
Structure and Intercalation of Cysteine–Asparagine–Serine Peptide into Montmorillonite as an Anti-Inflammatory Agent Preparation—A DFT Study
 , ,
, ,
Abstract

1. Introduction
2. Results and Discussion
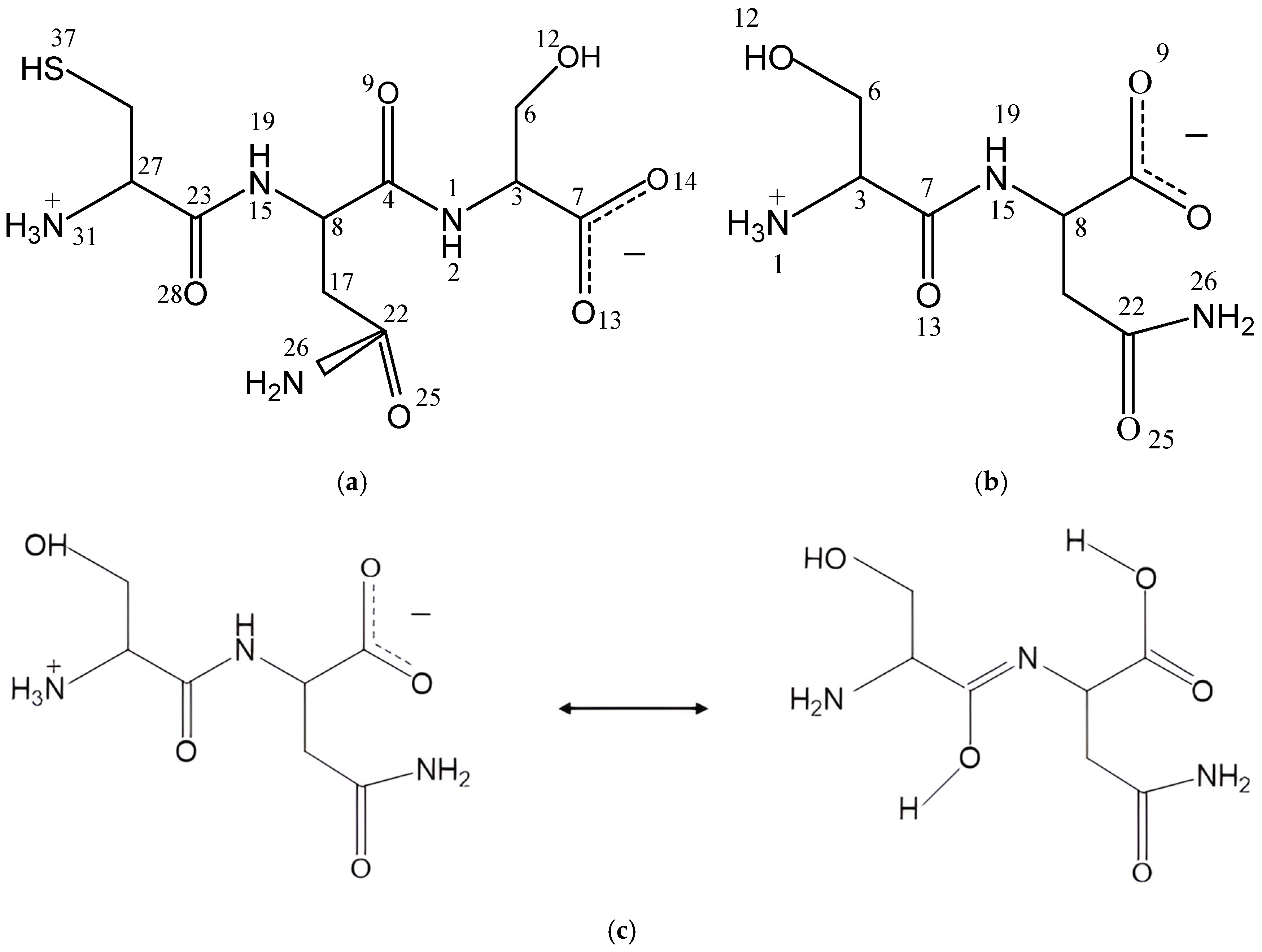
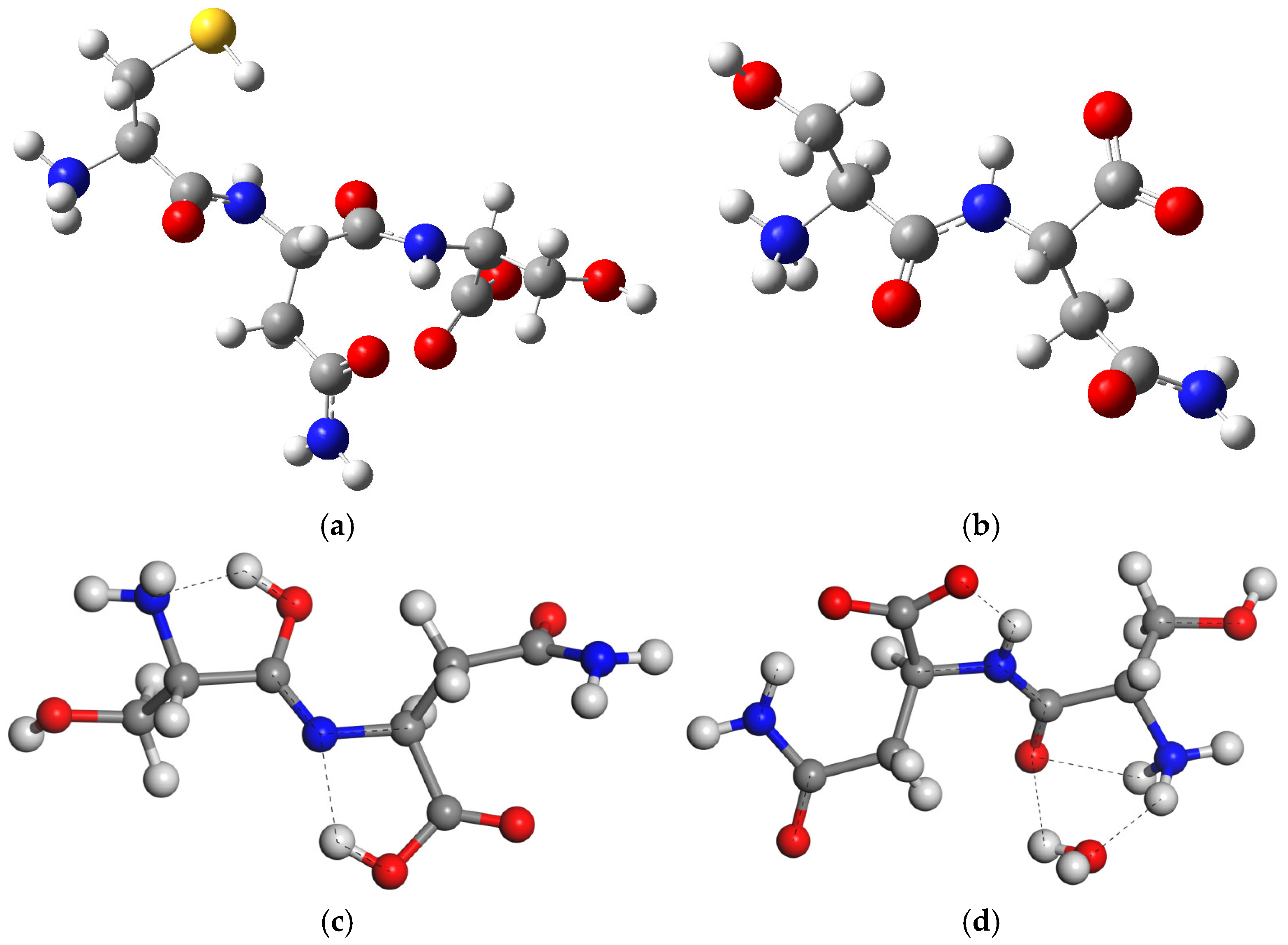
2.1. Molecular Structure of Peptides
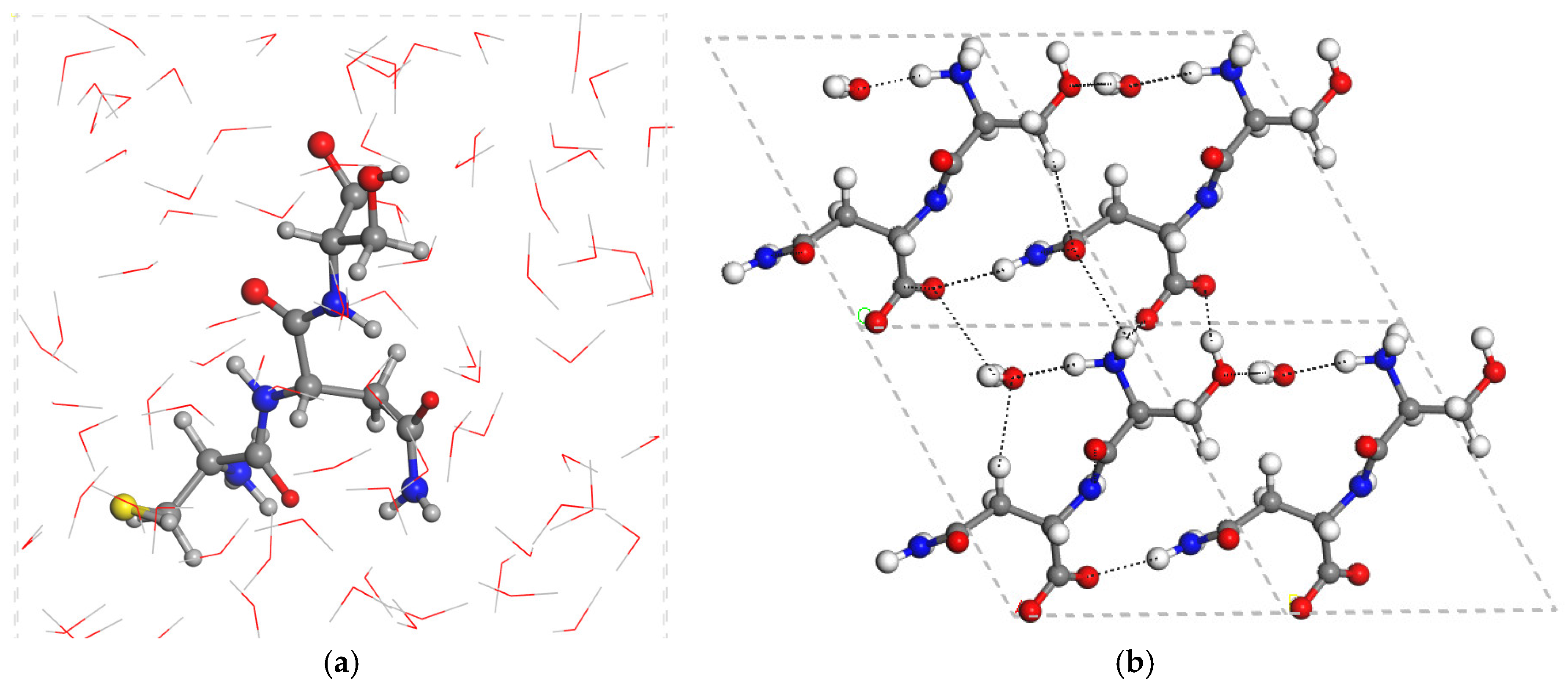
2.2. SN Crystal Structure
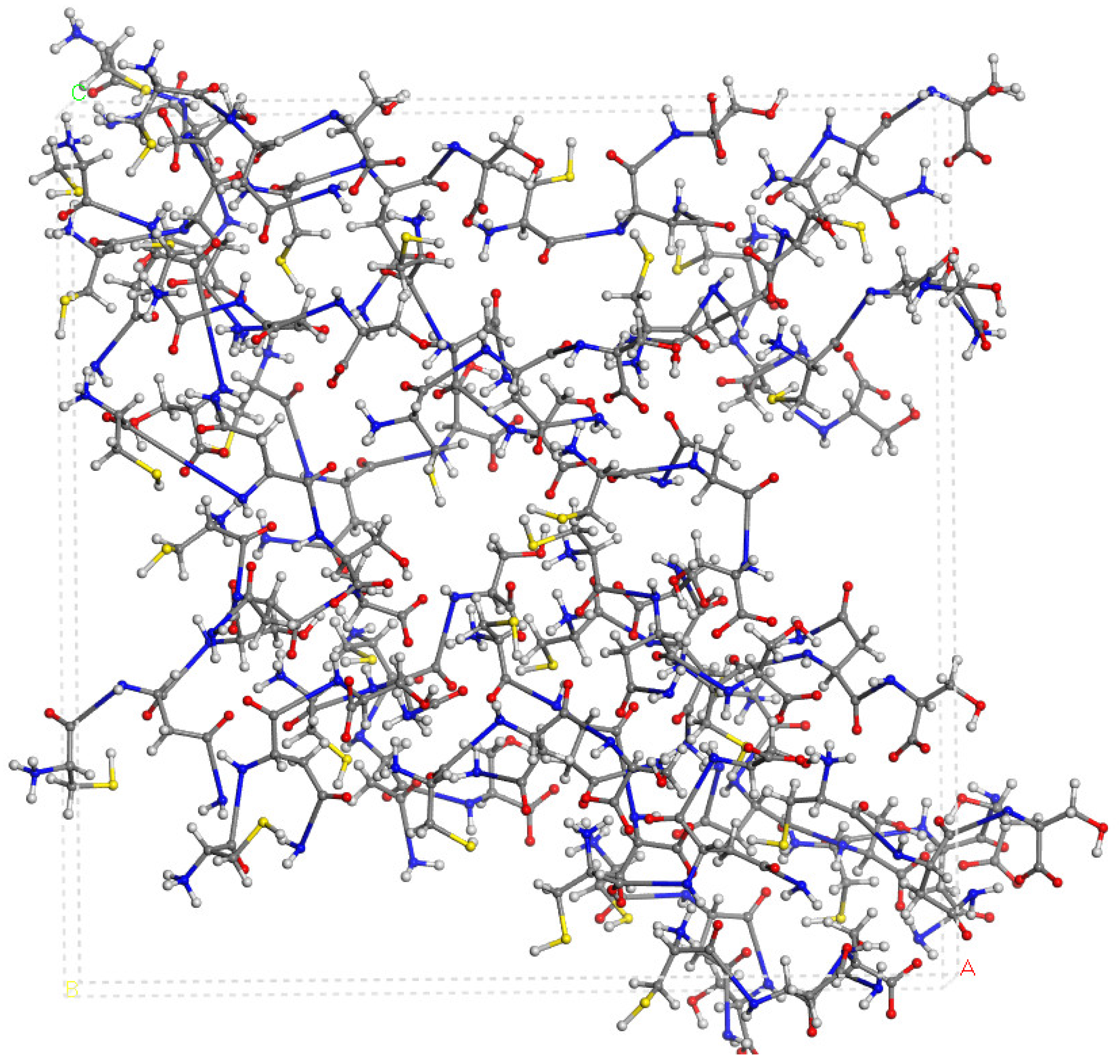
2.3. CNS Intercalated in Montmorillonite
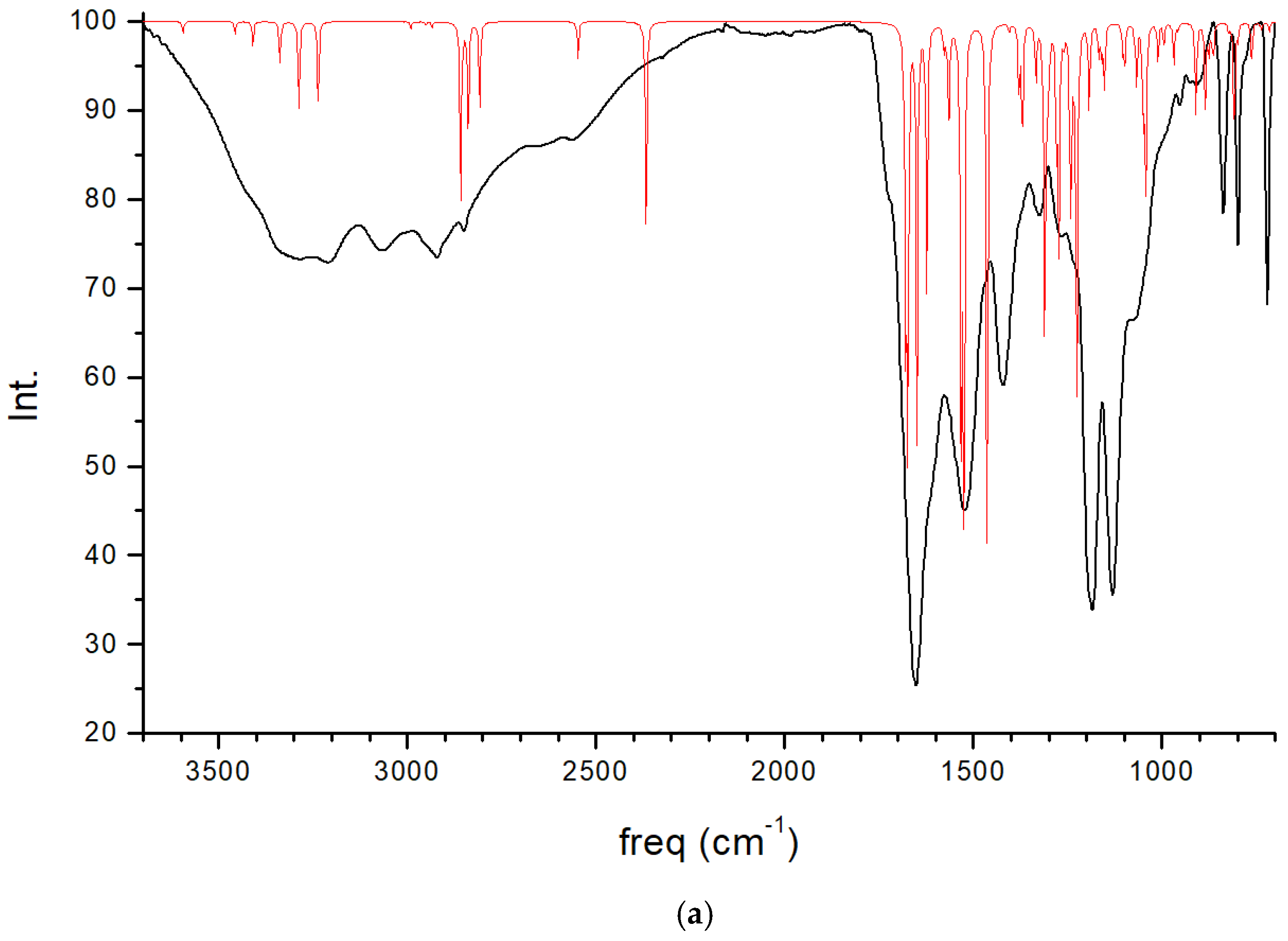
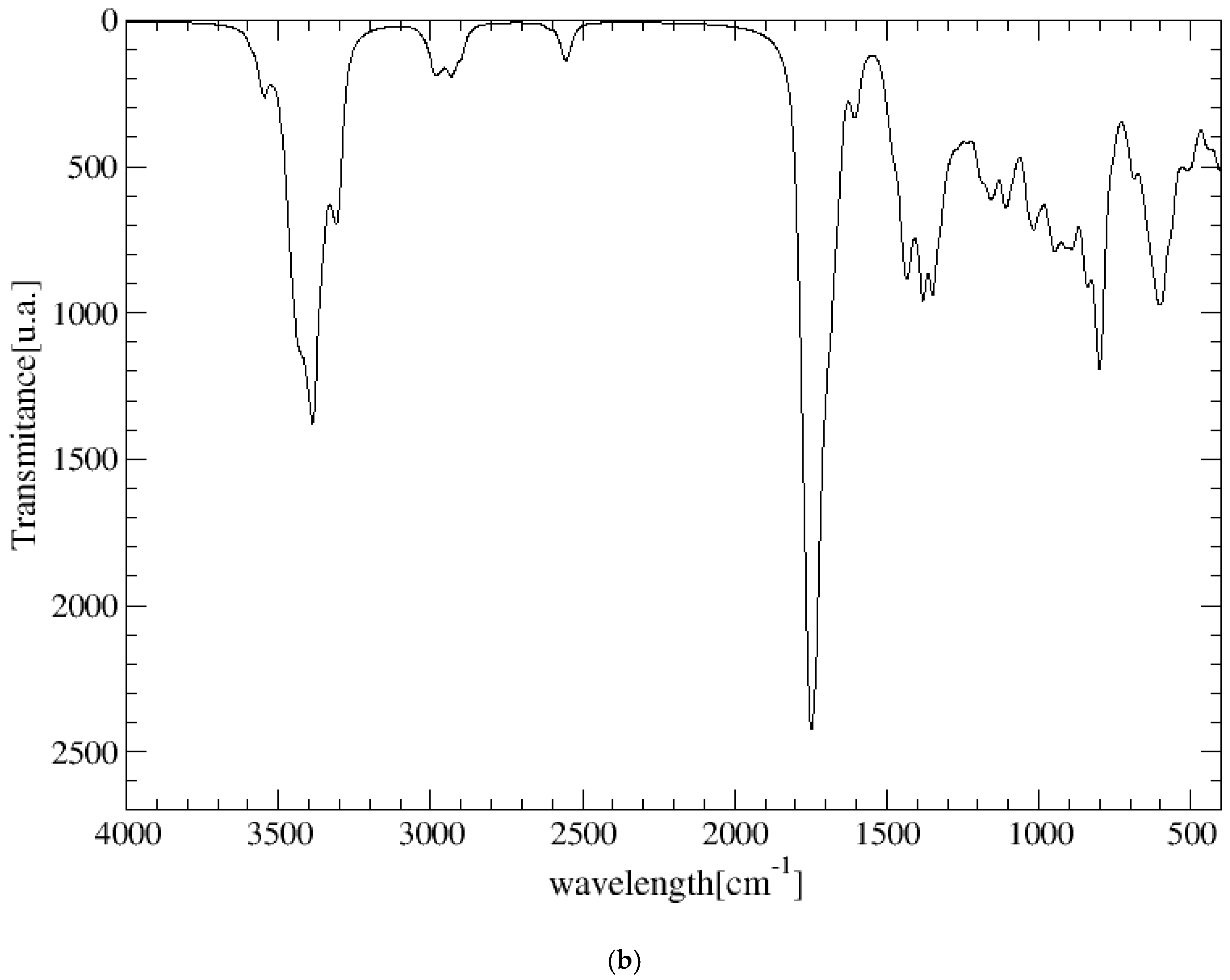
2.4. Spectroscopic Properties
3. Materials and Methods
3.1. Experimental Methods
3.2. Models and Calculation Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Cellular and Molecular Immunology, 8th ed.; Elsevier/Saunders: Philadelphia, PA, USA, 2014; ISBN 978-0-323-28645-9. [Google Scholar]
- Gudkov, A.V.; Komarova, E.A. P53 and the Carcinogenicity of Chronic Inflammation. Cold Spring Harb. Perspect. Med. 2016, 6, a026161. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory Responses and Inflammation-Associated Diseases in Organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef]
- Arnhold, J. Host-Derived Cytotoxic Agents in Chronic Inflammation and Disease Progression. Int. J. Mol. Sci. 2023, 24, 3016. [Google Scholar] [CrossRef] [PubMed]
- Barrientos-Salcedo, C.; Lara-Rodríguez, M.; Campos-Fernández, L.; Legorreta-Herrera, M.; Soto-Cruz, I.; Soriano-Correa, C. DFT Calculations of Electronic Structure Evaluation and Intermolecular Interactions of P53-Derived Peptides with Cytotoxic Effect on Breast Cancer. Theor. Chem. Acc. 2021, 140, 121. [Google Scholar] [CrossRef]
- Soriano-Correa, C.; Vichi-Ramírez, M.M.; Herrera-Valencia, E.E.; Barrientos-Salcedo, C. The Role of ETFS Amino Acids on the Stability and Inhibition of P53-MDM2 Complex of Anticancer P53-Derivatives Peptides: Density Functional Theory and Molecular Docking Studies. J. Mol. Graph. Model. 2023, 122, 108472. [Google Scholar] [CrossRef]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 6th ed.; W.H. Freeman and Company: New York, NY, USA, 2006. [Google Scholar]
