

Force Fields, Quantum-Mechanical- and Molecular-Dynamics-Based Descriptors of Radiometal–Chelator Complexes
 , , ,
, , , 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
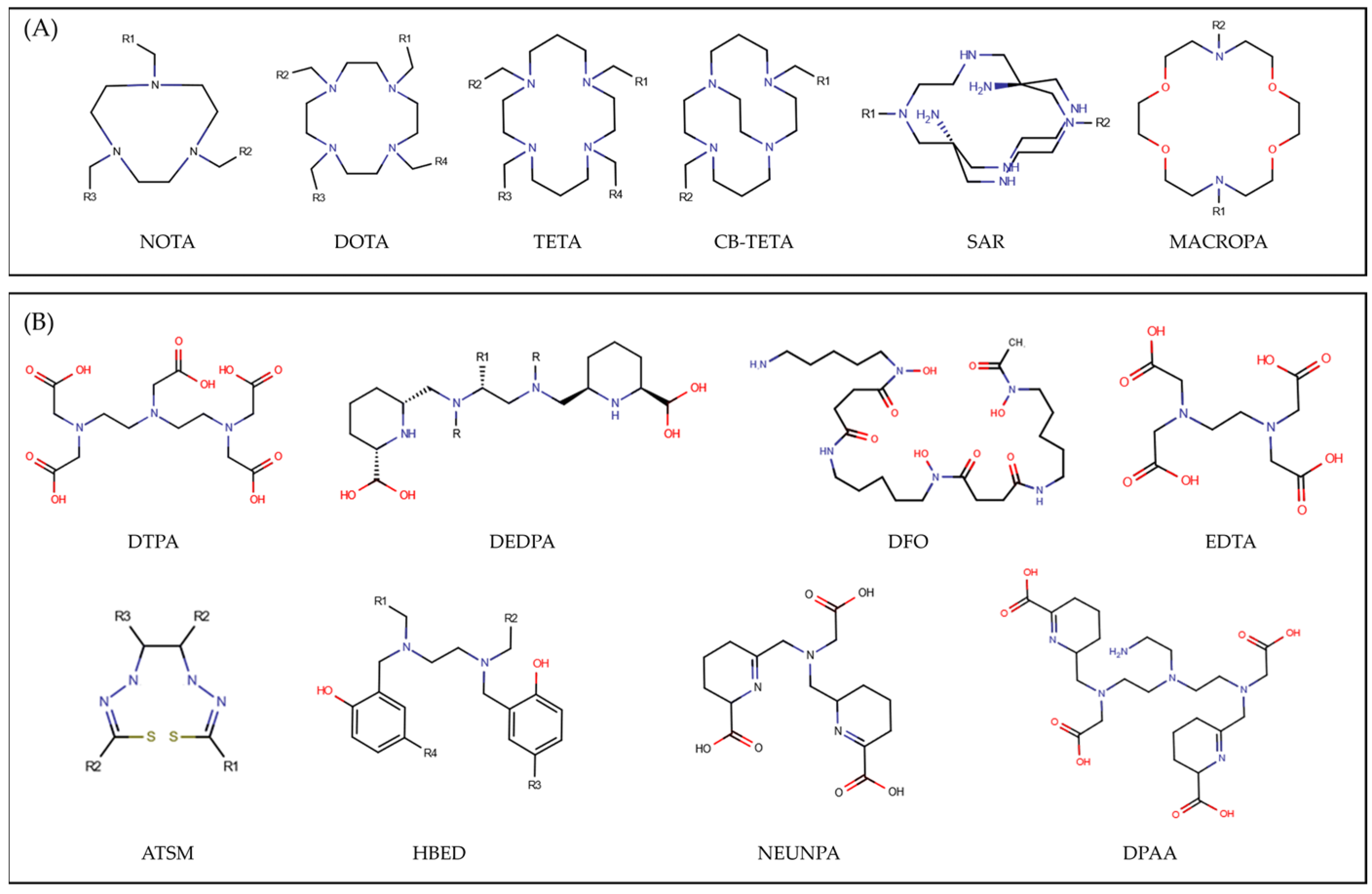
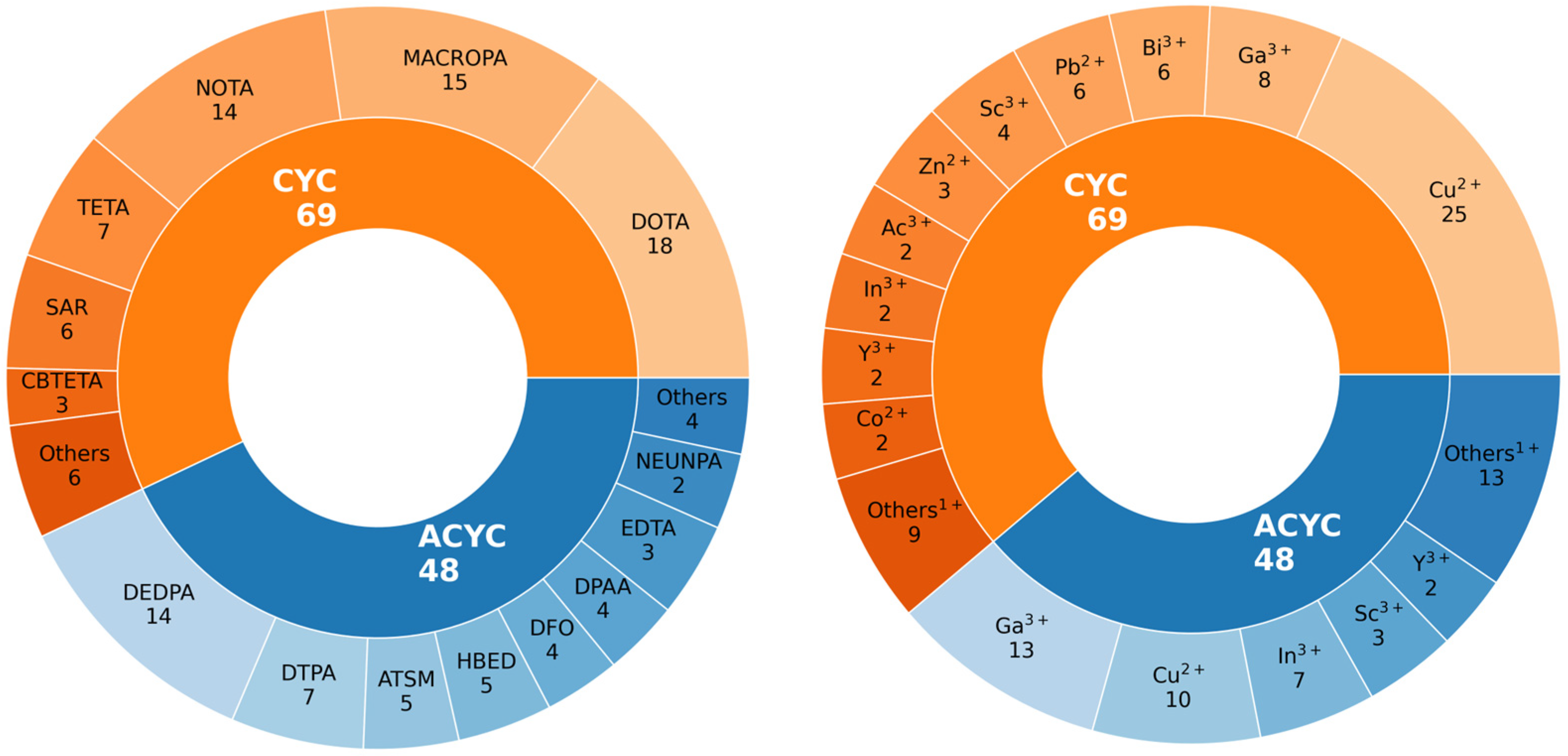
2.1. Sample of Compounds
2.2. Dataset Structure
2.2.1. Metal Center Parameter Builder Files
2.2.2. Quantum-Mechanical Data
2.2.3. Force Field Data
2.2.4. Molecular Dynamics Data
2.3. Sample of Computed Molecular Descriptors
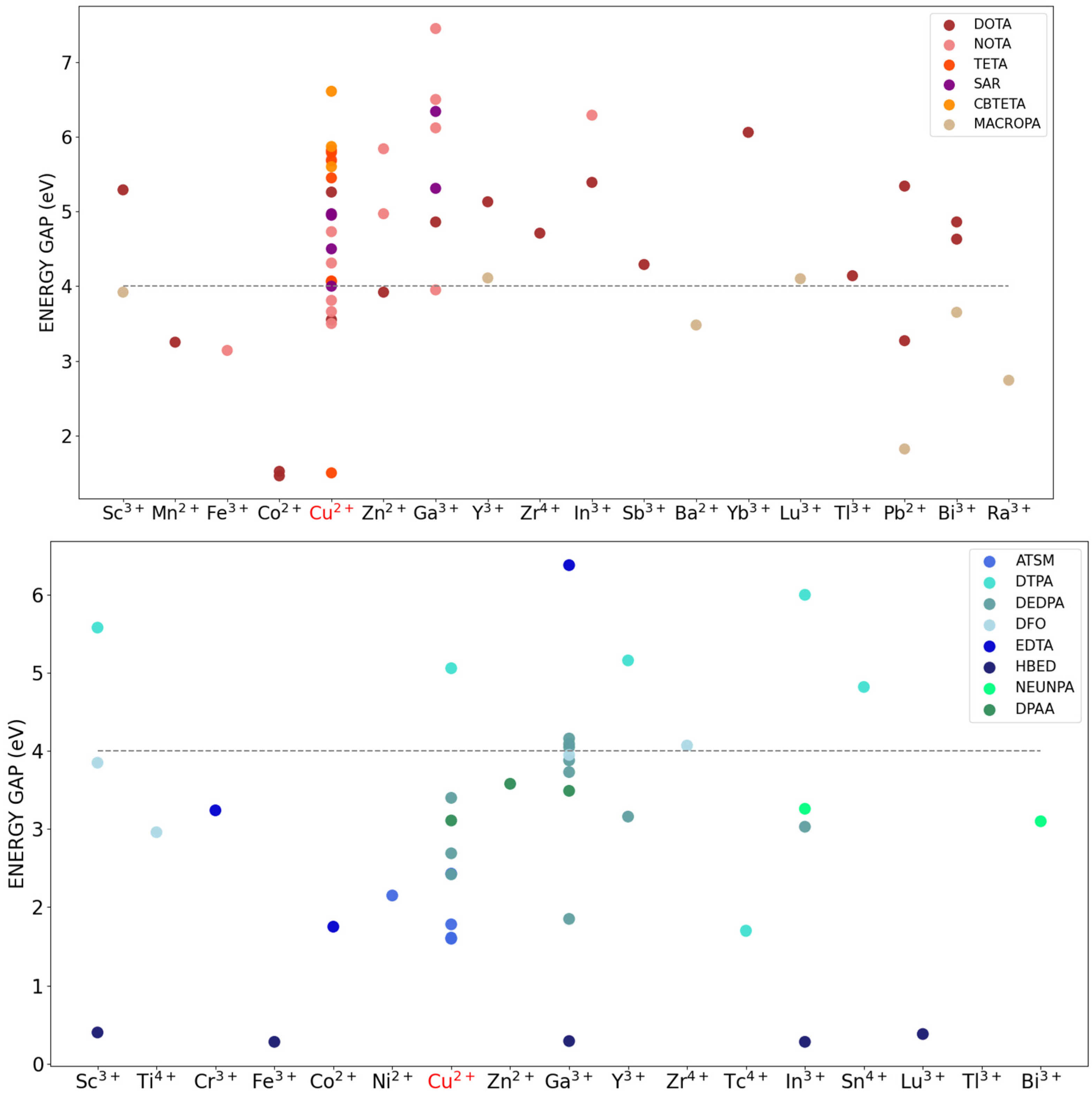
2.3.1. Computed DFT Energy-Gap as an Indicator of Stability
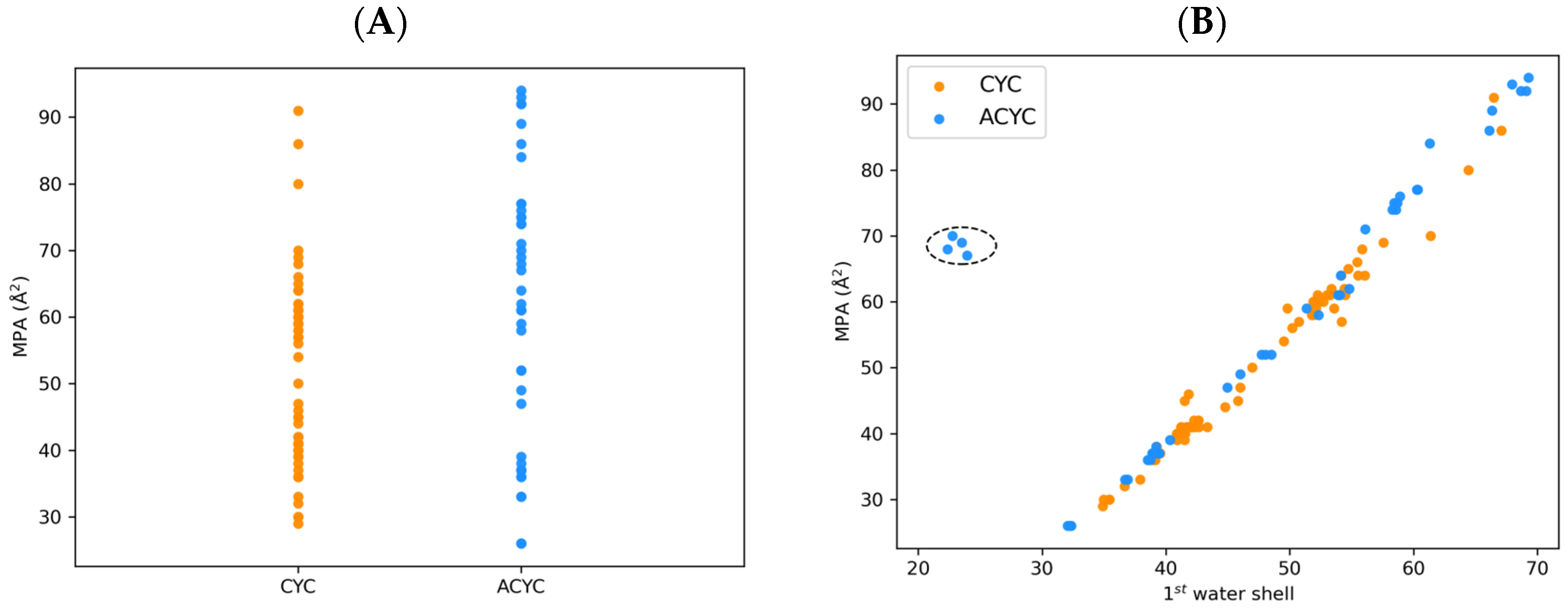
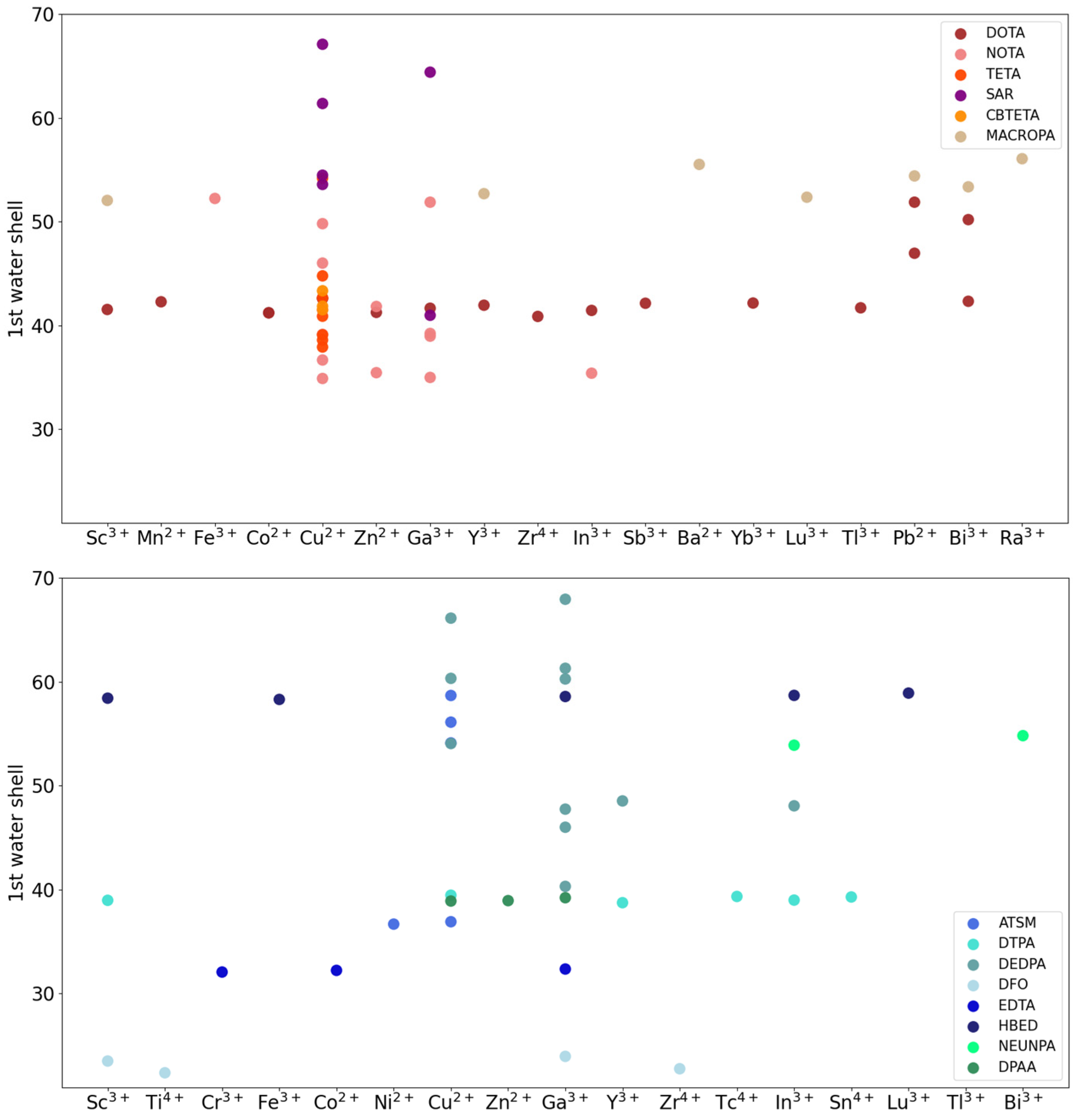
2.3.2. MD-Based Descriptors of Conformation and Solvation
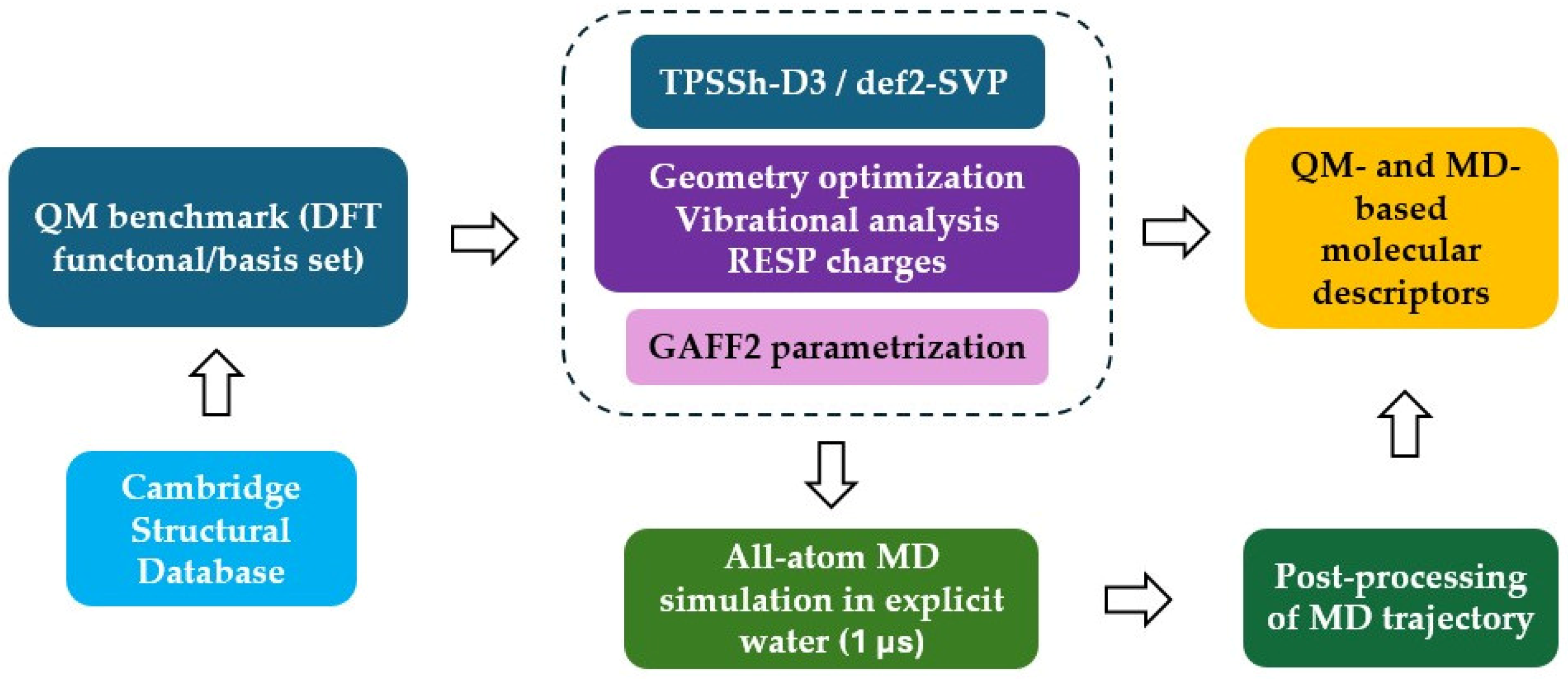
3. Computational Methods
3.1. QM Calculations
3.2. Force Field Parameter Generation
3.3. MD Simulations and Post-Processing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fani, M.; Peitl, P.; Velikyan, I. Current status of radiopharmaceuticals for the theranostics of neuroendocrine neoplasms. Pharmaceuticals 2017, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Shah, H.J.; Ruppell, E.; Bokhari, R.; Aland, P.; Lele, V.R.; Ge, C.; McIntosh, L.J. Current and upcoming radionuclide therapies in the direction of precision oncology: A narrative review. Eur. J. Radiol. Open 2023, 10, 100477. [Google Scholar] [CrossRef] [PubMed]
- Barca, C.; Griessinger, C.M.; Faust, A.; Depke, D.; Essler, M.; Windhorst, A.D.; Devoogdt, N.; Brindle, K.M.; Schäfers, M.; Zinnhardt, B.; et al. Expanding theranostic radiopharmaceuticals for tumor diagnosis and therapy. Pharmaceuticals 2022, 15, 13. [Google Scholar] [CrossRef]
- Price, E.W.; Orvig, C. Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef] [PubMed]
- Dhoundiyal, S.; Srivastava, S.; Kumar, S.; Singh, G.; Ashique, S.; Pal, R.; Mishra, N.; Taghizadeh-Hesary, F. Radiopharmaceuticals: Navigating the frontier of precision medicine and therapeutic innovation. Eur. J. Med. Res. 2024, 29, 26. [Google Scholar] [CrossRef] [PubMed]
