2.1. Conformation Changes in TKI-Xs Due to Halogen Substitution and Strain Energy (SE)
Constraining molecular conformation can be employed to enhance the comprehension of particular interactions between drugs and receptors. Even in the scenario where a medication substance selectively attaches to a singular receptor, there are still potential benefits associated with utilizing conformationally constrained analogs. Constraining conformational flexibility aids in the mapping of receptor-binding sites and has the potential to yield more potent compounds [
15]. Hence, conformations of TKIs-X, where X represents hydrogen or halogens, will be investigated in a DMSO solvent to elucidate the influence of halogens on the conformational distribution of TKIs.
The IUPAC name of TKI-H is 6,7-dimethoxy-N-phenylquinazolin-4-amine, of TKI-F is N-(3-fluorophenyl)-6,7-dimethoxyquinazolin-4-amine, of TKI-Cl (AG1478) is N-(3-chlorophenyl)-6,7-dimethoxyquinazolin-4-amine, of TKI-Br (PD153035) is N-(3-bromophenyl)-6,7-dimethoxyquinazolin-4-amine, and of TKI-I is N-(3-iodophenyl)-6,7-dimethoxyquinazolin-4-amine. The nomenclature is additionally provided in
Figure S1 of the Supplementary Material.
Accurate determination of electronic states in conformers with low energy could be achieved exclusively through the application of quantum-mechanical techniques, which were utilized to compute the molecular characteristics of TKIs-X conformers, along with their UV–visible spectra. The forthcoming discourse will primarily focus on the four most energetically favourable local minimum conformers, each with an energy level of approximately SE < 3.0 kcal mol
−1. The determined lowest-energy (global-minimum) conformation of TKI-H, TKI-F, AG1478, PD153035, and TKI-I, in a DMSO solvent environment were characterized with total energies −933.798880 E
h, −1033.081728 E
h, −1393.425183 E
h, −3507.379134 E
h, and −1230.996026 E
h, respectively (see
Table S2), using B3LYP/def2TZVP model in DMSO solvent.
Strain energy (SE) is the excess energy of a conformer above its global minimum structure of an inhibitor.
Figure 1 depicts the observed pattern in the strain energies (SEs) of the five low-energy conformations within the DMSO solvent for each TKI-X, in comparison to their corresponding individual global minimum structure, along with the structures retrieved from the EGFR database (
http://crdd.osdd.net/raghava/egfrindb/, accessed on 2 February 2023). The energies of the final structures (EGFR-DB) were optimized with the torsion angles held constant [
16]. Furthermore,
Figure 1 illustrates the considerable diversity in SE for the EGFR database structures. For example, the SE is given by 8.193 kcal/mol for TKI-Cl (AG1478) and 8.279 kcal/mol for TKI-Br (PD153035). In contrast, there is a relatively minor increase in SE for TKI-H, TKI-F, and TKI-I, amounting to 1.346, 0.876, and 1.040 kcal/mol, respectively.
Table 1 compares molecular properties between the global minimum conformer and the EGFR database structures of TKIs-X, determined through calculations using the same DFT B3LYP/def2TZVP model in DMSO solvent. The distribution of the population is influenced by the degeneracy of conformational states. For instance, at room temperature, TKI-H conformer 1 manifests approximately 89.7% of the population, while TKI-F shows a population of 61.2% for conformer 1. TKI-Cl (AG1478) exhibits a population of 63.5% for conformer 1, TKI-Br (PD153035) shows 62.7%, and TKI-I displays a population of 54.6% for the same conformer.