- Soriano-Correa, C.; Barrientos-Salcedo, C.; Campos-Fernández, L.; Alvarado-Salazar, A.; Esquivel, R.O. Importance of Asparagine on the Conformational Stability and Chemical Reactivity of Selected Anti-Inflammatory Peptides. Chem. Phys. 2015, 457, 180–187. [Google Scholar] [CrossRef]
- Sun, C.-L.; Wang, C.-S. Estimation on the Intramolecular Hydrogen-Bonding Energies in Proteins and Peptides by the Analytic Potential Energy Function. J. Mol. Struct. THEOCHEM 2010, 956, 38–43. [Google Scholar] [CrossRef]
- Esquivel, R.O.; Molina-Espíritu, M.; Salas, F.; Soriano, C.; Barrientos, C.; Dehesa, J.S.; Dobado, J.A. Decoding the Building Blocks of Life from the Perspective of Quantum Information; IntechOpen: London, UK, 2013; ISBN 978-953-51-1089-7. [Google Scholar]
- Pérez de Tudela, R.; Marx, D. Water-Induced Zwitterionization of Glycine: Stabilization Mechanism and Spectral Signatures. J. Phys. Chem. Lett. 2016, 7, 5137–5142. [Google Scholar] [CrossRef]
- Soriano-Correa, C.; Olivares del Valle, F.J.; Muñoz-Losa, A.; Fdez Galván, I.; Martín, M.E.; Aguilar, M.A. Theoretical Study of the Competition between Intramolecular Hydrogen Bonds and Solvation in the Cys-Asn-Ser Tripeptide. J. Phys. Chem. B 2010, 114, 8961–8970. [Google Scholar] [CrossRef]
- Zhu, C.; Wang, Q.; Huang, X.; Yun, J.; Hu, Q.; Yang, G. Adsorption of Amino Acids at Clay Surfaces and Implication for Biochemical Reactions: Role and Impact of Surface Charges. Colloids Surf. B Biointerfaces 2019, 183, 110458. [Google Scholar] [CrossRef]
- Borrego-Sánchez, A.; Muñoz-Santiburcio, D.; Viseras, C.; Hernández-Laguna, A.; Sainz-Díaz, C.I. Melatonin/Nanoclay Hybrids for Skin Delivery. Appl. Clay Sci. 2022, 218, 106417. [Google Scholar] [CrossRef]
