
Implementation of a Novel Method for Processing Proteins from Acetic Acid Bacteria via Liquid Chromatography Coupled with Tandem Mass Spectrometry
 ,
,  , , , , , and
, , , , , and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
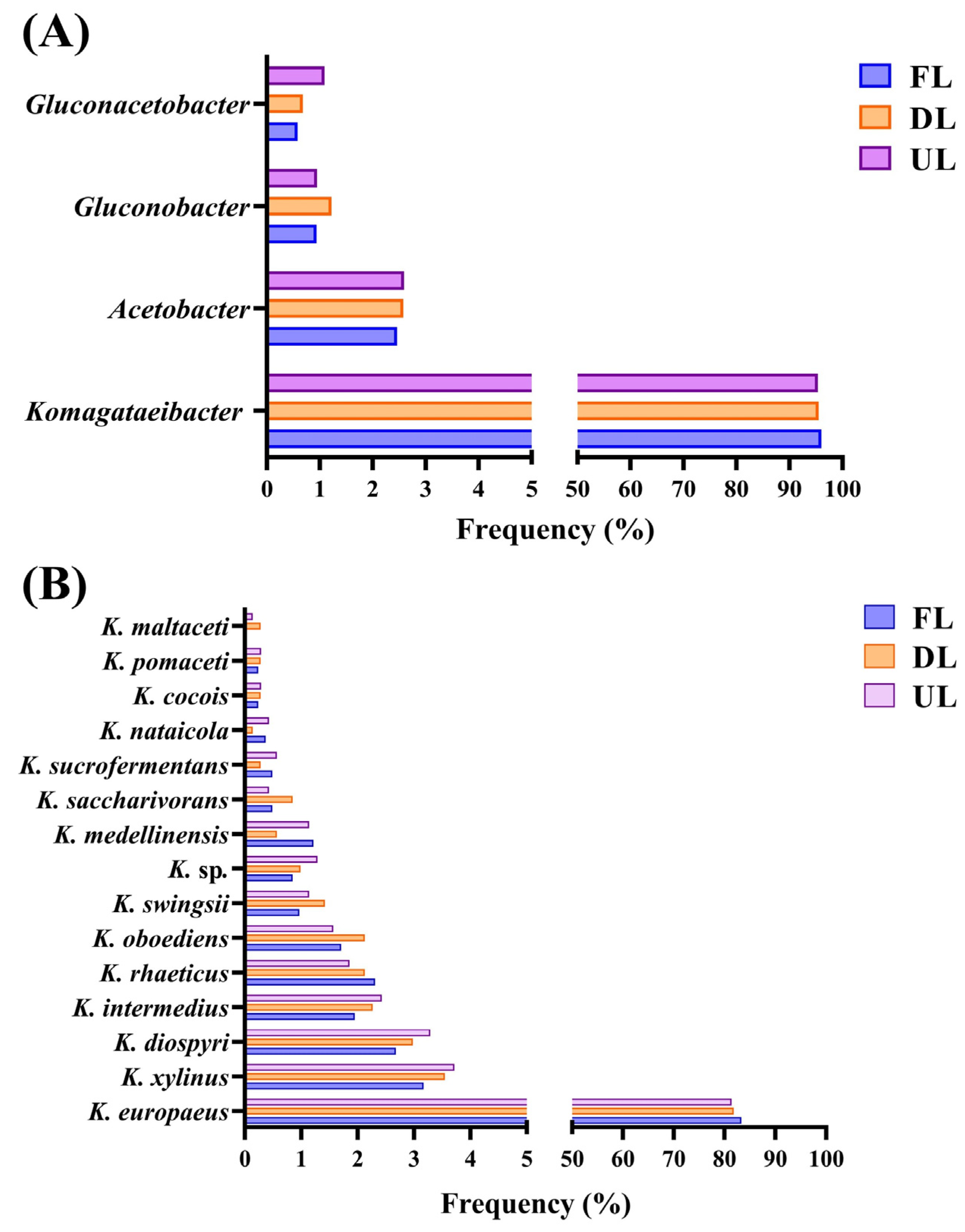
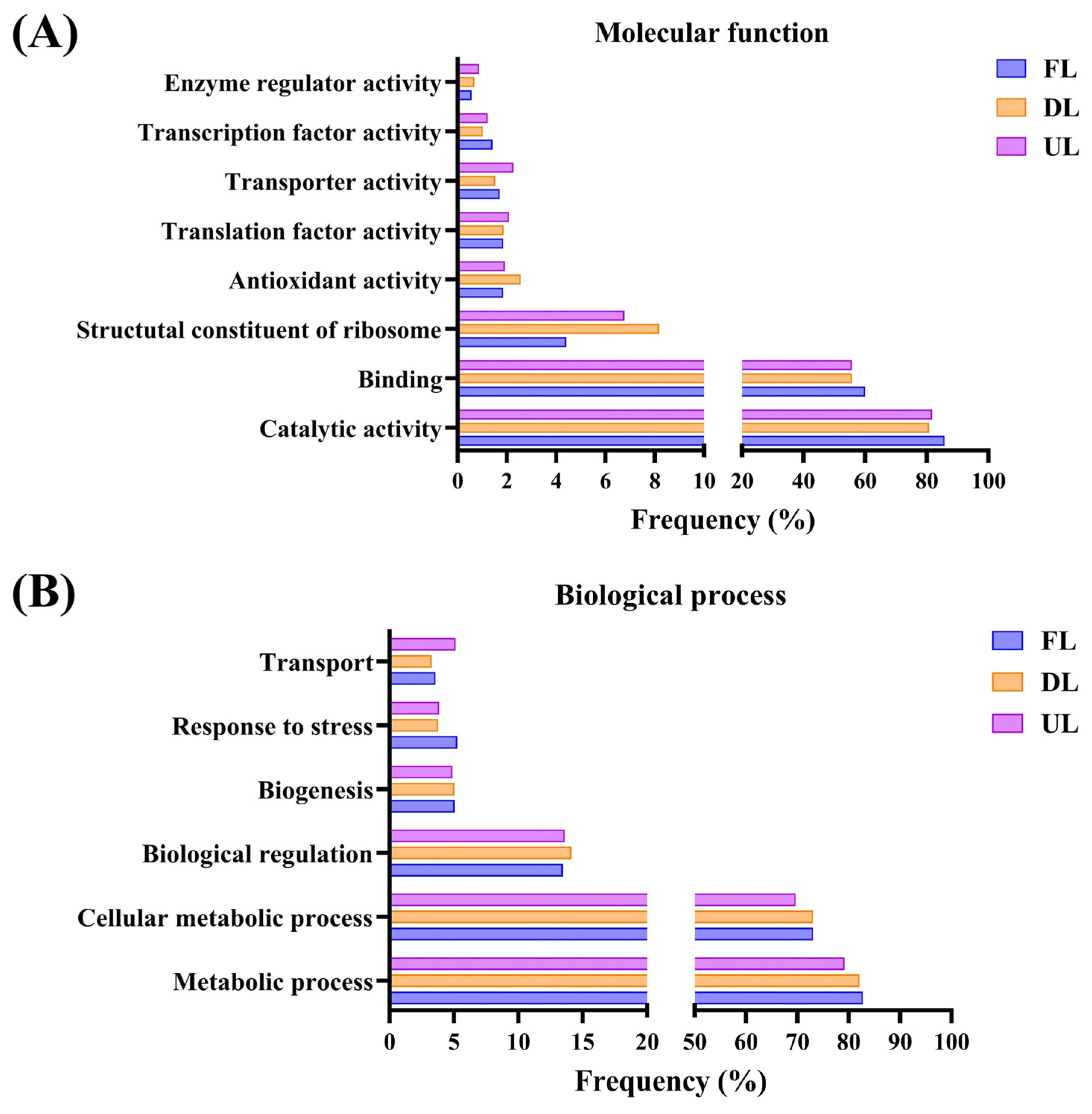
2.1. Qualitative Metaproteomics
2.2. Quantitative Proteomics
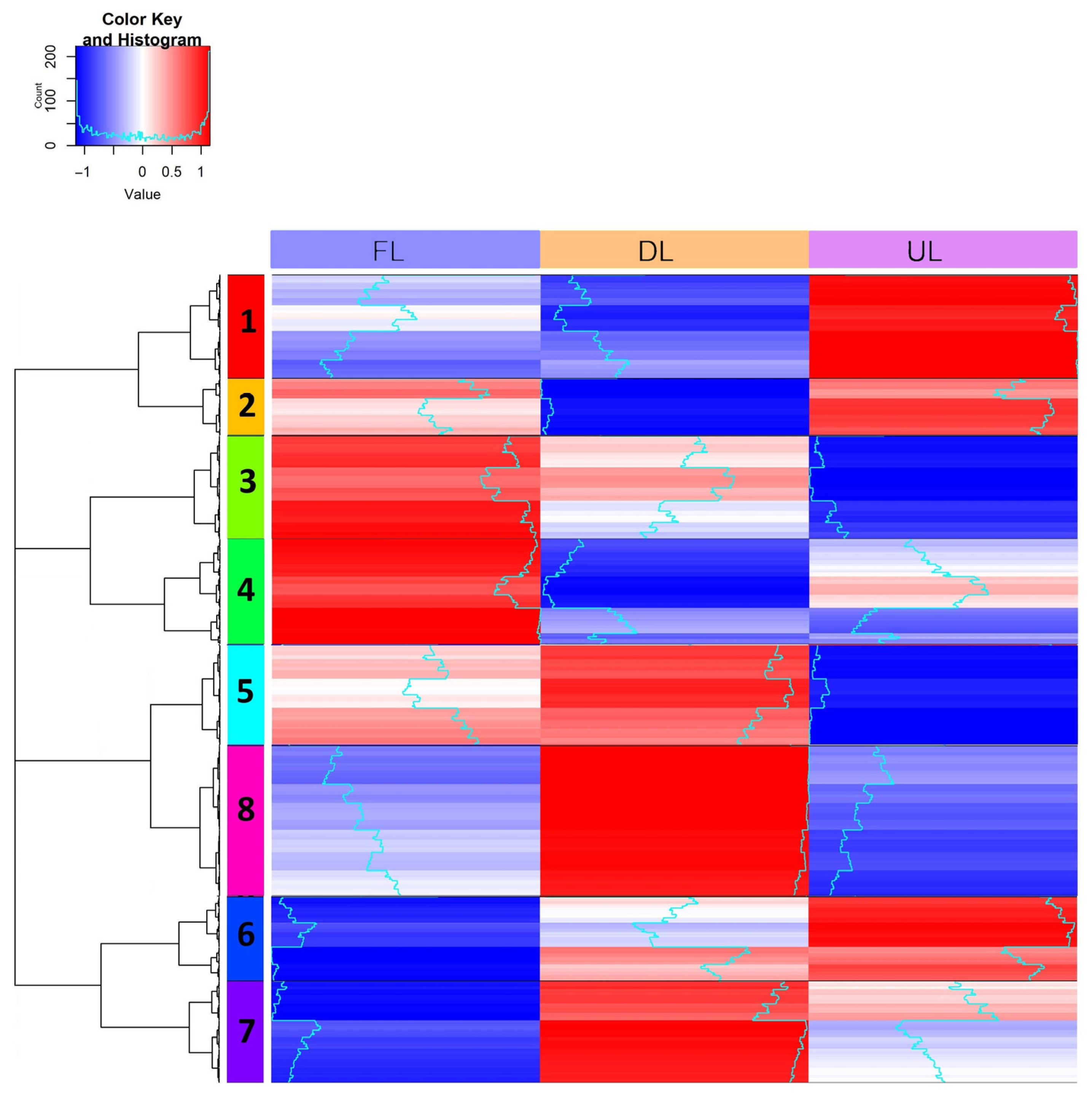
2.3. Proteomic Comparative Studies
3. Discussion
4. Materials and Methods
4.1. Raw Material
4.2. Starter Inoculum
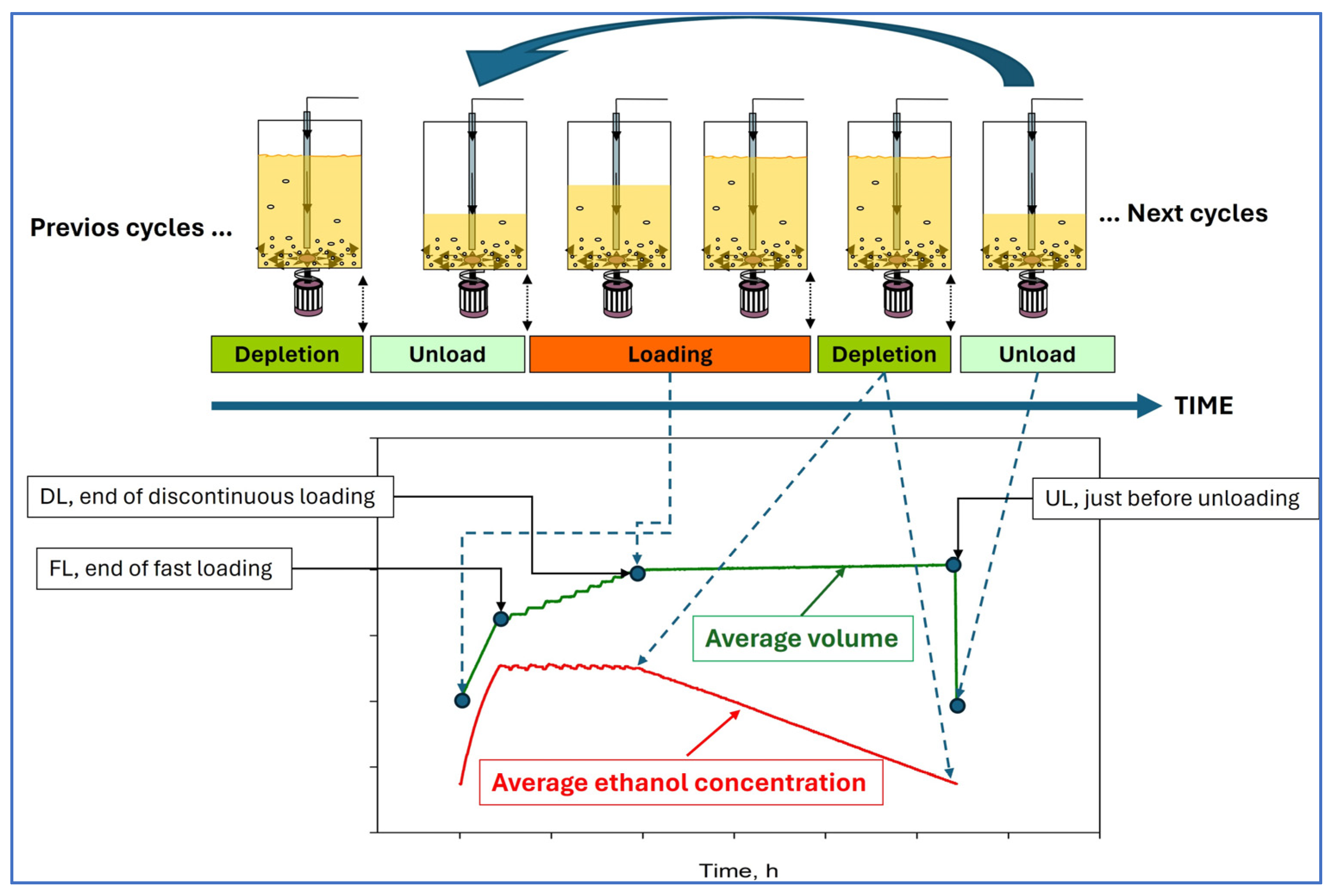
4.3. Fermentation Conditions
4.4. Sampling
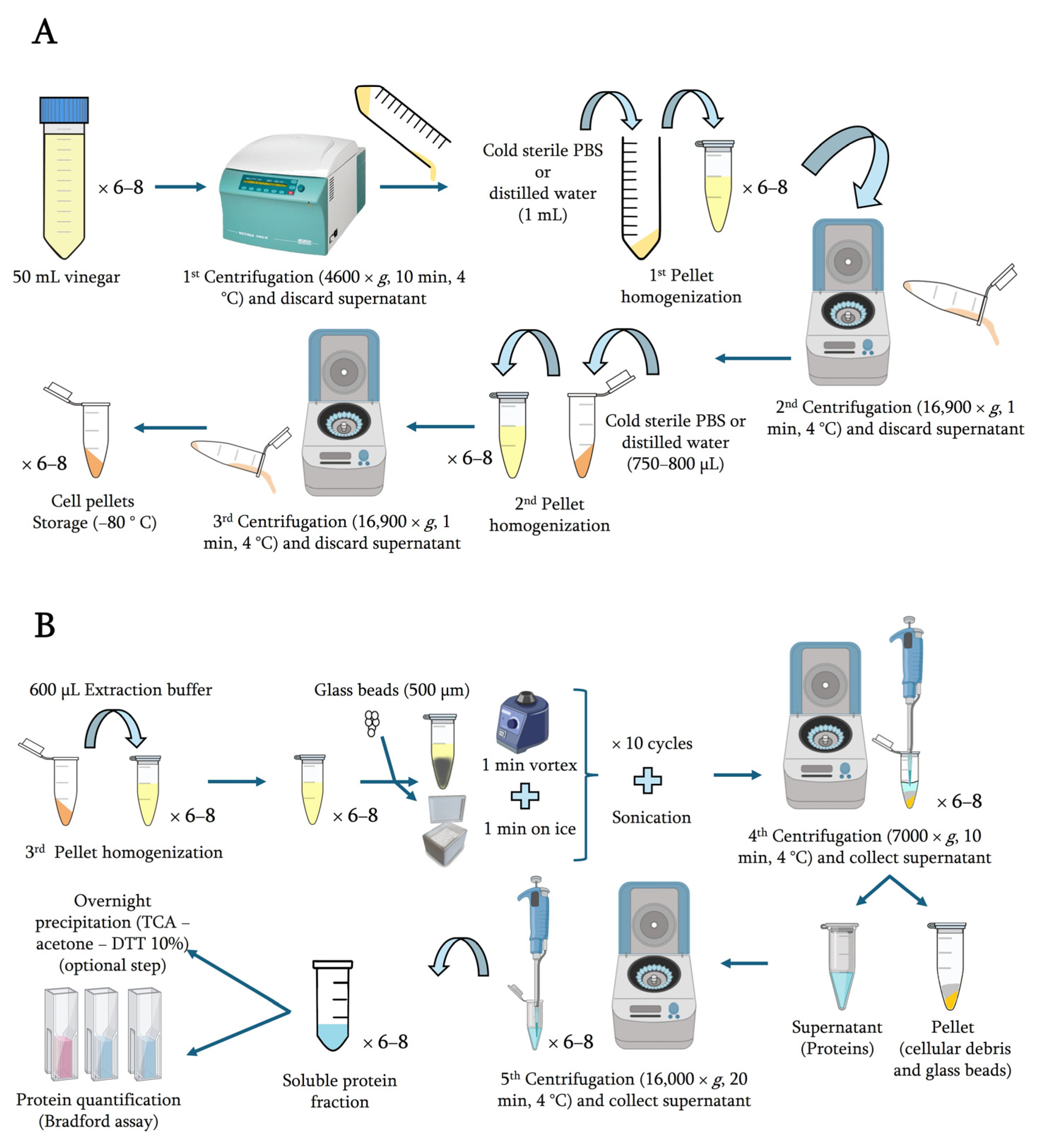
4.5. Cellular Collection
4.6. Protein Extraction
4.7. LC–MS/MS Analysis
4.8. Raw Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guzman, J.; Vilcinskas, A. Genome Analysis Suggests the Bacterial Family Acetobacteraceae Is a Source of Undiscovered Specialized Metabolites. Antonie Leeuwenhoek 2022, 115, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Komagata, K.; Iino, T.; Yamada, Y. The Family Acetobacteraceae. In The Prokaryotes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 3–78. ISBN 978-3-642-30196-4. [Google Scholar]
- Wang, B.; Shao, Y.; Chen, F. Overview on Mechanisms of Acetic Acid Resistance in Acetic Acid Bacteria. World J. Microbiol. Biotechnol. 2015, 31, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Gomes, R.J.; de Fatima Borges, M.; de Freitas Rosa, M.; Castro-Gómez, R.J.H.; Spinosa, W.A. Acetic Acid Bacteria in the Food Industry: Systematics, Characteristics and Applications. Food Technol. Biotechnol. 2018, 56, 139. [Google Scholar] [CrossRef]