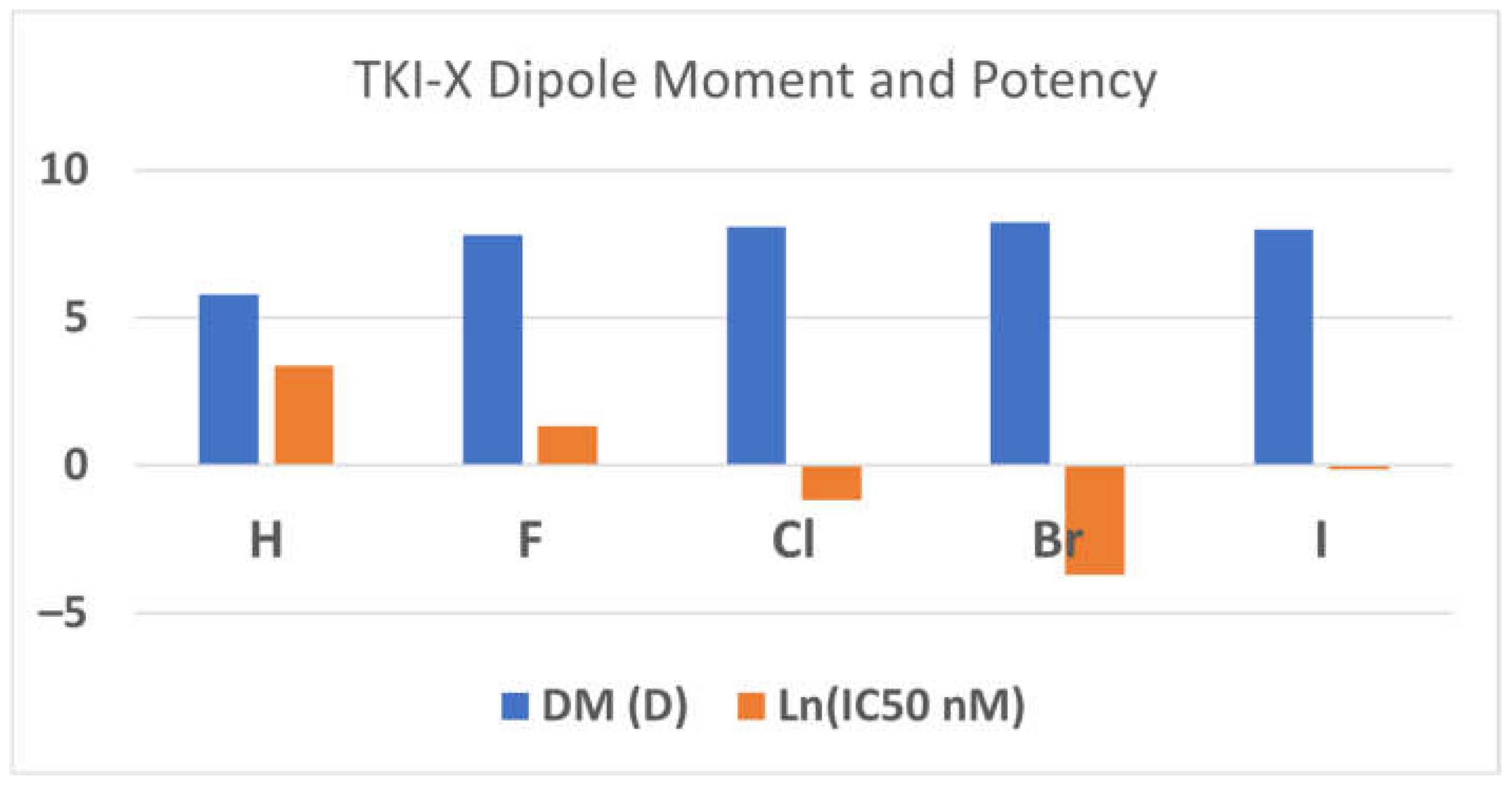
The dipole moments (DMs) of the global conformers
1 seem associated with the TKI potency. The order of the dipole moments of the TKI-Xs is the same as the order of the potency, the IC
50 values in (nM) [
3]. For example, the dipole moments of TKI-H, F, Cl, Brand I are 5.80D, 7.79D, 8.08D, 8.23D, and 7.99D, corresponding with the measured IC
50 values of 29 nM, 3.8 nM, 0.31 nM, 0.025 nM, and 0.89 nM, respectively [
3].
Figure 2 reports the relationship between dipole moments (DMs) and potency (ln(IC
50 in nM). The relationship between the dipole moment and electronegativity typically aligns in a manner where electronegativity tends to lead to larger dipole moments. Following this, elements like fluorine, chlorine, and bromine, which boast higher electronegativities compared to hydrogen, display an expected increase in their dipole moments. However, the observed decreases in the dipole moment for iodine, despite its larger size, could potentially be attributed to a multifaceted interplay of factors. This may include a reduction in electronegativity relative to smaller halogens, coupled with probable electronic influences within the molecular structure, which might counteract the anticipated rise in the dipole moment linked with size and passing through the aromatic 4-anilinoquinazoline conjugate system. Furthermore, alterations in atomic polarizability, signifying the ease of electron cloud distortion under an external electric field, could also contribute to the atypical trend noticed in the dipole moments of the conformers.
Additional properties of the various conformational forms are collected in
Table S1.1 in the Supplementary Materials. It should be noted that the halogen series, conformers 1, 3, and 5, exhibit comparatively higher dipole moments, measuring around 8D, 7D, and 9D, respectively. In contrast, conformers 2 and 4 demonstrate notably smaller dipole moments, approximately around 4.5D. The increased dipole moment of configuration 5 could potentially enhance the favourable binding interaction with the EGFR.