- Borrego-Sánchez, A.; Viseras, C.; Aguzzi, C.; Sainz-Díaz, C.I. Molecular and Crystal Structure of Praziquantel. Spectroscopic Properties and Crystal Polymorphism. Eur. J. Pharm. Sci. 2016, 92, 266–275. [Google Scholar] [CrossRef]
- Brandão-Lima, L.C.; Silva, F.C.; Costa, P.V.C.G.; Alves-Júnior, E.A.; Viseras, C.; Osajima, J.A.; Bezerra, L.R.; De Moura, J.F.P.; De, A.; Silva, A.G.; et al. Clay Mineral Minerals as a Strategy for Biomolecule Incorporation: Amino Acids Approach. Materials 2021, 15, 64. [Google Scholar] [CrossRef] [PubMed]
- Soriano-Correa, C.; Pérez De La Luz, A.; Sainz-Díaz, C.I. Adsorption of Capsaicin into the Nanoconfined Interlayer Space of Montmorillonite by DFT Calculations. J. Pharm. Sci. 2023, 112, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Feuillie, C.; Daniel, I.; Michot, L.J.; Pedreira-Segade, U. Adsorption of Nucleotides onto Fe–Mg–Al Rich Swelling Clays. Geochim. Cosmochim. Acta 2013, 120, 97–108. [Google Scholar] [CrossRef]
- Moreno-Domínguez, E.; Borrego-Sánchez, A.; Sánchez-Espejo, R.; Viseras, C.; Sainz-Díaz, C.I. Experimental and Computational Study for the Design of Sulfathiazole Dosage Form with Clay Mineral. Pharmaceutics 2023, 15, 575. [Google Scholar] [CrossRef]
- Pérez De La Luz, A.; Soriano-Correa, C.; Francisco-Márquez, M.; Barrientos-Salcedo, C.; Hernández-Laguna, A.; Sainz-Díaz, C.I. Intercalation of Sulfonamides in Montmorillonite by Molecular Dynamics and DFT Calculations for Bioavailability Control. J. Mol. Struct. 2023, 1291, 136085. [Google Scholar] [CrossRef]
- Baker, J.C.; Grabowska-Olszewska, B.; Uwins, P.J.R. ESEM Study of Osmotic Swelling of Bentonite from Radzionkow (Poland). Appl. Clay Sci. 1995, 9, 465–469. [Google Scholar] [CrossRef]
