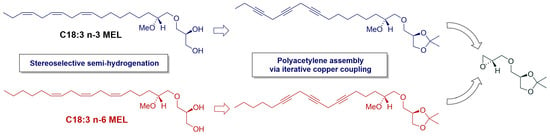
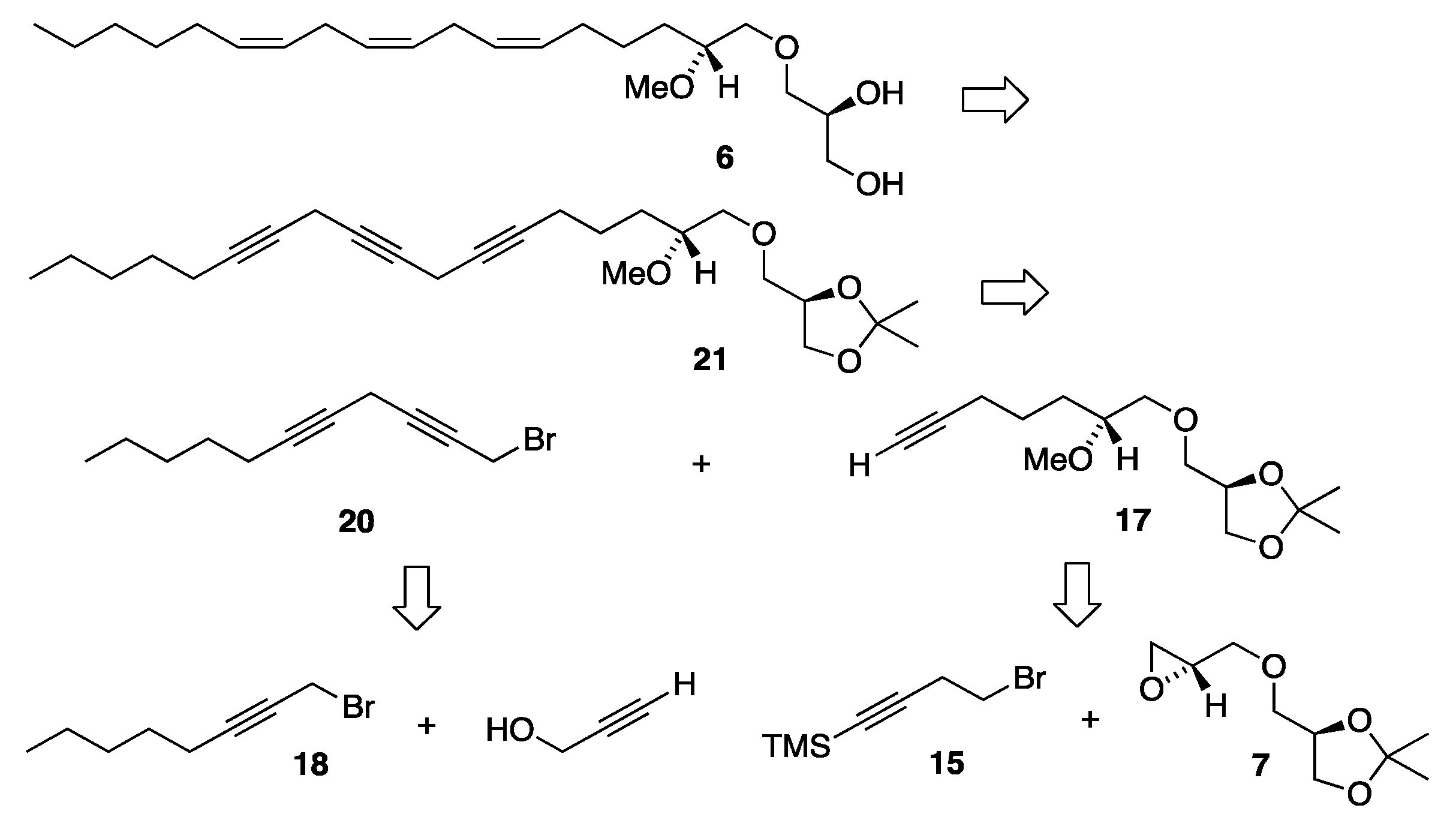
3.2. Synthesis of MEL 5
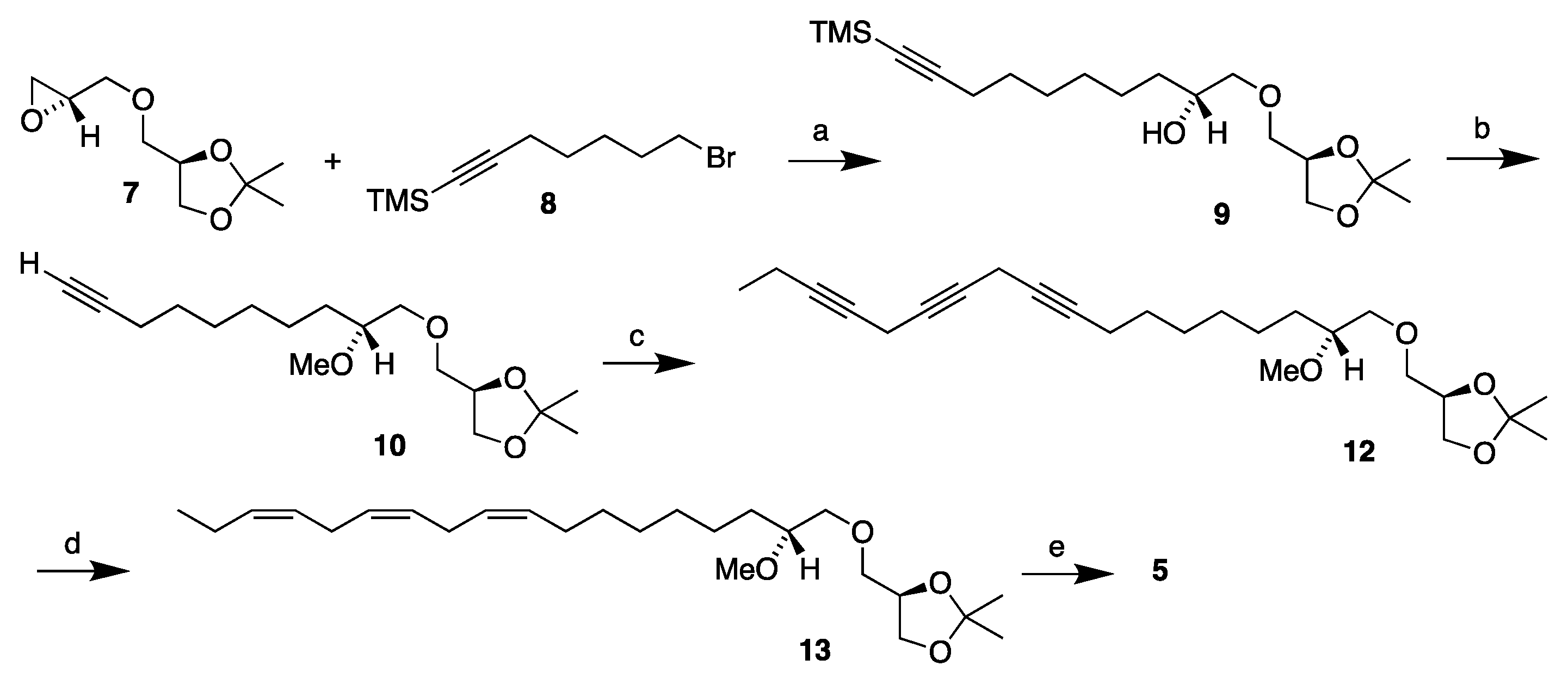
3.2.1. Synthesis of (2′R)-1-O-(2′-Methoxyocta-9′,12′,15′-triyn-1-yl)-2,3-O-isopropylidene-sn-glycerol, 12
To a stirred suspension of cuprous iodide (0.360 g, 1.89 mmol), sodium iodide (0.703 g, 4.73 mmol), and cesium carbonate (1.230 g, 3.78 mmol) in dry DMF (5 mL) under nitrogen atmosphere, 1-bromoocta-2,5-diyne
11 [
14] (0.350 g, 1.89 mmol) was added in DMF (2 mL), followed by the methoxylated monoyne
10 [
12] (0.282 g, 0.945 mmol) in DMF (2 mL). The resulting mixture was stirred vigorously for 41 h at room temperature. Then, the reaction mixture was quenched with a saturated ammonium chloride solution and stirred for 10 min. The mixture was then extracted three times with diethyl ether and the combined ether extracts washed with brine, dried over MgSO
4, filtered, then concentrated in vacuo. The crude concentrate was purified via flash column chromatography using ethyl acetate/petroleum ether (1:3) as eluent affording to the triyne product