- Garcia-Garcia, I.; Gullo, M.; Chen, F.; Garcia-Martinez, T. Editorial: Acetic Acid Bacteria. Front. Microbiol. 2023, 14, 1142659. [Google Scholar] [CrossRef] [PubMed]
- Adachi, O.; Tayama, K.; Shinagawa, E.; Matsushita, K.; Ameyama, M. Purification and Characterization of Membrane-Bound Aldehyde Dehydrogenase from Gluconobacter suboxydans. Agric. Biol. Chem. 1980, 44, 503–515. [Google Scholar] [CrossRef]
- Ameyama, M.; Adachi, O. Alcohol Dehydrogenase from Acetic Acid Bacteria, Membrane-Bound. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1982; Volume 89, pp. 450–457. ISBN 978-0-12-181989-7. [Google Scholar]
- He, Y.; Xie, Z.; Zhang, H.; Liebl, W.; Toyama, H.; Chen, F. Oxidative Fermentation of Acetic Acid Bacteria and Its Products. Front. Microbiol. 2022, 13, 879246. [Google Scholar] [CrossRef] [PubMed]
- Gullo, M.; Verzelloni, E.; Canonico, M. Aerobic Submerged Fermentation by Acetic Acid Bacteria for Vinegar Production: Process and Biotechnological Aspects. Process Biochem. 2014, 49, 1571–1579. [Google Scholar] [CrossRef]
- Santos-Dueñas, I.M.; Jimenez-Hornero, J.E.; Cañete-Rodriguez, A.M.; Garcia-Garcia, I. Modeling and Optimization of Acetic Acid Fermentation: A Polynomial-Based Approach. Biochem. Eng. J. 2015, 99, 35–43. [Google Scholar] [CrossRef]