- Koleman, H.A.; van Zyl, R.; Steyn, N.; Boneschans, B.; Steyn, H.S. Influence of Montmorillonite on the Dissolution and Bioavailablity of Phenyton. Drug Dev. Ind. Pharm. 2008, 16, 791–805. [Google Scholar] [CrossRef]
- Park, J.-H.; Shin, H.-J.; Kim, M.H.; Kim, J.-S.; Kang, N.; Lee, J.-Y.; Kim, K.-T.; Lee, J.I.; Kim, D.-D. Application of Montmorillonite in Bentonite as a Pharmaceutical Excipient in Drug Delivery Systems. J. Pharm. Investig. 2016, 46, 363–375. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [PubMed]
- Cristiani, C.; Finocchio, E.; Rossi, L.; Giromini, C.; Dell’Anno, M.; Panseri, S.; Bellotto, M. Natural Clays as Potential Amino Acids Carriers for Animal Nutrition Application. Appl. Sci. 2021, 11, 5669. [Google Scholar] [CrossRef]
- Mallakpour, S.; Dinari, M. Insertion of Novel Optically Active Poly(Amide-Imide) Chains Containing Pyromellitoyl-Bis-l-Phenylalanine Linkages into the Nanolayered Silicates Modified with l-Tyrosine through Solution Intercalation. Polymer 2011, 52, 2514–2523. [Google Scholar] [CrossRef]
- Morales-Martínez, M.E.; Silva-García, R.; Soriano-Correa, C.; Giménez-Scherer, J.A.; Rojas-Dotor, S.; Blanco-Favela, F.; Rico-Rosillo, G. The Cys-Asn-Ser Carboxyl-Terminal End Group Is the Pharmacophore of the Amebic Anti-Inflammatory Monocyte Locomotion Inhibitory Factor (MLIF). Mol. Biochem. Parasitol. 2008, 158, 46–51. [Google Scholar] [CrossRef]
- Awad, M.E.; Borrego-Sánchez, A.; Escamilla-Roa, E.; Hernández-Laguna, A.; Sainz-Díaz, C.I. Modeling of the Adsorption of a Protein-Fragment on Kaolinite with Potential Antiviral Activity. Appl. Clay Sci. 2020, 199, 105865. [Google Scholar] [CrossRef]
- Escamilla-Roa, E.; Huertas, F.J.; Hernández-Laguna, A.; Sainz-Díaz, C.I. A DFT Study of the Adsorption of Glycine in the Interlayer Space of Montmorillonite. Phys. Chem. Chem. Phys. 2017, 19, 14961–14971. [Google Scholar] [CrossRef]