- Gervasoni, S.; Öztürk, I.; Guccione, C.; Bosin, A.; Ruggerone, P.; Malloci, G. Interaction of Radiopharmaceuticals with Somatostatin Receptor 2 Revealed by Molecular Dynamics Simulations. J. Chem. Inf. Model. 2023, 63, 4924–4933. [Google Scholar] [CrossRef]
- Gervasoni, S.; Guccione, C.; Fanti, V.; Bosin, A.; Cappellini, G.; Golosio, B.; Ruggerone, P.; Malloci, G. Molecular simulations of SSTR2 dynamics and interaction with ligands. Sci. Rep. 2023, 13, 4768. [Google Scholar] [CrossRef]
- Boros, E.; Packard, A.B. Radioactive Transition Metals for Imaging and Therapy. Chem. Rev. 2019, 119, 870–901. [Google Scholar] [CrossRef]
- Kostelnik, T.I.; Orvig, C. Radioactive Main Group and Rare Earth Metals for Imaging and Therapy. Chem. Rev. 2019, 119, 902–956. [Google Scholar] [CrossRef]
- Morgan, K.A.; Rudd, S.E.; Noor, A.; Donnelly, P.S. Theranostic Nuclear Medicine with Gallium-68, Lutetium-177, Copper-64/67, Actinium-225, and Lead-212/203 Radionuclides. Chem. Rev. 2023, 123, 12004–12035. [Google Scholar] [CrossRef]
- Decristoforo, C.; Neels, O.; Patt, M. Emerging Radionuclides in a Regulatory Framework for Medicinal Products—How Do They Fit? Front. Med. 2021, 8, 678452. [Google Scholar] [CrossRef]
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical therapy in cancer: Clinical advances and challenges. Nat. Rev. Drug Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef] [PubMed]
- Harrison, A.; Walker, C.A.; Parker, D.; Jankowski, K.J.; Cox, J.P.L.; Craig, A.S.; Sansom, J.M.; Beeley, N.R.A.; Boyce, R.A.; Chaplin, L.; et al. The in vivo release of 90Y from cyclic and acyclic ligand-antibody conjugates. Int. J. Radiat. Appl. Instrum. 1991, 18, 469–476. [Google Scholar] [CrossRef]
- Hancock, R.D. Chelate ring size and metal ion selection: The basis of selectivity for metal ions in open-chain ligands and macrocycles. J. Chem. Educ. 1992, 69, 615–621. [Google Scholar] [CrossRef]