The specific properties of the top five conformers of TKIs-X in the DMSO solvent are collected in
Table S1.1 in the Supplementary Materials. The global minimum structure
1 for each TKIs-X exhibits planar structures with a 3-X-Ph orientation; in the case of conformers
2, an in-plane rotation (rotational isomerism) has occurred, resulting in a 180-degree flip of the phenyl ring containing the X atom (5-X-Ph orientation). Conformer
3 maintains a planar configuration; however, there is a specific rotation within the plane for each TKIs-X. When X is a halogen, this rotation involves the oxygen atom (atom 28) of the methoxy group. Conversely, when X is hydrogen, the conformer transitions to a non-planar state, and the alteration involves the rotation of the phenyl ring into the same plane. Conformer
4 exhibits similar changes in configuration as observed in conformer
3, along with an in-plane rotation leading to a 180-degree flip of the phenyl ring (5-X-Ph orientation) for each TKIs-X when X is a halogen. However, when X is hydrogen, the conformer transitions to a non-planar state, and the alteration involves the rotation of the phenyl ring out of the same plane. Conformer
5 presents a contrasting scenario compared to conformer
3; it retains a planar structure, yet there is a distinct rotation within the plane for each TKIs-X when X is a halogen or hydrogen, and this rotation affects the other oxygen atom (atom 27) of the methoxy group.
It is known that a potent inhibitor often takes a higher energy conformer rather than the global minimum structure. The alignment of the crystal structure and the global minimum of each TKI-X can be estimated using root mean square deviation (RMSD) (Å) values. The RMSD indicates how different the crystal structures of the TKI-X are from its global minimum structure. As a result, the TKI-X configurations from the EGFR database (
http://crdd.osdd.net/raghava/egfrindb/, accessed on 2 February 2023) are aligned with the global minimum conformers
1 of TKIs-X. All configurations are arranged in a coordinated manner based on the plane constituted by three atoms within the quinazoline core: C
(1), C
(4), and C
(10) [
12]. The deviations in conformation commence at the two methoxy (-OCH
3) side appendages situated on C
(1) and C
(6), associated with the methoxy (-OCH
3) groups. Subsequently, there is a reorientation of the X-phenyl ring observed in TKI-Cl and TKI-Br. The RMSD values of the TKI-X are 0.135 Å, 0.121 Å, 0.657 Å, 0.692 Å, and 0.124 Å for TKI-H, YKI-F, TKI-Cl, TKI-Br, and TKI-I, respectively. It is interesting to know that the crystal structures of the more potent TKI-Cl and TKI-Br are more distorted from their respective global minimum structures.
2.2. Changes in the UV–Visible Spectra of the TKIs-X Conformers
The absorption profile of a compound in the UV-Vis spectrum originates from the transition that takes place between the molecular orbitals that are filled (occupied) and those that are unfilled (virtual) [