- Soriano-Correa, C.; Barrientos-Salcedo, C.; Francisco-Márquez, M.; Sainz-Díaz, C.I. Computational Study of Substituent Effects on the Acidity, Toxicity and Chemical Reactivity of Bacteriostatic Sulfonamides. J. Mol. Graph. Model. 2018, 81, 116–124. [Google Scholar] [CrossRef]
- Görbitz, C.H.; Hartviksen, L.M. The Monohydrates of the Four Polar Dipeptides L-Seryl-L-Asparagine, L-Seryl-L-Tyrosine, L-Tryptophanyl-L-Serine and L-Tyrosyl-L-Tryptophan. Acta Crystallogr. C 2008, 64, o171–o176. [Google Scholar] [CrossRef]
- Blodgett, K.N.; Fischer, J.L.; Lee, J.; Choi, S.H.; Zwier, T.S. Conformation-Specific Spectroscopy of Asparagine-Containing Peptides: Influence of Single and Adjacent Asn Residues on Inherent Conformational Preferences. J. Phys. Chem. A 2018, 122, 8762–8775. [Google Scholar] [CrossRef]
- Wang, H.; Heger, M.; Al-Jabiri, M.H.; Xu, Y. Vibrational Spectroscopy of Homo- and Heterochiral Amino Acid Dimers: Conformational Landscapes. Molecules 2021, 27, 38. [Google Scholar] [CrossRef]
- Barth, A. The Infrared Absorption of Amino Acid Side Chains. Prog. Biophys. Mol. Biol. 2000, 74, 141–173. [Google Scholar] [CrossRef]
- Carneiro, C.E.A.; De Santana, H.; Casado, C.; Coronas, J.; Zaia, D.A.M. Adsorption of Amino Acids (Ala, Cys, His, Met) on Zeolites: Fourier Transform Infrared and Raman Spectroscopy Investigations. Astrobiology 2011, 11, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Kolev, T. Solid-state IR–LD Spectroscopic and Theoretical Analysis of Arginine-containing Peptides. Biopolymers 2006, 83, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Biovia Materials Studio, Dassault Systems: Vélizy-Villacoublay, France, 2016.
- Ortega-Castro, J.; Hernández-Haro, N.; Dove, M.T.; Hernández-Laguna, A.; Sainz-Díaz, C.I. Density Functional Theory and Monte Carlo Study of Octahedral Cation Ordering of Al/Fe/Mg Cations in Dioctahedral 2:1 Phyllosilicates. Am. Mineral. 2010, 95, 209–220. [Google Scholar] [CrossRef]