- Álvarez-Cáliz, C.M.; Santos-Dueñas, I.M.; Jiménez-Hornero, J.E.; García-García, I. Optimization of the Acetification Stage in the Production of Wine Vinegar by Use of Two Serial Bioreactors. Appl. Sci. 2021, 11, 1217. [Google Scholar] [CrossRef]
- Jiménez-Hornero, J.E.; Santos-Dueñas, I.M.; García-García, I. Modelling Acetification with Artificial Neural Networks and Comparison with Alternative Procedures. Processes 2020, 8, 749. [Google Scholar] [CrossRef]
- Qin, Z.; Yu, S.; Chen, J.; Zhou, J. Dehydrogenases of Acetic Acid Bacteria. Biotechnol. Adv. 2022, 54, 107863. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.; Mientus, M.; Kostner, D.; Daniel, R.; Liebl, W.; Ehrenreich, A. Expression of Membrane-Bound Dehydrogenases from a Mother of Vinegar Metagenome in Gluconobacter oxydans. Appl. Microbiol. Biotechnol. 2017, 101, 7901–7912. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yan, P.; Lei, Q.; Li, B.; Sun, Y.; Li, S.; Lei, H.; Xie, N. Metabolic Adaptability Shifts of Cell Membrane Fatty Acids of Komagataeibacter hansenii HDM1-3 Improve Acid Stress Resistance and Survival in Acidic Environments. J. Ind. Microbiol. Biotechnol. 2019, 46, 1491–1503. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhang, R.; Xia, M.; Bai, X.; Mou, J.; Zheng, Y.; Wang, M. Effect of Aspartic Acid and Glutamate on Metabolism and Acid Stress Resistance of Acetobacter pasteurianus. Microb. Cell Factories 2017, 16, 109. [Google Scholar] [CrossRef] [PubMed]