18]. The conjugated ring system present in 4-anilinoquinazoline TKIs-X renders them highly suitable for discovering their ultraviolet-visible (UV-Vis) spectral properties [
19].
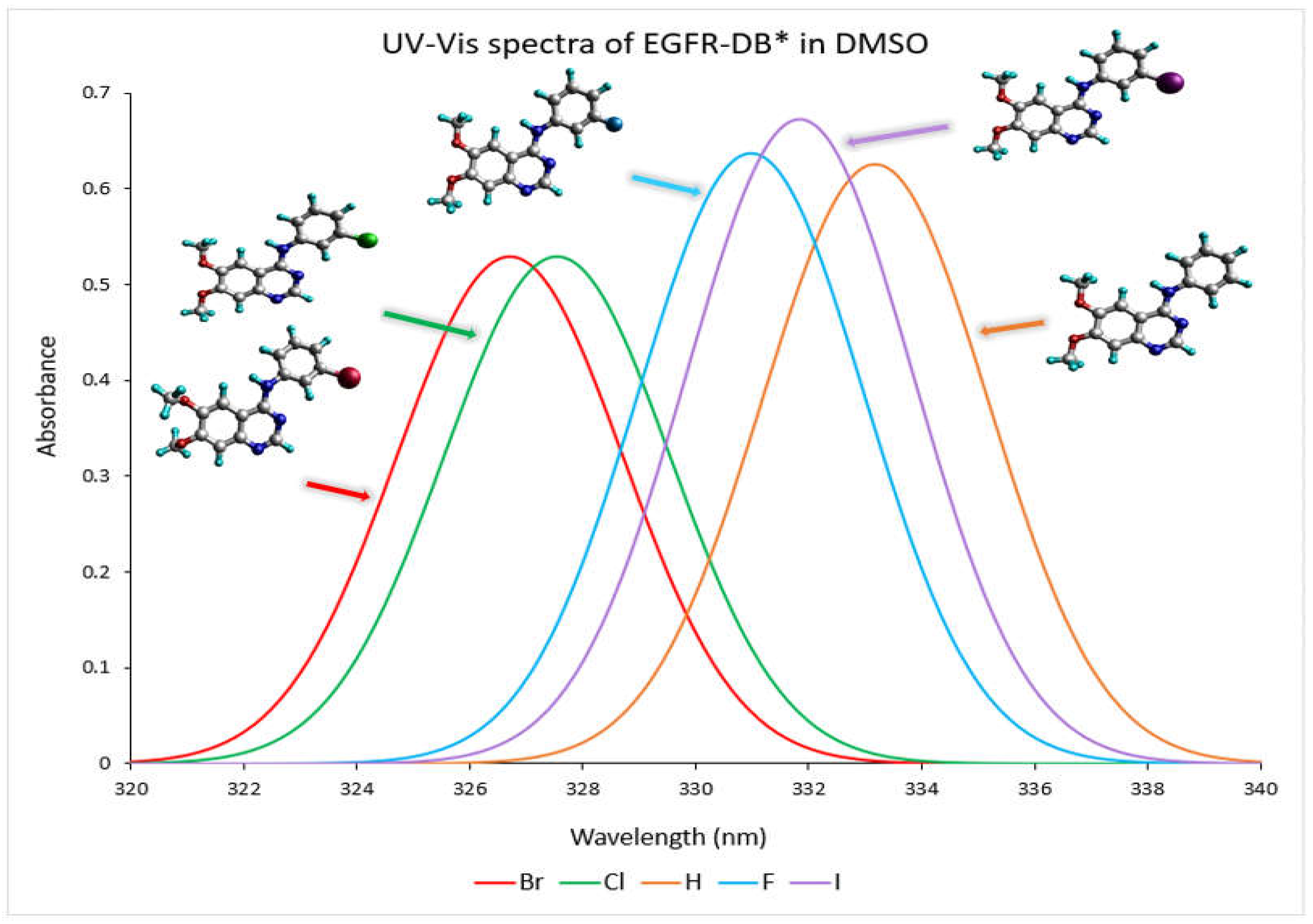
Figure 3 depicts the TD-DFT calculated UV–visible absorption spectrum spanning from a wavelength exceeding 320 nm to 340 nm for EGFR-DB in a DMSO solvent. As depicted in the spectra, the prominent spectral band with wavelengths exceeding 320 nm can be attributed to a robust, singular electronic transition. For instance, the transitions occurring at specific wavelengths, such as 333.17 nm with an oscillator strength of 0.62 in hydrogen, 330.98 nm with an oscillator strength of 0.64 in fluorine, 327.54 nm with an oscillator strength of 0.53 for chlorine, 326.71 nm with an oscillator strength of 0.53 for bromine, and 332.84 nm with an oscillator strength of 0.67 for iodine, are primarily dictated by the highest occupied molecular orbital to lowest unoccupied molecular orbital (HOMO-LUMO) transitions, each contributing substantially at 49%. In the UV-Vis spectra analysis, it is observed that the TKI-X, which is more potent, exhibits shorter wavelengths and a blueshift. For instance, when comparing the IC
50 values (nM), bromine with an IC
50 of 0.025 nM shows its maximum peak at 326.71 nm. Following this, chlorine with an IC
50 of 0.31 nM has its peak wavelength at 327.54 nm. Conversely, the hydrogen compound, which has the lowest IC
50 value of 29 nM, displays a rightward 7 nm red shift in its peak wavelength, measuring at 333.17 nm. Moreover, similar UV–visible wavelength peaks and spectra are observed between 210 and 330 nm for bromine and chlorine, which possess higher potencies compared to other TKIs-X with lower potency values. Refer to
Figure S2.1 in the Supplementary Materials for the comprehensive UV–visible spectra of EGFR-DB in a DMSO solvent.
The primary transitions, featuring an oscillator strength ƒ > 0.22 and electronic transition configurations with a contribution of more than 5%, are specified in
Table S4 for EGFR-DB structures and
Table S5 for the global minimum conformers in the
Supplementary Materials. Additional spectral bands observed at wavelengths below 320 nm are the result of numerous transitions involving frontier orbitals with low energy, like HOMO-n and LUMO+m, with n and m representing integers within a limited range (1, 2, 3, …, 9), indicating transitions associated with lower energy levels. The exact positions of the spectral bands in this range cannot be ascertained through the consideration of individual transitions alone. Instead, they require the utilization of the same fitting program employed in experimental measurements. Nonetheless, we have provisionally determined the positions by identifying the peaks of the highest points. For a comparative analysis of the UV-Vis absorption spectral maxima between the EGFR-DB and the five lowest energy conformers of TKIs-X, consult
Table S3 in the
Supplementary Materials.