- Ortega-Castro, J.; Hernández-Haro, N.; Hernández-Laguna, A.; Sainz-Díaz, C.I. DFT Calculation of Crystallographic Properties of Dioctahedral 2:1 Phyllosilicates. Clay Miner. 2008, 43, 351–361. [Google Scholar] [CrossRef]
- Sainz-Díaz, C.I.; Escamilla-Roa, E.; Hernández-Laguna, A. Quantum Mechanical Calculations of Trans-Vacant and Cis-Vacant Polymorphism in Dioctahedral 2:1 Phyllosilicates. Am. Mineral. 2005, 90, 1827–1834. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Rev. E.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Mardirossian, N.; Head-Gordon, M. Thirty Years of Density Functional Theory in Computational Chemistry: An Overview and Extensive Assessment of 200 Density Functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Ab Initio Molecular Orbital Theory; Hehre, W.J., Ed.; A Wiley-Interscience Publication: New York, NY, USA, 1986; ISBN 978-0-471-81241-8. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-Principles Simulation: Ideas, Illustrations and the CASTEP Code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Baroni, S.; de Gironcoli, S.; Dal Corso, A.; Giannozzi, P. Phonons and Related Crystal Properties from Density-Functional Perturbation Theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef]
- Delley, B. Hardness Conserving Semilocal Pseudopotentials. Phys. Rev. B 2002, 66, 155125. [Google Scholar] [CrossRef]
- Heinz, H.; Ramezani-Dakhel, H. Simulations of inorganic–bioorganic interfaces to discover new materials: Insights, com-parisons to experiment, challenges, and opportunities. Chem. Soc. Rev. 2016, 45, 412–448. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode | CNSexp g | CNSmol | MNT-CNS |
|---|---|---|---|
| ν(NH2) | 3520–3440, 3560–3505 a,m, 3393–3383s a,m, 3430s n | 3534 a (3594 a, 3456s a) | 3595 a, 3449s a |
| ν(NH3+) | 3420–3350, 3430 n, 2620–2580 | 3459 c, 3384 c, (3410as, 3337s) c, 2363 c,f (2367 c,f) | 3401 c, 3154 c,d, 2652 c,e |