- Sankuan, X.; Cuimei, Z.; Bingqian, F.; Yu, Z.; Menglei, X.; Linna, T.; Jia, S.; Xinyi, Z.; Min, W. Metabolic Network of Ammonium in Cereal Vinegar Solid-State Fermentation and Its Response to Acid Stress. Food Microbiol. 2021, 95, 103684. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Zhang, Y.; Hong, H. Classification of Acetic Acid Bacteria and Their Acid Resistant Mechanism. AMB Express 2021, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Xia, K.; Zang, N.; Zhang, J.; Zhang, H.; Li, Y.; Liu, Y.; Feng, W.; Liang, X. New Insights into the Mechanisms of Acetic Acid Resistance in Acetobacter pasteurianus Using iTRAQ-Dependent Quantitative Proteomic Analysis. Int. J. Food Microbiol. 2016, 238, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Román-Camacho, J.J.; García-García, I.; Santos-Dueñas, I.M.; Ehrenreich, A.; Liebl, W.; García-Martínez, T.; Mauricio, J.C. Combining Omics Tools for the Characterization of the Microbiota of Diverse Vinegars Obtained by Submerged Culture: 16S rRNA Amplicon Sequencing and MALDI-TOF MS. Front. Microbiol. 2022, 13, 1055010. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, F.; Zhang, C.; Yang, L.; Fan, G.; Xu, Y.; Sun, B.; Li, X. Dynamic Microbial Succession of Shanxi Aged Vinegar and Its Correlation with Flavor Metabolites during Different Stages of Acetic Acid Fermentation. Sci. Rep. 2018, 8, 8612. [Google Scholar] [CrossRef]
- Trček, J.; Mahnič, A.; Rupnik, M. Diversity of the Microbiota Involved in Wine and Organic Apple Cider Submerged Vinegar Production as Revealed by DHPLC Analysis and Next-Generation Sequencing. Int. J. Food Microbiol. 2016, 223, 57–62. [Google Scholar] [CrossRef]