12 as a yellow oil (0.344 g, 91% yield). [α]
D20 = −3.80 (c 2.0, ethanol). IR (NaCl, ν
max/cm
−1): 2981, 2935, 2860, 2213.
1H NMR (400 MHz, CDCl
3) δ
H: 4.29–4.23 (m, 1H, CH
sn-2), 4.05 (dd,
J = 8.3, 6.4 Hz, 1H, CH
2 sn-3), 3.75 (dd,
J = 8.3, 6.3 Hz, 1H, CH
2 sn-3), 3.56 (dd,
J = 10.0, 5.4 Hz, 1H, CH
2 sn-1), 3.52 (dd,
J = 10.3, 5.8 Hz, 1H, CH
2-1′), 3.49 (dd,
J = 10.0, 5.7 Hz, 1H, CH
2 sn-1), 3.47 (dd,
J = 10.3, 4.2 Hz, 1H, CH
2-1′), 3.40 (s, 3H, OCH
3), 3.35–3.28 (m, 1H, CH-2′), 3.15–3.12 (m, 4H, ≡CC
H2C≡), 2.20–2.12 (m, 4H, ≡CC
H2CH
2/CH
3), 1.53–1.25 (m, 10H, C
H2), 1.42 (s, 3H, C(CH
3)
2), 1.35 (s, 3H, C(CH
3)
2), 1.12 (t,
J = 7.5 Hz, 3H, CH
2C
H3) ppm.
13C{H} NMR (101 MHz, CDCl
3) δ
C: 109.5, 82.3, 80.9, 80.3, 75.01, 74.96, 74.8, 73.94, 73.91, 73.3, 67.0, 57.8, 31.5, 29.4, 29.0, 28.8, 26.9, 25.6, 25.4, 18.8, 14.0, 12.5, 10.0, 9.9 ppm. HRMS (ESI)
m/
z: [M + Na]
+ calcd for C
25H
38O
4Na 425.2668; found, 425.2674.