- Hu, A.; Wilson, J.J. Advancing Chelation Strategies for Large Metal Ions for Nuclear Medicine Applications. Acc. Chem. Res. 2022, 55, 904–915. [Google Scholar] [CrossRef]
- Boros, E.; Holland, J.P. Chemical aspects of metal ion chelation in the synthesis and application antibody-based radiotracers. J. Label. Compd. Radiopharm. 2018, 61, 652–671. [Google Scholar] [CrossRef]
- Holik, H.A.; Ibrahim, F.M.; Elaine, A.A.; Putra, B.D.; Achmad, A.; Kartamihardja, A.H.S. The Chemical Scaffold of Theranostic Radiopharmaceuticals: Radionuclide, Bifunctional Chelator, and Pharmacokinetics Modifying Linker. Molecules 2022, 27, 3062. [Google Scholar] [CrossRef]
- Blei, M.K.; Waurick, L.; Reissig, F.; Kopka, K.; Stumpf, T.; Drobot, B.; Kretzschmar, J.; Mamat, C. Equilibrium Thermodynamics of Macropa Complexes with Selected Metal Isotopes of Radiopharmaceutical Interest. Inorg. Chem. 2023, 62, 20699–20709. [Google Scholar] [CrossRef]
- Wadas, T.J.; Wong, E.H.; Weisman, G.R.; Anderson, C.J. Coordinating radiometals of copper, gallium, indium, yttrium, and zirconium for PET and SPECT imaging of disease. Chem. Rev. 2010, 110, 2858–2902. [Google Scholar] [CrossRef]
- Macpherson, D.S.; Fung, K.; Cook, B.E.; Francesconi, L.C.; Zeglis, B.M. A brief overview of metal complexes as nuclear imaging agents. Dalt. Trans. 2019, 48, 14547–14565. [Google Scholar] [CrossRef]
- Akter, A.; Lyons, O.; Mehra, V.; Isenman, H.; Abbate, V. Radiometal chelators for infection diagnostics. Front. Nucl. Med. 2022, 2, 1058388. [Google Scholar] [CrossRef]
- Brandt, M.; Cardinale, J.; Aulsebrook, M.L.; Gasser, G.; Mindt, T.L. An overview of PET radiochemistry, part 2: Radiometals. J. Nucl. Med. 2018, 59, 1500–1506. [Google Scholar] [CrossRef] [PubMed]
- Isert, C.; Atz, K.; Jiménez-Luna, J.; Schneider, G. QMugs, quantum mechanical properties of drug-like molecules. Sci. Data 2022, 9, 273. [Google Scholar] [CrossRef]
- Balcells, D.; Skjelstad, B.B. TmQM Dataset—Quantum Geometries and Properties of 86k Transition Metal Complexes. J. Chem. Inf. Model. 2020, 60, 6135–6146. [Google Scholar] [CrossRef] [PubMed]