- Mamlouk, D.; Gullo, M. Acetic Acid Bacteria: Physiology and Carbon Sources Oxidation. Indian J. Microbiol. 2013, 53, 377–384. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, C.; Dong, L.; Huan, Y.; Yoshimoto, M.; Zhu, Y.; Tada, I.; Wang, X.; Zhao, S.; Zhang, F.; et al. Comprehensive Metabolomic Comparison of Five Cereal Vinegars Using Non-Targeted and Chemical Isotope Labeling LC-MS Analysis. Metabolites 2022, 12, 427. [Google Scholar] [CrossRef]
- Andrés-Barrao, C.; Saad, M.M.; Cabello Ferrete, E.; Bravo, D.; Chappuis, M.-L.; Ortega Pérez, R.; Junier, P.; Perret, X.; Barja, F. Metaproteomics and Ultrastructure Characterization of Komagataeibacter spp. Involved in High-Acid Spirit Vinegar Production. Food Microbiol. 2016, 55, 112–122. [Google Scholar] [CrossRef]
- Wu, L.-H.; Lu, Z.-M.; Zhang, X.-J.; Wang, Z.-M.; Yu, Y.-J.; Shi, J.-S.; Xu, Z.-H. Metagenomics Reveals Flavour Metabolic Network of Cereal Vinegar Microbiota. Food Microbiol. 2017, 62, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hong, H.; Zhang, C.; Huang, Z.; Guo, H. Transcriptome Analysis of Komagataeibacter europaeus CGMCC 20445 Responses to Different Acidity Levels During Acetic Acid Fermentation. Pol. J. Microbiol. 2021, 70, 305–313. [Google Scholar] [CrossRef]
- Heyer, R.; Schallert, K.; Zoun, R.; Becher, B.; Saake, G.; Benndorf, D. Challenges and Perspectives of Metaproteomic Data Analysis. J. Biotechnol. 2017, 261, 24–36. [Google Scholar] [CrossRef]