3.2.2. Synthesis of (2′R)-1-O-[(9′Z,12′Z,15′Z)-2′-Methoxyocta-9′,12′,15′-trien-1-yl]-2,3-O-isopropylidene-sn-glycerol, 13
The Lindlar catalyst (0.500g) was placed into a dry two-necked round-bottom flask equipped with a magnetic stirrer under nitrogen at room temperature and the flask was sealed with a septum. Toluene (20 mL) was added to the flask with a syringe. A balloon filled with hydrogen gas was then mounted on a syringe and stuck through the septum. The mixture was then stirred while the hydrogen gas was blown through the flask to replace the nitrogen atmosphere with hydrogen. Then quinoline (0.055 g, 0.438 mmol) and triyne 12 (0.353 g, 0.877 mmol), dissolved in toluene (2 mL), were added with a syringe and the reaction was stirred vigorously while being monitored with TLC. When the reaction came to completion according to the TLC (75 min), the flask was promptly opened and the reaction mixture was filtered through a celite layer on a fritted disk using dichloromethane as the eluent. After removing the solvent in vacuo, the crude concentrate was purified via flash column chromatography using ethyl acetate/petroleum ether (1:4) as the eluent, affording the crude product 13 as a light-yellow liquid (0.280 g, 78% yield). Part of the product (0.043 g) was then applied to a 10% silver nitrate-impregnated silica gel column using acetone/petroleum ether (1:4) as the eluent, resulting in the purified product 12 as a faintly yellow liquid (0.022 g, 51% yield; overall in 40% yield). [α]D20 = −4.25 (c 1.6, ethanol). IR (NaCl, νmax/cm−1): 3011, 2932, 2857, 846. 1H NMR (400 MHz, CDCl3) δH: 5.43–5.28 (m, 6H, =CH), 4.29–4.23 (m, 1H, CH sn-2), 4.05 (dd, J = 8.3, 6.4 Hz, 1H, CH2 sn-3), 3.76 (dd, J = 8.3, 6.3 Hz, 1H, CH2 sn-3), 3.56 (dd, J = 10.0, 5.4 Hz, 1H, CH2 sn-1), 3.52 (dd, J = 10.3, 5.8 Hz, 1H, CH2-1′), 3.49 (dd, J = 10.0, 5.8 Hz, 1H, CH2 sn-1), 3.48 (dd, J = 10.3, 4.4 Hz, 1H, CH2-1′), 3.40 (s, 3H, OCH3), 3.35–3.28 (m, 1H, CH-2′), 2.81 (t, J = 6.0 Hz, 4H, =CHCH2CH=), 2.11–2.03 (m, 4H, =CHCH2CH2/=CHCH2CH3), 1.52–1.45 (m, 2H, CH2-3′), 1.42 (s, 3H, C(CH3)2), 1.36 (s, 3H, C(CH3)2), 1.40–1.25 (m, 8H, CH2), 0.97 (t, J = 7.5 Hz, 3H, CH2CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 132.1, 130.4, 128.43, 128.41, 127.9, 127.3, 109.5, 80.3, 74.8, 74.0, 72.5, 67.0, 57.7, 31.6, 29.9, 29.8, 29.4, 27.4, 26.9, 25.8, 25.7, 25.6, 25.5, 20.7, 14.4 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C25H44O4Na 431.3137; found, 431.3132.
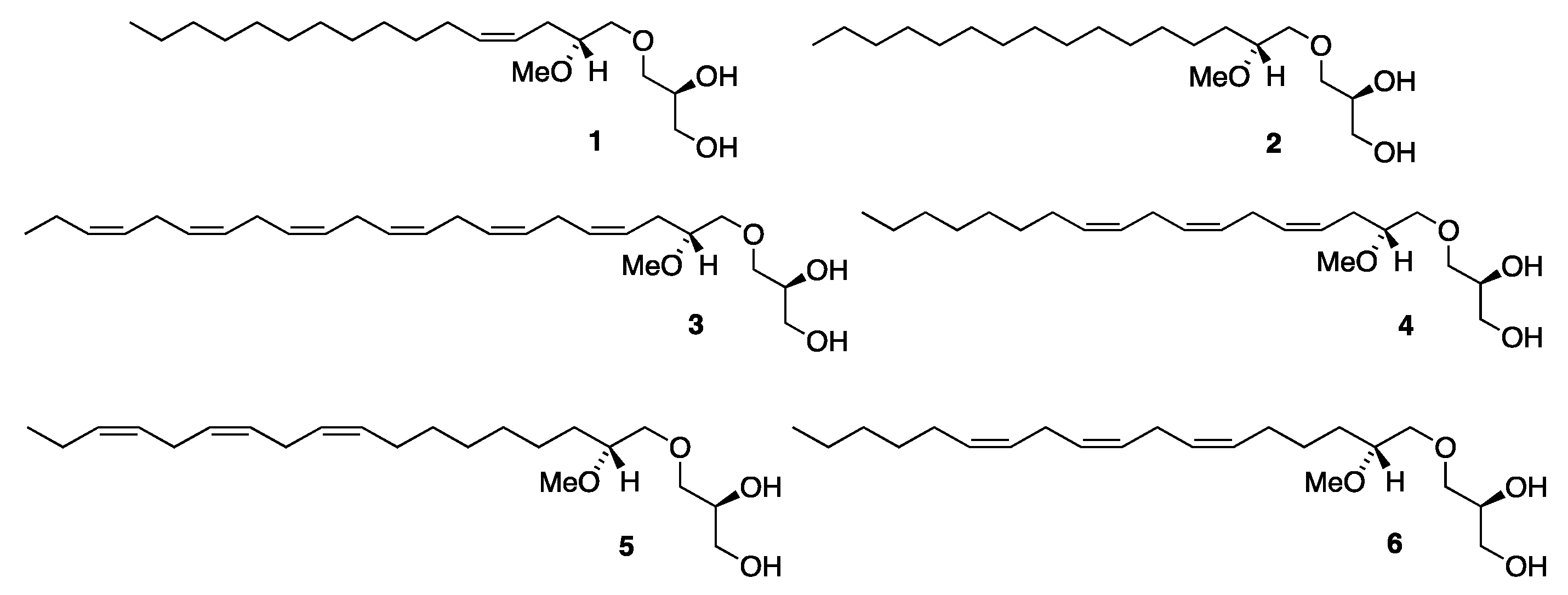
3.2.3. Synthesis of (2′R)-1-O-[(9′Z,12′Z,15′Z)-2′-Methoxyocta-9′,12′,15′-trien-1-yl]-sn-glycerol, 5
Triene 13 (0.020 g, 0.049 mmol) in ethanol (2.7 mL) was loaded into a two-necked round-bottom flask equipped with a magnetic stirrer and a reflux condenser under nitrogen. To that solution, wet Amberlyst-15® (0.007 g) was added and the reaction was heated to reflux. After refluxing for 3 h, the mixture was allowed to cool, the Amberlyst-15® was filtered off, and the solvent was removed in vacuo. The crude concentrate was then purified via flash column chromatography, eluting first with ethyl acetate/petroleum ether (1:2) and then with pure ethyl acetate to afford the final product MEL 5 as a yellow oil (0.016 g, 89%). [α]D20 = +4.27 (c 1.1, ethanol). IR (NaCl, νmax/cm−1): 3403, 3010, 2926, 2855, 1093, 928. 