| ν(NH) | 3280–3200, 3293–3238 o | 3469 b, 3212 a,c, 3108 a,f (3287 b, 3236 a) | 3494 b, 3385, |
| ν(CH) | 3100–3063 | 3087 c, 3071 b, 3070 c, 3061 b, 3016 a, 2822 b (3022–3008 c, 2991 b) | 3057 c, 3052 b, 3013 a |
| ν(CH2) | 3063–2950 | 3078 a, 3004s a,c (2990–2975 a, 2951s c, 2934 b, 2839 a, 2808 b) | 3106 c, 3078 b, 3065 a, 3042s c, 3016s b, 3004s a |
| ν(OH)ser | 2840 | 2895 b (2858) | 3493 |
| ν(SH) | 2620–2580, 2551 j, 2563 k | 2622 (2548) | 2656 |
| ν(C=O) | 1720–1600, 1704–1678 h, 1709–1683 m, 1657–1648 o | 1732 a, 1713–1705 a,b (1674–1650 a) | 1701 c, 1671 a,b, 1654 a, 1608 b,f |
| δ(OH)ser | 1570–1500 b | 1659 b, 1550 b (1624, 1525) b | 1446–1406 b |
| δ(NH3) | 1570–1500, 1616–1585 m | 1604–1600 c (1578, 1571) | 1678 c, 1621 c, 1537s c |
| δ(NH2) | 1550–1500, 1612 h | 1599 a (1565s a) | 1572s a, |
| δ(NH) | 1470sh, 1514 m, 1533 o | 1519 a,b, 1479 a,c, 1177 (1680 c, 1533 b, 1464 a) | 1510 a,b, 1458 a,c, 1179 |
| δ(CH2) | 1423, 1467–1450 i, 1424 j | 1473s b, 1440s a, 1433s c (1448s b, 1405s c, 1380s a, 1335 b) | 1484s b, 1422s c, 1404–1393s a |
| δ(CH) | 1324, 1341 j | 1353, 1340 a, 1338 c, 1299–1256 (1311 c, 1304 a, 1274 b) | 1344–1224 |
| γ(CH) | 1184 | 1256–1254 | 1201–1191, 1120 c, 994 b |
| ν(C-O) | 1131–1050, 1030 i | 1058 b (1044 b) | |
| γ(NH) | 800, 740–721 o | (809–799 b) | 1082 a, 1108 c |
| δ(SH) | 838 | 998 (995) | 914 c |
| γ(OH) | 799–722 | 947 (886) |
| Mode | Exp | SN | SN Monohydrate | SN Crystal |
|---|---|---|---|---|
| ν(NH2) | 3566 a, 3467 b, 3438s a, 3384sb | 3520 a, 3353 b, 3059 b,g, 3015 b,h, 2920 a,g, 2683 c | 3390 a, 3284 a,g, 3249s a, 2946–2934 b, 2844–2822 b | |
| ν(OH) | 3740 b, 3203 a,f, 3098 b,d | 3780 k, 3736 b, 3629 k | 3533 k, 3406 k, 2867 b | |
| ν(CH2) | 3030 a, 2969 b, 2963s a, 2955sb | 3004 a, 2968 b | 3026 a, 2958s a | |
| ν(CH) | 2924 a, 2893 b | 3022 b, 2962–2932 a, | 3017 b, 3004 a,b | |