- Elviri, L.; Mattarozzi, M. Food Proteomics. In Chemical Analysis of Food: Techniques and Applications; Elsevier: Amsterdam, The Netherlands, 2012; pp. 519–537. ISBN 978-0-12-384862-8. [Google Scholar]
- Winnik, W.M.; DeKroon, R.M.; Jeong, J.S.Y.; Mocanu, M.; Robinette, J.B.; Osorio, C.; Dicheva, N.N.; Hamlett, E.; Alzate, O. Analysis of Proteins Using DIGE and MALDI Mass Spectrometry. In Difference Gel Electrophoresis (DIGE); Cramer, R., Westermeier, R., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; Volume 854, pp. 47–66. ISBN 978-1-61779-572-5. [Google Scholar]
- Trček, J.; Barja, F. Updates on Quick Identification of Acetic Acid Bacteria with a Focus on the 16S–23S rRNA Gene Internal Transcribed Spacer and the Analysis of Cell Proteins by MALDI-TOF Mass Spectrometry. Int. J. Food Microbiol. 2015, 196, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Malachová, A.; Stránská, M.; Václavíková, M.; Elliott, C.T.; Black, C.; Meneely, J.; Hajšlová, J.; Ezekiel, C.N.; Schuhmacher, R.; Krska, R. Advanced LC–MS-Based Methods to Study the Co-Occurrence and Metabolization of Multiple Mycotoxins in Cereals and Cereal-Based Food. Anal. Bioanal. Chem. 2018, 410, 801–825. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhang, P.; Xie, M.; An, F.; Qiu, B.; Wu, R. Metaproteomics of Microbiota in Naturally Fermented Soybean Paste, Da-jiang. J. Food Sci. 2018, 83, 1342–1349. [Google Scholar] [CrossRef]