1H NMR (400 MHz, CDCl3) δH: 5.43–5.28 (m, 6H, =CH), 3.89-3.84 (m, 1H, CH sn-2), 3.71 (dd, J = 11.4, 3.9 Hz, 1H, CH2 sn-3), 3.64 (dd, J = 11.4, 5.2 Hz, 1H, CH2 sn-3), 3.61 (dd, J = 10.0, 4.0 Hz, 1H, CH2 sn-1), 3.56 (dd, J = 10.5, 3.5 Hz, 1H, CH2-1′), 3.54 (dd, J = 10.0, 6.4 Hz, 1H, CH2 sn-1), 3.48 (dd, J = 10.5, 6.0 Hz, 1H, CH2-1′), 3.40 (s, 3H, OCH3), 3.35–3.29 (m, 1H, CH-2′), 2.98 (br s, 1H, OH), 2.81 (t, J = 6.1 Hz, 4H, =CHCH2CH=), 2.32 (br s, 1H, OH), 2.11–2.03 (m, 4H, =CHCH2CH2/=CHCH2CH3), 1.54–1.42 (m, 2H, CH2-3′), 1.41–1.22 (m, 8H, CH2), 0.97 (t, J = 7.5 Hz, 3H, CH2CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 132.1, 130.4, 128.44, 128.39, 127.9, 127.3, 80.5, 73.8, 73.5, 70.7, 64.2, 57.5, 31.1, 29.8, 29.7, 29.4, 27.4, 25.8, 25.7, 25.5, 20.7, 14.4 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C22H40O4Na 391.2824; found, 391.2819.
3.3. Synthesis of MEL 6
3.3.1. Synthesis of 4-(Trimethylsilyl)but-3-yn-1-ol, 14
Into a dried round-bottom flask under nitrogen atmosphere containing but-3-yn-1-ol (0.385 g, 5.50 mmol) in tetrahydrofuran (18 mL) at −78 °C, n-butyllithium in cyclohexane (6.05 mL, 2 M, 12.10 mmol) was added with a syringe. After 60 min at −78 °C, trimethylsilyl chloride (1.315 g, 12.10 mmol) was added dropwise from a syringe. Five minutes after that, the reaction was allowed to reach room temperature. After 6 h, 2 M HCl (12 mL) was added and the reaction stirred vigorously for 10 min, then a saturated solution of sodium bicarbonate was added and the solution was extracted three times with diethyl ether, dried over MgSO4, filtered, and then the solvent evaporated under reduced pressure. The extract was then applied to the silica gel chromatography using ethyl acetate/petroleum ether (1:3) as the eluent, affording the product 14 as a colorless liquid (0.579 g, 74% yield). IR (NaCl, νmax/cm−1): 3347, 2960, 2900, 2177. 1H NMR (400 MHz, CDCl3) δH: 3.71 (q, J = 6.3 Hz, 2H, HOCH2), 2.51 (t, J = 6.2 Hz, 2H, ≡CCH2), 1.75 (t, J = 6.4 Hz, 1H, OH), 0.16 (s, 9H, TMS) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 103.5, 87.2, 61.0, 24.4, 0.20 (3) ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C7H14OSiNa 165.0712; found, 165.0706.
3.3.2. Synthesis of 4-Bromo-1-(trimethylsilyl)but-1-yne, 15
Into a dry round-bottom flask equipped with a magnetic stirrer, TMS-protected but-3-yn-1-ol 14 (0.202 g, 1.60 mmol) dissolved in dichloromethane (20 mL) was placed. The flask was then cooled to 0–4 °C (ice/water), carbon tetrabromide (1.427 g, 4.30 mmol) was added, and the resulting mixture was stirred for 10 min. Then triphenylphosphine (0.939 g, 3.58 mmol) was added, the reaction was stirred for further 10 min, and then more triphenylphosphine (0.376 g, 1.42 mmol) was added in small doses every 30 min until the reaction turned yellow. The reaction mixture was then diluted with diethyl ether (50 mL) and the white precipitate was filtered through a short silica layer (2 cm) using diethyl ether as the eluent. The solvent was removed in vacuo, resulting in a colorless oil which was then applied to the silica gel flash chromatography using ethyl acetate/petroleum ether (1:6) as the eluent, affording the product 15 as a colorless oil (0.710 g, 96% yield). IR (NaCl, νmax/cm−1): 3019, 2960, 2924, 2899, 2854, 2178. 1H NMR (400 MHz, CDCl3) δH: 3.43 (t, J = 7.5 Hz, 2H, BrCH2), 2.77 (t, J = 7.5 Hz, 2H, ≡CCH2), 0.16 (s, 9H, TMS) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 103.3, 87.2, 29.3, 24.5, 0.1 (3) ppm. Too volatile for HRMS measurement.