Additionally, consult
Figure S2.2 for a comparative analysis of the UV-vis spectra of EGFR-DB. Notably, the structural resemblance among these compounds, characterized by sharing the common base of 6,7-dimethoxy-N-phenylquinazolin-4-amine, except for the variance in the atom (24) of the phenyl ring, contributes significantly to their spectral similarity. Meanwhile, differences observed in the spectra are attributed to the effects stemming from the substitution of halogens at the atom (24). For example, there are distinctions at the onset of the spectra. For instance, the iodine spectrum is more confined, commencing after 200 nm and concluding around 340 nm, while fluorine and hydrogen spectra commence at 180 nm, chlorine at 190 nm, and bromine at 195 nm. The key factors differentiating halogens and their effects on UV-vis spectra include their atomic structure, electronic configuration, and electronegativity. Variations in UV-Vis spectra among halogens are attributable to their unique electronic structures [
20]. This can manifest in discrepancies such as the intensity and position of absorption peaks and the range of wavelengths at which transitions occur, which might vary due to the different electronic configurations and bonding interactions involving halogens. The specific spectral transitions and features observed are influenced by the individual electronic properties of the halogen atoms and their interactions within the molecular environment.
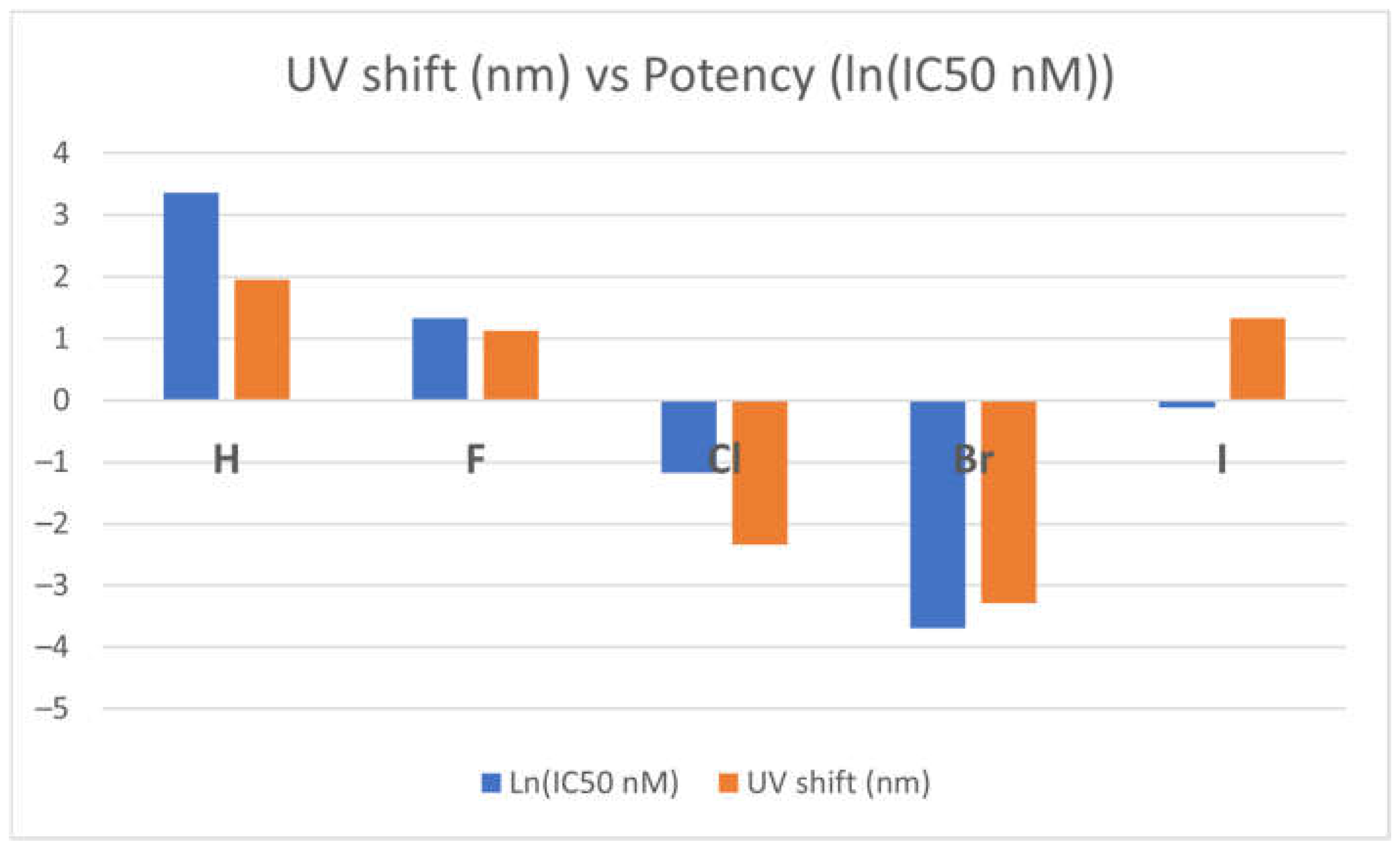
The potency of the TKI-X inhibitors seems to relate to the absorption optical shift.