- Malloci, G.; Vargiu, A.V.; Serra, G.; Bosin, A.; Ruggerone, P.; Ceccarelli, M. A database of force-field parameters, dynamics, and properties of antimicrobial compounds. Molecules 2015, 20, 13997–14021. [Google Scholar] [CrossRef]
- Kabylda, A.; Vassilev-Galindo, V.; Chmiela, S.; Poltavsky, I.; Tkatchenko, A. Efficient interatomic descriptors for accurate machine learning force fields of extended molecules. Nat. Commun. 2023, 14, 3562. [Google Scholar] [CrossRef]
- Li, P.; Merz, K.M. Metal Ion Modeling Using Classical Mechanics. Chem. Rev. 2017, 117, 1564–1686. [Google Scholar] [CrossRef]
- Gervasoni, S.; Malloci, G.; Bosin, A.; Vargiu, A.V.; Zgurskaya, H.I.; Ruggerone, P. AB-DB: Force-Field parameters, MD trajectories, QM-based data, and Descriptors of Antimicrobials. Sci. Data 2022, 9, 148. [Google Scholar] [CrossRef]
- Öztürk, I.; Gervasoni, S.; Guccione, C.; Bosin, A.; Vargiu, A.V.; Ruggerone, P.; Malloci, G. Force fFelds, Quantum-Mechanical- and Molecular-Dynamics-Based Descriptors of Radiometal-Chelator Complexes. Figshare Dataset 2024. [Google Scholar] [CrossRef]
- Hu, A.; Brown, V.; Macmillan, S.N.; Radchenko, V.; Yang, H.; Wharton, L.; Ramogida, C.F.; Wilson, J.J. Chelating the Alpha Therapy Radionuclides 225Ac3+ and 213Bi3+ with 18-Membered Macrocyclic Ligands Macrodipa and Py-Macrodipa. Inorg. Chem. 2022, 61, 801–806. [Google Scholar] [CrossRef]
- Frimpong, E.; Skelton, A.A.; Honarparvar, B. DFT study of the interaction between DOTA chelator and competitive alkali metal ions. J. Mol. Graph. Model. 2017, 76, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sevilla, M.D. SOMO-HOMO Level Inversion in Biologically Important Radicals. J. Phys. Chem. B 2018, 122, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Kubíček, V.; Böhmová, Z.; Ševčíková, R.; Vaněk, J.; Lubal, P.; Poláková, Z.; Michalicová, R.; Kotek, J.; Hermann, P. NOTA Complexes with Copper(II) and Divalent Metal Ions: Kinetic and Thermodynamic Studies. Inorg. Chem. 2018, 57, 3061–3072. [Google Scholar] [CrossRef] [PubMed]
