMR chemical shifts and spin–spin coupling constants in trans-and cis-decalins. Magn. Reson. Chem. 2005, 43, 639–646. [Google Scholar] [CrossRef]
- Haeggström, J.Z.; Funk, C.D. Lipoxygenase and Leukotriene Pathways: Biochemistry, Biology, and Roles in Disease. Chem. Rev. 2011, 111, 5866–5898. [Google Scholar] [CrossRef]
- Serhan, C.N.; Petasis, N.A. Resolvins and Protectins in Inflammation Resolution. Chem. Rev. 2011, 111, 5922–5943. [Google Scholar] [CrossRef]
- Kobe, M.J.; Neau, D.B.; Mitchell, C.E.; Bartlett, S.G.; Newcomer, M.E. The structure of human 15-lipoxygenase-2 with a substrate mimic. J. Biol. Chem. 2014, 289, 8562–8569. [Google Scholar] [CrossRef]
- Krey, G.; Braissant, O.; L’Horset, F.; Kalkhoven, E.; Perroud, M.; Parker, M.G.; Wahli, W. Fatty acids, eicosanoids, and hypolipidemic agents identified as ligands of peroxisome proliferator-activated receptors by coactivator-dependent receptor ligand assay. Mol. Endocrinol. 1997, 11, 779–791. [Google Scholar] [CrossRef]
- Gronemeyer, H.; Gustafsson, J.-Å.; Laudet, V. Principles for modulation of the nuclear receptor superfamily. Nat. Rev. Drug Discov. 2004, 3, 950–964. [Google Scholar] [CrossRef] [PubMed]
- Sæther, T.; Paulsen, S.M.; Tungen, J.E.; Vik, A.; Aursnes, M.; Holen, T.; Hansen, T.V.; Nebb, H.I. Synthesis and biological evaluations of marine oxohexadecenoic acids: PPARα/γ dual agonism and anti-diabetic target gene effects. Eur. J. Med. Chem. 2018, 155, 736–753. [Google Scholar] [CrossRef] [PubMed]
- Arnesen, H.; Haj-Yasein, N.N.; Tungen, J.E.; Soedling, H.; Matthews, J.; Paulsen, S.M.; Nebb, H.I.; Sylte, I.; Hansen, T.V.; Sæther, T. Molecular modelling, synthesis, and biological evaluations of a 3, 5-disubstituted isoxazole fatty acid analogue as a PPARα-selective agonist. Bioorganic Med. Chem. 2019, 27, 4059–4068. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Bobiński, R.; Dutka, M. Self-regulation of the inflammatory response by peroxisome proliferator-activated receptors. Inflamm. Res. 2019, 68, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.K.; Zhuang, Y.; Wahli, W. Synthetic and natural Peroxisome Proliferator-Activated Receptor (PPAR) agonists as candidates for the therapy of the metabolic syndrome. Expert Opin. Ther. Targets 2017, 21, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Ju, Z.; Su, M.; Hong, J.; Ullah, S.; La Kim, E.; Zhao, C.-H.; Moon, H.R.; Kim, S.; Jung, J.H. Design of PPAR-γ agonist based on algal metabolites and the endogenous ligand 15-deoxy-Δ12, 14-prostaglandin J2. Eur. J. Med. Chem. 2018, 157, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Moldes-Anaya, A.; Sæther, T.; Uhlig, S.; Nebb, H.I.; Larsen, T.; Eilertsen, H.C.; Paulsen, S.M. Two Isomeric C16 Oxo-Fatty Acids from the Diatom Chaetoceros karianus Show Dual Agonist Activity towards Human Peroxisome Proliferator-Activated Receptors (PPARs) α/γ. Mar. Drugs 2017, 15, 148. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Waku, T.; Shiraki, T.; Oyama, T.; Morikawa, K. Atomic structure of mutant PPARgamma LBD complexed with 15d-PGJ2: Novel modulation mechanism of PPARgamma/RXRalpha function by covalently bound ligands. FEBS Lett. 2009, 583, 320–324. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef]