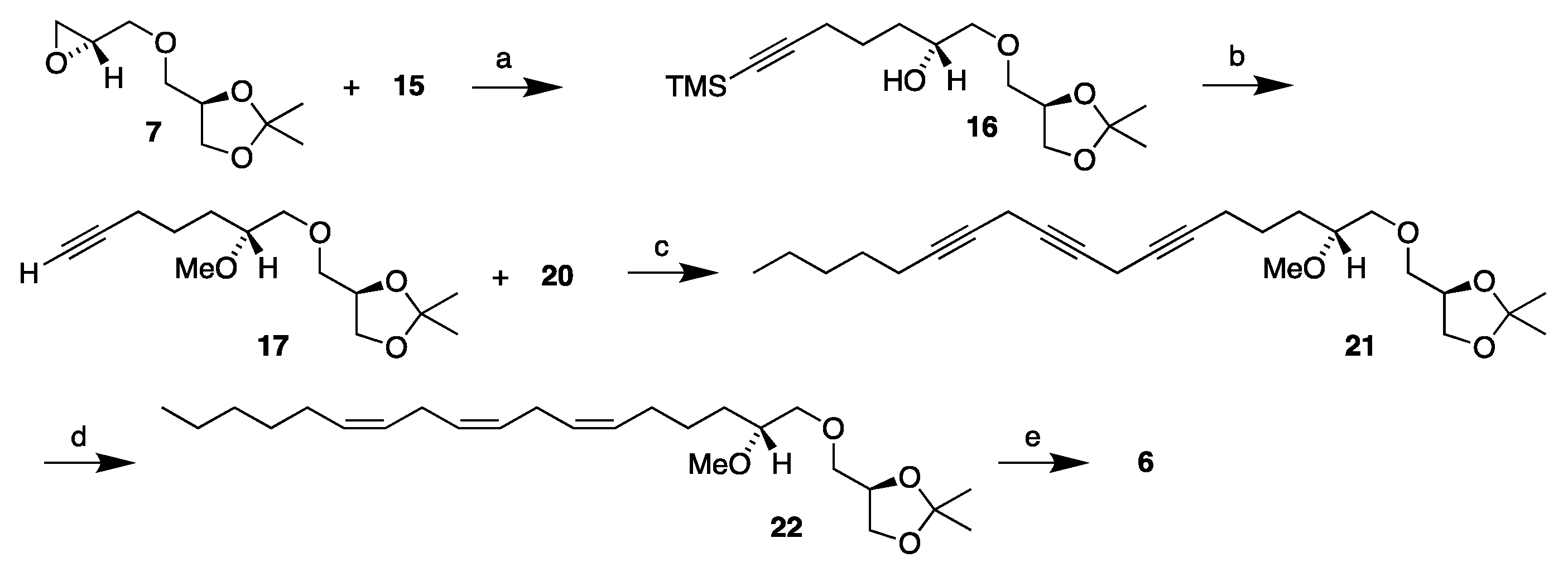
3.3.3. (2′R)-1-O-[7′-(Trimethylsilyl)-2′-hydroxyhept-6′-yn-1-yl]-2,3-O-isopropylidene-sn-glycerol, 16
To a dried two-necked round-bottom flask equipped with a reflux condenser, magnesium (0.078 g, 3.20 mmol) was added under nitrogen atmosphere. To the flask, dry THF (8 mL) and TMS-protected monoyne bromide
15 (0.586 g, 2.86 mmol) in THF (2 mL) was then added via syringe. Heat was applied and the reaction was allowed to reflux until most of the magnesium metal had disappeared. The reaction was then cooled to about −10 °C (ice/acetone) and cuprous bromide (0.046 g, 0.32 mmol) and the head piece
7 [
13] (0.301 g, 1.60 mmol) in THF (1 mL) were added. The stirred reaction was allowed to warm up overnight and was then finally quenched with a saturated aqueous ammonium chloride solution, extracted three times with diethyl ether, and the combined ether extracts were washed once with a saturated sodium chloride solution and dried over MgSO
4. After filtering and evaporation of the solvent, the resulting concentrate was applied to the silica gel column chromatography using ethyl acetate/petroleum ether (1:2) as the eluent, affording the TMS-protected monoyne
16 as a colorless liquid (0.385 g, 76% yield). [α]
D20 = +1.7 (c 0.1, ethanol). IR (NaCl, ν
max/cm
−1): 3474, 2986, 2956, 2936, 2900, 2872, 2174.
1H NMR (400 MHz, CDCl
3) δ
H: 4.33–4.22 (m, 1H, CH
sn-2), 4.05 (dd,
J = 8.3, 6.5 Hz, 1H, CH
2 sn-3), 3.86–3.77 (m, 1H, CH-2′), 3.74 (dd,
J = 8.3, 6.4 Hz, 1H, CH
2 sn-3), 3.56 (d,
J = 5.3 Hz, 2H, CH
2 sn-1), 3.54 (dd,
J = 9.7, 3.0 Hz, 1H, CH
2-1′), 3.35 (dd,
J = 9.7, 7.9 Hz, 1H, CH
2-1′), 2.42 (d,
J = 3.4 Hz, 1H, OH), 2.26 (t,
J = 6.8 Hz, 2H, ≡CCH
2), 1.77–1.48 (m, 4H, CH
2-3′ and ≡CCH
2C
H2), 1.43 (s, 3H, C(CH
3)
2), 1.37 (s, 3H, C(CH
3)
2), 0.14 (s, 9H, TMS) ppm.