- Li, H.; Byrne, K.; Galiamov, R.; Mendoza-Porras, O.; Bose, U.; Howitt, C.A.; Colgrave, M.L. Using LC-MS to Examine the Fermented Food Products Vinegar and Soy Sauce for the Presence of Gluten. Food Chem. 2018, 254, 302–308. [Google Scholar] [CrossRef]
- Hu, C.; Wang, W.; Jo, J.; Garey, K.W. Development and Validation of LC-MS/MS for Quantifying Omadacycline from Stool for Gut Microbiome Studies. J. Chromatogr. B 2024, 1236, 124057. [Google Scholar] [CrossRef] [PubMed]
- Zayed, M.; El-Garawani, I.M.; El-Sabbagh, S.M.; Amr, B.; Alsharif, S.M.; Tayel, A.A.; AlAjmi, M.F.; Ibrahim, H.M.S.; Shou, Q.; Khalifa, S.A.M.; et al. Structural Diversity, LC-MS-MS Analysis and Potential Biological Activities of Brevibacillus laterosporus Extract. Metabolites 2022, 12, 1102. [Google Scholar] [CrossRef] [PubMed]
- Román-Camacho, J.J.; Santos-Dueñas, I.M.; García-García, I.; Moreno-García, J.; García-Martínez, T.; Mauricio, J.C. Metaproteomics of Microbiota Involved in Submerged Culture Production of Alcohol Wine Vinegar: A First Approach. Int. J. Food Microbiol. 2020, 333, 108797. [Google Scholar] [CrossRef]
- Román-Camacho, J.J.; Mauricio, J.C.; Santos-Dueñas, I.M.; García-Martínez, T.; García-García, I. Functional Metaproteomic Analysis of Alcohol Vinegar Microbiota during an Acetification Process: A Quantitative Proteomic Approach. Food Microbiol. 2021, 98, 103799. [Google Scholar] [CrossRef] [PubMed]