| ν(C=O) | 1704–1677 i | 1756 e, 1700 a | 1680, 1670 a | |
| ν(C-N) | 1690 | 1090 a, 1058 b | ||
| δ(NH) | 1622–1612 i | 1599s b, 1570s a | 1663 a, 1625 b, 1585 a, 1504 b | 1649–1639 b, 1630 a,1609 b, 1577s a, 1551–1534, 1506s b |
| δ(CH2) | 1467–1450 j | 1450s b, 1419s a | 1455 b, 1402 a | |
| δ(OH) | 1420–1181 j | 1404 g, 1401 b | 1596 k, 1190 b | 1617 k, 1469 b |
| δ(CH) | 1334 a, 1309 b | 1334 b, 1302–1256 a | 1432s b, 1419s a, 1362s b, 1390–1272 | |
| γ(CH) | 1382–1170 j | 1261, 1171 | ||
| γ(NH) | 1226, 1113 | |||
| ν(C-O) | 1030 j | 1051 b | ||
| γ(OH) | 940 j | 982 b |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barrientos-Salcedo, C.; Soriano-Correa, C.; Hernández-Laguna, A.; Sainz-Díaz, C.I. Structure and Intercalation of Cysteine–Asparagine–Serine Peptide into Montmorillonite as an Anti-Inflammatory Agent Preparation—A DFT Study. Molecules 2024, 29, 4250. https://doi.org/10.3390/molecules29174250
Barrientos-Salcedo C, Soriano-Correa C, Hernández-Laguna A, Sainz-Díaz CI. Structure and Intercalation of Cysteine–Asparagine–Serine Peptide into Montmorillonite as an Anti-Inflammatory Agent Preparation—A DFT Study. Molecules. 2024; 29(17):4250. https://doi.org/10.3390/molecules29174250
Chicago/Turabian StyleBarrientos-Salcedo, Carolina, Catalina Soriano-Correa, Alfonso Hernández-Laguna, and Claro Ignacio Sainz-Díaz. 2024. "Structure and Intercalation of Cysteine–Asparagine–Serine Peptide into Montmorillonite as an Anti-Inflammatory Agent Preparation—A DFT Study" Molecules 29, no. 17: 4250. https://doi.org/10.3390/molecules29174250
APA StyleBarrientos-Salcedo, C., Soriano-Correa, C., Hernández-Laguna, A., & Sainz-Díaz, C. I. (2024). Structure and Intercalation of Cysteine–Asparagine–Serine Peptide into Montmorillonite as an Anti-Inflammatory Agent Preparation—A DFT Study. Molecules, 29(17), 4250. https://doi.org/10.3390/molecules29174250