Table 2 compares the optical properties with respect to the calculated maximum absorption wavelength (λ
max) of the global minimum structures
1 and EGFR-DB of the TKIs-X with the oscillator strengths, ƒ ≥ 0.5. The substitution of X leads to optical spectral shifts (λ
max) from the crystal structure to the global minimum structures
1, depending on the X atoms on the TKI. When X becomes hydrogen (H), fluorine (F), and iodine (I), the λ
max of their optical spectra of TKI-X exhibits a small red shift with 1.96 nm, 1.13 nm, and 1.33 nm, respectively. In contrast, TKI-X with X as chlorine (Cl) and bromine (Br) show redshift as −2.34 nm and −3.28 nm, respectively (highlighted in the table). Interestingly, Cl and Br show a blue shift from their EGFR-DB spectrum compared to the calculated global minimum structure spectrum, while the others exhibit a red-shift peak. This finding agrees with prior studies conducted using DFT-B3LYP. In our study, the absorption peak wavelengths of the global minimum structures for Br and Cl are 329.99 and 329.88, respectively, compared to 331.77 for Br and 331.02 for Cl in the previous study [
16,
19].
Figure 4 presents the correlation between optical shift and the potency of the TKI-X. As shown in the figure, the more blue shifts in their optical spectra (the blue columns in the figure) between the global minimum structures and their corresponding crystal structures from the EGFR database, the more potent it seems to be. Note that the relationship is more obvious when using the natural logarithm function ln(IC
50) function, as the potency indicator is usually very small.
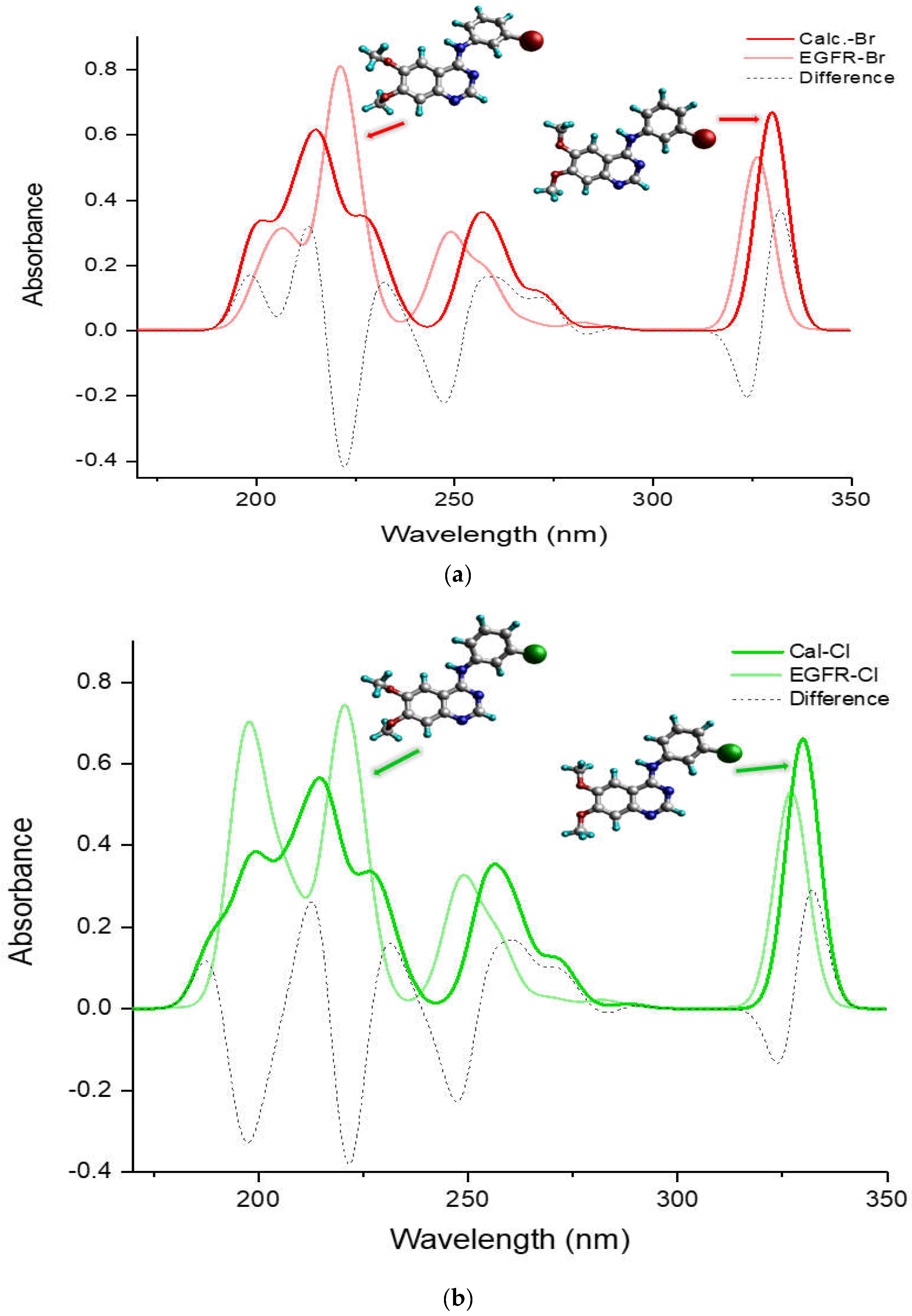
Figure 5 compares the entire UV-Vis absorption spectrum in a DMSO solvent ranging from 180 to 350 nm for the more potent TKIs, (a) TKI-Br and (b) TKI-Cl, which exhibit a different pattern compared to the others, H, F, and I. It presents spectra of the calculated global minimum structure, EGFR-DB structure, and the UV-Vis difference spectra produced by subtracting the spectrum of the calculated global minimum with the UV-Vis spectrum of the EGFR-DB (λcal−λEGFR-DB) for both TKI-Br and TKI-Cl (dash spectra). In