- Kovács, A. Metal-Ligand Interactions in Scandium Complexes with Radiopharmaceutical Applications. Inorg. Chem. 2023, 62, 20733–20744. [Google Scholar] [CrossRef]
- Voss, S.D.; Smith, S.V.; Dibartolo, N.; McIntosh, L.J.; Cyr, E.M.; Bonab, A.A.; Dearling, J.L.J.; Carter, E.A.; Fischman, A.J.; Treves, S.T.; et al. Positron emission tomography (PET) imaging of neuroblastoma and melanoma with 64Cu-SarAr immunoconjugates. Proc. Natl. Acad. Sci. USA 2007, 104, 17489–17493. [Google Scholar] [CrossRef]
- Di Bartolo, N.; Sargeson, A.M.; Smith, S.V. New 64Cu PET imaging agents for personalised medicine and drug development using the hexa-aza cage, SarAr. Org. Biomol. Chem. 2006, 4, 3350–3357. [Google Scholar] [CrossRef]
- Ma, M.T.; Karas, J.A.; White, J.M.; Scanlon, D.; Donnelly, P.S. A new bifunctional chelator for copper radiopharmaceuticals: A cage amine ligand with a carboxylate functional group for conjugation to peptides. Chem. Commun. 2009, 22, 3237–3239. [Google Scholar] [CrossRef]
- Máté, G.; Šimeček, J.; Pniok, M.; Kertész, I.; Notni, J.; Wester, H.J.; Galuska, L.; Hermann, P. The influence of the combination of carboxylate and phosphinate pendant arms in 1,4,7-triazacyclononane-based chelators on their 68Ga labelling properties. Molecules 2015, 20, 13112–13126. [Google Scholar] [CrossRef]
- Liu, S.; Li, Z.; Conti, P.S. Development of multi-functional chelators based on sarcophagine cages. Molecules 2014, 19, 4246–4255. [Google Scholar] [CrossRef]
- Gai, Y.; Sun, L.; Lan, X.; Zeng, D.; Xiang, G.; Ma, X. Synthesis and Evaluation of New Bifunctional Chelators with Phosphonic Acid Arms for Gallium-68 Based PET Imaging in Melanoma. Bioconjug. Chem. 2018, 29, 3483–3494. [Google Scholar] [CrossRef]