- Lyckander, I.M.; Malterud, K.E. Lipophilic flavonoids from Orthosiphon spicatus prevent oxidative inactivation of 15-lipoxygenase. Prostaglandins Leukot. Essent. Fat. Acids 1996, 54, 239–246. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Accession Number | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|---|
| ABCA1 | NM_005502.3 | TGTCCAGTCCAGTAATGGTTCTGT | CGAGATATGGTCCGGATTGC |
| ABCG8 | NM_001357321.2 | CCCGAGTCCTACGAAGATGC | CAGGTCTCGGAAGTCGTTGG |
| ACOX1 | NM_004035.6 | CTTCAACCCGGAGCTGCTTA | ATGTTCTCGATCTCTCGGCG |
| ANGPTL4 | NM_139314.2 | TCCACCGACCTCCCGTTAG | GGCCACCTTGTGGAAGAGTT |
| CPT1A | NM_001876.3 | CAGGAGACAGAGTTCCCTGG | TCTAACGTCACGAAGAACGCT |
| CYP1A1 | NM_000499.5 | TGGTCTCCCTTCTCTACACTCTTGT | ATTTTCCCTATTACATTAAATCAATGGTTCT |
| FASN | NM_004104.4 | CTTCAAGGAGCAAGGCGTGA | ACTGGTACAACGAGCGGATG |
| HMGCR | XM_011543357.1 | CCGAATCCTGTAACTCAGAGGG | CAGCGACTGTGAGCATGAAC |
| LXRB | NM_007121.5 | ACAACCACGAGACAGAGTGTA | AACTCGAAGATGGGGTTGATG |
| PPARG | NM_015869.5 | AAATGCCTTGCAGTGGGGA | GCTTCTCCTTCTCGGCCTG |
| SCD | NM_005063.4 | ACACCCAGCTGTCAAAGAGA | GCCAGGTTTGTAGTACCTCCTC |
| TBP | NM_003194.4 | TTGTACCGCAGCTGCAAAAT | TATATTCGGCGTTTCGGGCA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nolsøe, J.M.J.; Underhaug, J.; Sørskar, Å.M.; Antonsen, S.G.; Malterud, K.E.; Gani, O.; Fan, Q.; Hjorth, M.; Sæther, T.; Hansen, T.V.; et al. Biological Evaluations, NMR Analyses, Molecular Modeling Studies, and Overview of the Synthesis of the Marine Natural Product (−)-Mucosin. Molecules 2024, 29, 994. https://doi.org/10.3390/molecules29050994
Nolsøe JMJ, Underhaug J, Sørskar ÅM, Antonsen SG, Malterud KE, Gani O, Fan Q, Hjorth M, Sæther T, Hansen TV, et al. Biological Evaluations, NMR Analyses, Molecular Modeling Studies, and Overview of the Synthesis of the Marine Natural Product (−)-Mucosin. Molecules. 2024; 29(5):994. https://doi.org/10.3390/molecules29050994
Chicago/Turabian StyleNolsøe, Jens M. J., Jarl Underhaug, Åshild Moi Sørskar, Simen Gjelseth Antonsen, Karl E. Malterud, Osman Gani, Qiong Fan, Marit Hjorth, Thomas Sæther, Trond V. Hansen, and et al. 2024. "Biological Evaluations, NMR Analyses, Molecular Modeling Studies, and Overview of the Synthesis of the Marine Natural Product (−)-Mucosin" Molecules 29, no. 5: 994. https://doi.org/10.3390/molecules29050994
APA StyleNolsøe, J. M. J., Underhaug, J., Sørskar, Å. M., Antonsen, S. G., Malterud, K. E., Gani, O., Fan, Q., Hjorth, M., Sæther, T., Hansen, T. V., & Stenstrøm, Y. H. (2024). Biological Evaluations, NMR Analyses, Molecular Modeling Studies, and Overview of the Synthesis of the Marine Natural Product (−)-Mucosin. Molecules, 29(5), 994. https://doi.org/10.3390/molecules29050994