Figure 5a for bromine, the transparent-red spectrum represents TKI-Br with a peak absorption at 326.71 nm, and the red spectrum represents the calculated global minimum structure of TKI-Br with a blue-shifted peak at 329.99 nm. The figure illustrates that in the lower transition bands within the wavelength region of 180–230 nm, the transparent-red UV-Vis spectrum of TKI-Br shows one sharp peak with a strong absorption intensity at 220 nm shifted slightly from the 216 nm in the red spectrum, contrasting with the two moderate peaks observed in the red UV-Vis spectrum of the calculated global minimum structure of Br at 199 nm and 216 nm. Additionally, the onset of the red spectrum occurs at 199 nm, while for the transparent-red spectrum, the onset exhibits a blue shift at 201 nm. Difference absorbance spectra presented in a dashed line show the highest difference value between the calculated and EGFR spectra of TKI-Br occurred at 222 nm position bands, following the difference peak at 324 nm. In
Figure 5b, the transparent-green spectrum represents TKI-Cl, showing its highest absorption peak at 327.54 nm. In contrast, the green spectrum depicts the calculated global minimum structure of TKI-Cl, which has a slightly shifted peak at 329.88 nm. Within the wavelength range of 180–230 nm, the transparent-green UV-Vis spectrum of TKI-Cl displays two sharp peaks with strong absorption at 196 nm and 220 nm, contrasting with the two moderately intense peaks observed in the green UV-Vis spectrum at 197 nm and 216.5 nm. Moreover, the transparent-green spectrum begins its absorption at 189 nm, while the green spectrum starts at a slightly higher wavelength of 195 nm. The difference absorbance spectra highlight the greatest disparities between the calculated and EGFR spectra of TKI-Cl at the positions of 222 nm, 197 nm, and 247 nm, following the difference peak at 324 nm.