- Rinne, S.S.; Dahlsson Leitao, C.; Gentry, J.; Mitran, B.; Abouzayed, A.; Tolmachev, V.; Ståhl, S.; Löfblom, J.; Orlova, A. Increase in negative charge of 68Ga/chelator complex reduces unspecific hepatic uptake but does not improve imaging properties of HER3-targeting affibody molecules. Sci. Rep. 2019, 9, 17710. [Google Scholar] [CrossRef]
- Skulska, M.; Falborg, L. A Simple Kit for the Good-Manufacturing-Practice Production of [68Ga]Ga-EDTA. Molecules 2023, 28, 6131. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Dohm, S.; Hansen, A.; Steinmetz, M.; Grimme, S.; Checinski, M.P. Comprehensive Thermochemical Benchmark Set of Realistic Closed-Shell Metal Organic Reactions. J. Chem. Theory Comput. 2018, 14, 2596–2608. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, M.; Grimme, S. Benchmark study of the performance of density functional theory for bond activations with (Ni,Pd)-based transition-metal catalysts. ChemistryOpen 2013, 2, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Iron, M.A.; Janes, T. Evaluating Transition Metal Barrier Heights with the Latest Density Functional Theory Exchange-Correlation Functionals: The MOBH35 Benchmark Database. J. Phys. Chem. A 2019, 123, 3761–3781. [Google Scholar] [CrossRef]
- Jiang, W.; Laury, M.L.; Powell, M.; Wilson, A.K. Comparative study of single and double hybrid density functionals for the prediction of 3d transition metal thermochemistry. J. Chem. Theory Comput. 2012, 8, 4102–4111. [Google Scholar] [CrossRef]
- Kovács, A. Metal-ligand bonding in bispidine chelate complexes for radiopharmaceutical applications. Struct. Chem. 2023, 34, 5–15. [Google Scholar] [CrossRef]
- Kovács, A. Theoretical Study of Complexes of Tetravalent Actinides with DOTA. Symmetry 2022, 14, 2451. [Google Scholar] [CrossRef]
- Hansen, A.; Bannwarth, C.; Grimme, S.; Petrović, P.; Werĺ, C.; Djukic, J.P. The thermochemistry of London dispersion-driven transition metal reactions: Getting the “right answer for the right reason”. ChemistryOpen 2014, 3, 177–189. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Lin, Y.S.; Li, G.D.; Mao, S.P.; Chai, J. Da Long-range corrected hybrid density functionals with improved dispersion corrections. J. Chem. Theory Comput. 2013, 9, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Valdés, Á.; Prosmiti, R.; Villarreal, P.; Delgado-Barrio, G. HeBr2 complex: Ground-state potential and vibrational dynamics from ab initio calculations. Mol. Phys. 2004, 102, 2277–2283. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to Isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Singh, U.C.; Kollman, P.A. An approach to computing electrostatic charges for molecules. J. Comput. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Jones, R.O.; Gunnarsson, O. The density functional formalism, its applications and prospects. Rev. Mod. Phys. 1989, 61, 689. [Google Scholar] [CrossRef]
- Adeowo, F.Y.; Honarparvar, B.; Skelton, A.A. Density Functional Theory Study on the Complexation of NOTA as a Bifunctional Chelator with Radiometal Ions. J. Phys. Chem. A 2017, 121, 6054–6062. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016; Wallingford CT 2016, Revision A.03. [Google Scholar]
- Li, P.; Merz, K.M. MCPB. py: A Python Based Metal Center Parameter Builder. J. Chem. Inf. Model. 2016, 56, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Seminario, J.M. Calculation of Intramolecular Force Fields from Second-Derivative Tensors. Int. J. Quantum. Chem. 1996, 60, 1271–1277. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Berryman, J.T.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cisneros, G.A.; Cruzeiro, V.W.D.; et al. Amber 2022; University of California: San Francisco, CA, USA, 2022. [Google Scholar]
- Izadi, S.; Anandakrishnan, R.; Onufriev, A.V. Building Water Models: A Different Approach. J. Phys. Chem. Lett. 2014, 5, 3863–3871. [Google Scholar]
- Sengupta, A.; Li, Z.; Song, L.F.; Li, P.; Merz, K.M. Parameterization of Monovalent Ions for the OPC3, OPC, TIP3P-FB, and TIP4P-FB Water Models. J. Chem. Inf. Model. 2021, 61, 869–880. [Google Scholar] [CrossRef]