13C{H} NMR (101 MHz, CDCl
3) δ
C: 109.7, 107.2, 100.1, 85.0, 76.2, 74.9, 72.5, 70.0, 69.9, 66.7, 32.2, 26.9, 25.5, 24.8, 20.0, 0.3 ppm. HRMS (ESI)
m/z: [M + Na]
+ calcd for C
16H
30O
4SiNa 337.1811; found, 337.1806.
3.3.4. (2′R)-1-O-(2′-Methoxydec-6′-yn-1-yl)-2,3-O-isopropylidene-sn-glycerol, 17
Into a flame-dried round-bottom 25 mL flask equipped with a magnetic stirrer, the TMS-protected alkyne alcohol 16 (0.093 g, 0.296 mmol) dissolved in dry DMF (6 mL) was placed and molecular sieves were added (0.2 g). Into the stirred mixture, potassium hydroxide (0.066 g, 1.18 mmol) was added and stirred for 10 min. Subsequently methyl iodide (0.420 g, 2.96 mmol) was added. The reaction was monitored with TLC, and after two hours, more potassium hydroxide (0.015 g, 0.27 mmol) was added, and then also every 40 min until the reaction was nearly completed according to TLC. When the reaction came to a stop according to TLC, the reaction mixture was quenched with a saturated ammonium chloride solution and extracted three times with diethyl ether. The combined extracts were washed with brine, dried over MgSO4, filtered, then concentrated in vacuo. The resulting concentrate was purified via flash silica gel column chromatography using ethyl acetate/petroleum ether (1:1) as the eluent, affording the methoxylated monoyne product 16 as a colorless liquid (0.063 g, 83% yield). [α]D20 = −3.29 (c 0.7, ethanol). IR (NaCl, νmax/cm−1): 3290, 2986, 2934, 2872, 2828, 2117. 1H NMR (400 MHz, CDCl3) δH: 4.30–4.22 (m, 1H, CH sn-2), 4.05 (dd, J = 8.3, 6.4 Hz, 1H, CH2 sn-3), 3.75 (dd, J = 8.3, 6.3 Hz, 1H, CH2 sn-3), 3.56 (dd, J = 10.0, 5.4 Hz, 1H, CH2 sn-1), 3.54 (dd, J = 10.3, 5.6 Hz, 1H, CH2-1′), 3.49 (dd, J = 10.0, 5.7 Hz, 1H, CH2 sn-1), 3.48 (dd, J = 10.3, 5.7 Hz, 1H, CH2-1′), 3.40 (s, 3H, OCH3), 3.37–3.31 (m, 1H, CH-2′), 2.25–2.14 (m, 2H, ≡CCH2), 1.94 (t, J = 2.6 Hz, 1H, ≡CH), 1.71–1.51 (m, 4H, CH2-3′ and ≡CCH2CH2), 1.42 (s, 3H, C(CH3)2), 1.36 (s, 3H, C(CH3)2) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 109.5, 84.4, 79.7, 74.8, 73.8, 72.6, 68.6, 67.0, 57.8, 30.7, 26.9, 25.6, 24.5, 18.7 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C14H24O4Na 279.1572; found, 279.1567.
3.3.5. Synthesis of 1-Bromooct-2-yne, 18
Into a dry two-necked round-bottom flask equipped with a magnetic stirrer, oct-2-yn-1-ol (0.500 g, 3.96 mmol) dissolved in dichloromethane (22 mL) was placed. The flask was then cooled to 0–4 °C (ice/water), carbon tetrabromide (1.58 g, 4.75 mmol) was added, and the resulting mixture was stirred for 10 min. Then triphenylphosphine (1.040 g, 3.97 mmol) was added and the reaction was stirred for a further 10 min. After 15 min and 30 min, more triphenylphosphine (0.208 g, 0.79 mmol) was added, at which point the reaction mixture turned yellow (in all 1.456 g, 5.55 mmol). The reaction mixture was then diluted with diethyl ether (50 mL) and the white precipitate was filtered through a short silica layer (2 cm) using diethyl ether as the eluent. The solvent was removed in vacuo, resulting in a colorless oil which was then applied to the silica gel flash chromatography using ethyl acetate/petroleum ether (1:6) as the eluent, affording the product 18 as a colorless oil (0.745 g, 100% yield). IR (NaCl, νmax/cm−1): 2957, 2932, 2871, 2860, 2311, 2234. 1H NMR (400 MHz, CDCl3) δH: 3.93 (t, J = 2.4 Hz, 2H, BrCH2), 2.23 (tt, J = 7.2, 2.4 Hz, 2H, ≡CCH2CH2), 1.58–1.46 (m, 2H, ≡CCH2CH2), 1.41–1.23 (m, 4H, CH2CH2CH3), 0.90 (t, J = 7.0 Hz, 3H, CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 88.5, 75.4, 31.2, 28.2, 22.3, 19.1, 16.0, 14.1 ppm. Too volatile for HRMS measurement.
3.3.6. Synthesis of Undeca-2,5-diyn-1-ol, 19
To a stirred suspension of cuprous iodide (0.236 g, 1.24 mmol), sodium iodide (0.184 g, 1.24 mmol), and cesium carbonate (0.403 g, 1.24 mmol) in dry DMF (5 mL) under nitrogen atmosphere was added 1-bromooct-2-yne 18 (0.234 g, 1.24 mmol) in DMF (2 mL), followed by propargyl alcohol (0.069 g, 1.24 mmol). The resulting mixture was stirred vigorously for 24 h at room temperature. Then, the reaction mixture was quenched with a saturated ammonium chloride solution and stirred for 10 min. The mixture was then extracted three times with diethyl ether and the combined ether extracts were washed with brine, dried over MgSO4, filtered, then concentrated in vacuo. The crude concentrate was purified via flash column chromatography using ethyl acetate/petroleum ether (1:3) as the eluent, affording the diyne alcohol product 19 as a yellow oil (0.150 g, 74% yield). IR (NaCl, νmax/cm−1): 3358, 2957, 2932, 2871, 2860, 2259. 1H NMR (400 MHz, CDCl3) δH: 4.26 (t, J = 2.2 Hz, 2H, HOCH2), 3.19 (quin, J = 2.3 Hz, 2H, ≡CCH2C≡), 2.15 (tt, J = 7.2, 2.4 Hz, 2H, ≡CCH2CH2), 1.55 (br s, 1H, OH) 1.49 (quin, J = 7.3 Hz, 2H, ≡CCH2CH2), 1.40–1.25 (m, 4H, CH2CH2CH3), 0.89 (t, J = 7.2 Hz, 3H, CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 81.4, 81.1, 78.5, 73.4, 51.5, 31.2, 28.6, 22.4, 18.8, 14.1, 10.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C11H16ONa 187.1099; found, 187.1093.