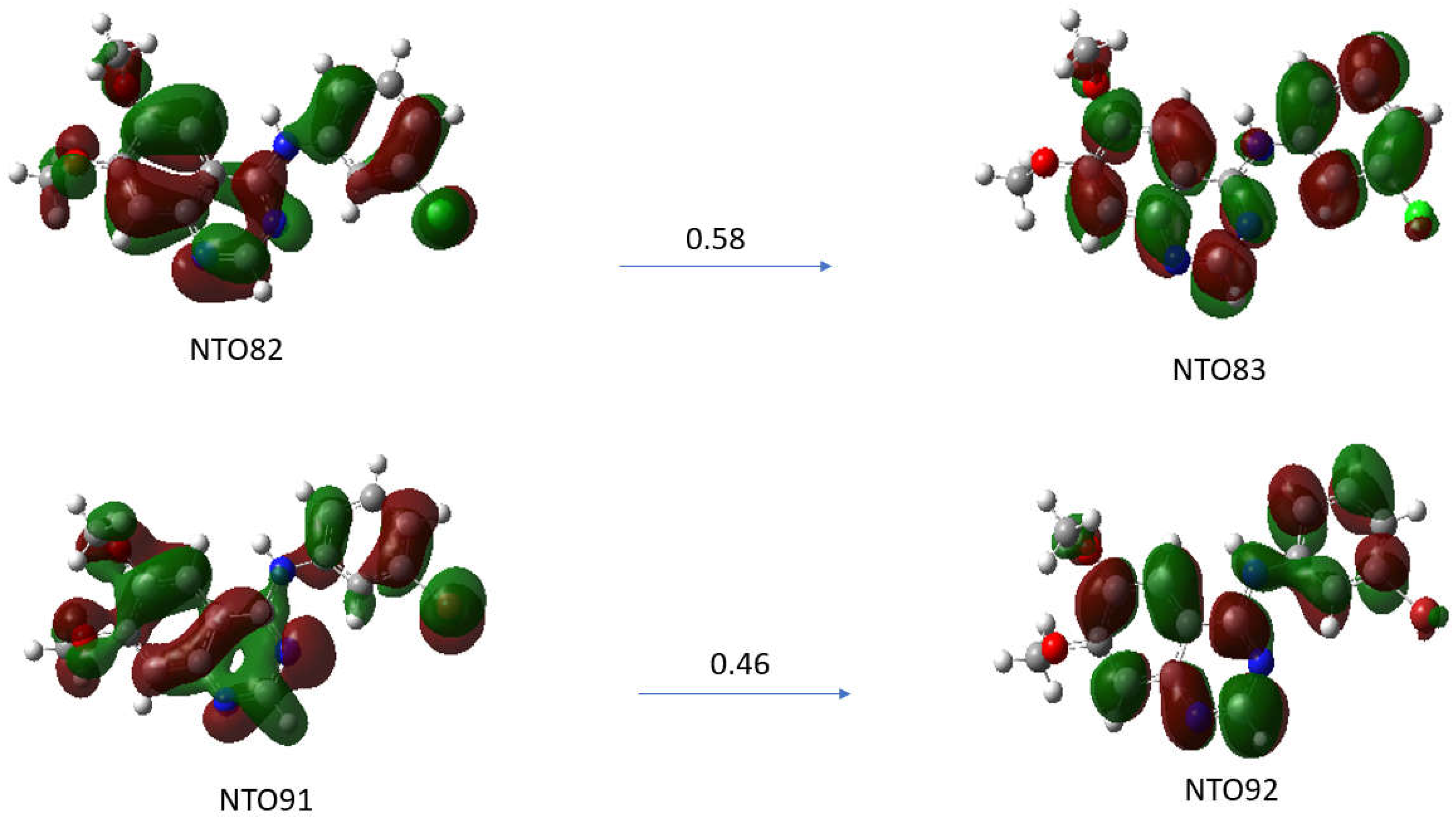
The λmax transition of the absorption spectra of the TKI-X are transitions between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). The LUMO-HOMO for TKI-Cl are orbital 82 and orbital 83, respectively, and for TKI-Br, are orbital 91 and orbital 92, respectively.
Figure 6 presents the natural transition orbital (NTO) method performing separate unitary transformations on occupied and virtual MOs, resulting in only one or a few dominant orbital pairs with significant contributions.
Natural transition orbitals (NTOs) illustrate the nature of optically active singlet excited states in the absorption bands, which help to recover an electron/hole picture while preserving phase information [
21,
22]. Specifically, the compound (a) TKI-Cl is examined for the NTO82→NTO83 with an oscillation strength of 0.5732 and the compound (b) TKI-Br for the transition of NTO91→NTO92 with an oscillation strength of 0.5542. As can be seen, although there are some similarities in the HOMOs and LUMOs of the TKI-Cl and TKI-Br, the electron distributions of the potent TKIs are not the same, reflecting the halogen impact. For example, the HOMO of TKI-Cl (NTO81) and the HOMO of TKI-Br (NTPO91) exhibit major differences in the quinazoline moieties and the phase where the HOMO (NTO91) of TKI-Br is more delocalized than the HOMO (NTO82) of TKI-Cl.
The dominant NTO pairs for the first four excited states of (a) TKI-Br and (b) TKI-Cl are shown in
Figure 7. The first excited state is at the top of the figure, the “hole” is on the left, and the “particle” is on the right for each compound and state. The associated eigenvalues λ for TKI-Br are 0.982423, 0.990344, 0.730883, and 0.861056, respectively. For (b) TKI-Cl, the associated eigenvalues λ are 0.983168, 0.980275, 0.729861, and 0.896348, respectively. This eigenvalue is >0.98 for the first two excited states, indicating that even the heavily mixed second state can be accurately described by a dominant excitation pair, which accounts for over 98% of the transition “percentage contribution to the electron excitation”.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}