- Boothroyd, S.; Behara, P.K.; Madin, O.C.; Hahn, D.F.; Jang, H.; Gapsys, V.; Wagner, J.R.; Horton, J.T.; Dotson, D.L.; Thompson, M.W.; et al. Development and Benchmarking of Open Force Field 2.0.0: The Sage Small Molecule Force Field. J. Chem. Theory Comput. 2023, 19, 3251–3275. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Loncharich, R.J.; Brooks, B.R.; Pastor, R.W. Langevin dynamics of peptides: The frictional dependence of isomerization rates of N-acetylalanyl-N′-methylamide. Biopolymers 1992, 32, 523–535. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Tanner, S.W.; Thompson, N.; Cheatham, T.E. Clustering molecular dynamics trajectories: 1. Characterizing the performance of different clustering algorithms. J. Chem. Theory Comput. 2007, 3, 2312–2334. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Öztürk, I.; Gervasoni, S.; Guccione, C.; Bosin, A.; Vargiu, A.V.; Ruggerone, P.; Malloci, G. Force Fields, Quantum-Mechanical- and Molecular-Dynamics-Based Descriptors of Radiometal–Chelator Complexes. Molecules 2024, 29, 4416. https://doi.org/10.3390/molecules29184416
Öztürk I, Gervasoni S, Guccione C, Bosin A, Vargiu AV, Ruggerone P, Malloci G. Force Fields, Quantum-Mechanical- and Molecular-Dynamics-Based Descriptors of Radiometal–Chelator Complexes. Molecules. 2024; 29(18):4416. https://doi.org/10.3390/molecules29184416
Chicago/Turabian StyleÖztürk, Işılay, Silvia Gervasoni, Camilla Guccione, Andrea Bosin, Attilio Vittorio Vargiu, Paolo Ruggerone, and Giuliano Malloci. 2024. "Force Fields, Quantum-Mechanical- and Molecular-Dynamics-Based Descriptors of Radiometal–Chelator Complexes" Molecules 29, no. 18: 4416. https://doi.org/10.3390/molecules29184416
APA StyleÖztürk, I., Gervasoni, S., Guccione, C., Bosin, A., Vargiu, A. V., Ruggerone, P., & Malloci, G. (2024). Force Fields, Quantum-Mechanical- and Molecular-Dynamics-Based Descriptors of Radiometal–Chelator Complexes. Molecules, 29(18), 4416. https://doi.org/10.3390/molecules29184416