3.3.7. Synthesis of 1-Bromoundeca-2,5-diyne, 20
Into a dry two-necked round-bottom flask equipped with a magnetic stirrer, undeca-2,5-diyn-1-ol 19 (0.168 g, 1.02 mmol) dissolved in dichloromethane (20 mL) was placed. The flask was then cooled to 0–4 °C (ice/water), carbon tetrabromide (0.407 g, 1.23 mmol) was added, and the resulting mixture was stirred for 10 min. Triphenylphosphine (0.268 g, 1.02 mmol) was added and the reaction was stirred for a further 40 min. Then, more triphenylphosphine (0.080 g, 0.30 mmol) was added, after which the reaction turned yellow within 20 min and showed completion according to TLC. The reaction mixture was then diluted with diethyl ether (50 mL) and the precipitate was filtered through a short silica layer (2 cm) using diethyl ether as the eluent. The solvent was removed in vacuo, resulting in a yellow oil which was applied to the silica gel flash chromatography using ethyl acetate/petroleum ether (1:6) as the eluent, affording the diyne product 20 as a yellow oil (0.207 g, 89% yield). IR (NaCl, νmax/cm−1): 3003, 2957, 2931, 2870, 2859, 2268, 2234. 1H NMR (400 MHz, CDCl3) δH: 3.91 (t, J = 2.3 Hz, 2H, BrCH2), 3.21 (quin, J = 2.4 Hz, 2H, ≡CCH2C≡), 2.15 (tt, J = 7.2, 2.4 Hz, 2H, ≡CCH2CH2), 1.49 (quin, J = 7.2 Hz, 2H, ≡CCH2CH2), 1.40–1.27 (m, 4H, CH2CH2CH3), 0.90 (t, J = 7.2 Hz, 3H, CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 82.2, 81.5, 75.2, 72.8, 31.1, 28.4, 22.2, 18.7, 14.9, 14.0, 10.1 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C11H15BrNa 249.0255; found, 249.0249.
3.3.8. Synthesis of (2′R)-1-O-(2′-Methoxyocta-6′,9′,12′-triyn-1-yl)-2,3-O-isopropylidene-sn-glycerol, 21
To a stirred suspension of cuprous iodide (0.119 g, 0.62 mmol), sodium iodide (0.232 g, 1.56 mmol) and cesium carbonate (0.407 g, 1.25 mmol) in dry DMF (3 mL) under nitrogen atmosphere was added 1-bromoundeca-2,5-diyne 20 (0.207 g, 0.91 mmol) in DMF (2 mL), followed by the methoxylated monoyne 17 (0.080 g, 0.312 mmol) in DMF (2 mL). The resulting mixture was stirred vigorously for 43 h at room temperature. Then, the reaction mixture was quenched with a saturated ammonium chloride solution and stirred for 10 min. The mixture was then extracted three times with diethyl ether and the combined ether extracts were washed with brine, dried over MgSO4, filtered, then concentrated in vacuo. The crude concentrate was purified via flash column chromatography using ethyl acetate/petroleum ether (4:11) as the eluent, affording the triyne product 21 as a yellow oil (0.119 g, 94% yield). [α]D20 = -3.14 (c 0.7, ethanol). IR (NaCl, νmax/cm−1): 2984, 2933, 2872, 2211, 1088. 1H NMR (400 MHz, CDCl3) δH: 4.30–4.22 (m, 1H, CH sn-2), 4.05 (dd, J = 8.3, 6.4 Hz, 1H, CH2 sn-3), 3.76 (dd, J = 8.3, 6.3 Hz, 1H, CH2 sn-3), 3.56 (dd, J = 10.0, 5.4 Hz, 1H, CH2 sn-1), 3.53 (dd, J = 10.3, 5.8 Hz, 1H, CH2-1′), 3.49 (dd, J = 10.0, 5.7 Hz, 1H, CH2 sn-1), 3.48 (dd, J = 10.3, 4.3 Hz, 1H, CH2-1′), 3.40 (s, 3H, OCH3), 3.37–3.30 (m, 1H, CH-2′), 3.15–3.09 (m, 4H, ≡CCH2C≡), 2.22–2.10 (m, 4H, ≡CCH2CH2), 1.68–1.44 (m, 6H, CH2), 1.42 (s, 3H, C(CH3)2), 1.36 (s, 3H, C(CH3)2), 1.38–1.23 (m, 4H, CH2CH2CH3), 0.89 (t, J = 7.1 Hz, 3H, CH2CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 109.5, 81.1, 80.5, 79.8, 75.1, 74.9, 74.8, 74.3, 73.84, 73.81, 72.6, 67.0, 57.8, 31.2, 30.8, 28.6, 26.9, 25.6, 24.7, 22.4, 19.0, 18.8, 14.1, 9.9 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C25H38O4Na 425.2668; found, 425.2662.
3.3.9. Synthesis of (2′R)-1-O-[(6′Z,9′Z,12′Z)-2′-Methoxyocta-6′,9′,12′-trien-1-yl]-2,3-O-isopropylidene-sn-glycerol, 22
The Lindlar catalyst (0.117 g) was placed into a dry two-necked round-bottom flask equipped with a magnetic stirrer under nitrogen atmosphere at room temperature and the flask was sealed with a septum. Toluene (5 mL) was added to the flask with a syringe. A balloon filled with hydrogen gas was then mounted on a syringe and stuck through the septum. The mixture was then stirred while the hydrogen gas was blown through the flask to replace the nitrogen atmosphere with hydrogen. Then quinoline (0.018 g, 0.145 mmol) and triyne 21 (0.109 g, 0.290 mmol), dissolved in toluene (2 mL), were added with a syringe and the reaction was stirred vigorously while being monitored with TLC. When the reaction came to completion according to the TLC (40 min), the flask was promptly opened and the reaction mixture was filtered through a celite layer on a fritted disk using dichloromethane as the eluent. After removing the solvent in vacuo, the crude concentrate was purified via flash column chromatography using ethyl acetate/petroleum ether (1:4) as the eluent, affording the crude product 22 as a light-yellow liquid (0.087 g, 78% yield). The crude product was then applied to a 10% silver nitrate-impregnated silica gel column using an ethyl acetate/petroleum ether gradient from 1:7 to 7:9 as the eluent, resulting in the purified triene product 22 as a faintly yellow liquid (0.037 g, 43% yield; overall in a 33% yield). [α]D20 = −6.88 (c 0.9, ethanol). IR (NaCl, νmax/cm−1): 3010, 2985, 2930, 2860, 1652, 975. 1H NMR (400 MHz, CDCl3) δH: 5.45–5.28 (m, 6H, =CH), 4.32–4.21 (m, 1H, CH sn-2), 4.05 (dd, J = 8.3, 6.4 Hz, 1H, CH2 sn-3), 3.76 (dd, J = 8.3, 6.3 Hz, 1H, CH2 sn-3), 3.57 (dd, J = 10.0, 5.3 Hz, 1H, CH2 sn-1), 3.55–3.48 (m, 2H, CH2-1′ and CH2 sn-1), 3.48 (dd, J = 10.3, 4.4 Hz, 1H, CH2-1′), 3.40 (s, 3H, OCH3), 3.36–3.29 (m, 1H, CH-2′), 2.80 (t, J = 5.8 Hz, 4H, =CHCH2CH=), 2.13-2.01 (m, 4H, =CHCH2), 1.55–1.43 (m, 4H, CH2-3′ and CH2-4′), 1.42 (s, 3H, C(CH3)2), 1.36 (s, 3H, C(CH3)2), 1.40–1.24 (m, 6H, CH2), 0.89 (t, J = 6.8 Hz, 3H, CH2CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 130.5, 129.9, 128.4, 128.3, 128.1, 127.6, 109.5, 80.1, 74.7, 73.8, 72.4, 66.9, 57.7, 31.5, 31.1, 29.4, 27.3, 27.2, 26.8, 25.7 (2), 25.5, 25.4, 22.6, 14.1 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C25H44O4Na 431.3137; found, 431.3132.
3.3.10. Synthesis of (2′R)-1-O-[(6′Z,9′Z,12′Z)-2′-Methoxyocta-6′,9′,12′-trien-1-yl]-sn-glycerol, 6
Triene 22 (0.017 g, 0.042 mmol) in ethanol (3 mL) was loaded into a two-necked round-bottom flask equipped with a magnetic stirrer and a reflux condenser under nitrogen. To that solution, wet Amberlyst-15® (0.007 g) was added and the reaction was heated to reflux. After refluxing for 3 h, the mixture was allowed to cool, the Amberlyst-15® was filtered off and the solvent was removed in vacuo. The crude concentrate was then purified via flash column chromatography, eluting first with ethyl acetate/petroleum ether (1:2) and then with pure ethyl acetate to afford the final product MEL 6 as a yellow oil (0.015 g, 100%). [α]D20 = +3.26 (c 1.5, ethanol). IR (NaCl, νmax/cm−1): 3404, 3011, 2927, 2858, 1651. 1H NMR (400 MHz, CDCl3) δH: 5.44–5.28 (m, 6H, =CH), 3.89–3.84 (br m, 1H, CH sn-2), 3.73–3.69 (br dd, 1H, CH2 sn-3), 3.66–3.63 (br dd, 1H, CH2 sn-3), 3.61 (dd, J = 10.0, 3.9 Hz, 1H, CH2 sn-1), 3.56 (dd, J = 10.5, 3.6 Hz, 1H, CH2-1′), 3.54 (dd, J = 10.0, 6.3 Hz, 1H, CH2 sn-1), 3.48 (dd, J = 10.5, 6.0 Hz, 1H, CH2-1′), 3.40 (s, 3H, OCH3), 3.36–3.30 (m, 1H, CH-2′), 2.95 (br s, 1H, OH), 2.80 (t, J = 5.7 Hz, 4H, =CHCH2CH=), 2.28 (br s, 1H, OH), 2.14–2.00 (m, 4H, =CHCH2CH2), 1.58–1.22 (m, 10H, CH2), 0.89 (t, J = 6.5 Hz, 3H, CH2CH3) ppm. 13C{H} NMR (101 MHz, CDCl3) δC: 130.6, 129.8, 128.6, 128.5, 128.2, 127.7, 80.4, 73.8, 73.5, 70.7, 64.2, 57.6, 31.7 30.7, 29.5 27.4, 27.4, 25.8 (2), 25.5, 22.7, 14.2 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C22H40O4Na 391.2824; found, 391.2819.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}