- Román-Camacho, J.J.; Mauricio, J.C.; Santos-Dueñas, I.M.; García-Martínez, T.; García-García, I. Unraveling the Role of Acetic Acid Bacteria Comparing Two Acetification Profiles From Natural Raw Materials: A Quantitative Approach in Komagataeibacter europaeus. Front. Microbiol. 2022, 13, 840119. [Google Scholar] [CrossRef]
- Román-Camacho, J.J.; García-García, I.; Santos-Dueñas, I.M.; García-Martínez, T.; Mauricio, J.C. Latest Trends in Industrial Vinegar Production and the Role of Acetic Acid Bacteria: Classification, Metabolism, and Applications—A Comprehensive Review. Foods 2023, 12, 3705. [Google Scholar] [CrossRef]
- Lasko, D.R.; Schwerdel, C.; Bailey, J.E.; Sauer, U. Acetate-Specific Stress Response in Acetate-Resistant Bacteria: An Analysis of Protein Patterns. Biotechnol. Prog. 1997, 13, 519–523. [Google Scholar] [CrossRef]
- Nakano, S.; Fukaya, M.; Horinouchi, S. Putative ABC Transporter Responsible for Acetic Acid Resistance in Acetobacter aceti. Appl. Environ. Microbiol. 2006, 72, 497–505. [Google Scholar] [CrossRef]
- Nakano, S.; Fukaya, M.; Horinouchi, S. Enhanced Expression of Aconitase Raises Acetic Acid Resistance in Acetobacter aceti. FEMS Microbiol. Lett. 2004, 235, 315–322. [Google Scholar] [CrossRef]
- Andrés-Barrao, C.; Saad, M.M.; Chappuis, M.-L.; Boffa, M.; Perret, X.; Ortega Pérez, R.; Barja, F. Proteome Analysis of Acetobacter pasteurianus during Acetic Acid Fermentation. J. Proteom. 2012, 75, 1701–1717. [Google Scholar] [CrossRef]
- Wieme, A.D.; Spitaels, F.; Aerts, M.; De Bruyne, K.; Van Landschoot, A.; Vandamme, P. Effects of Growth Medium on Matrix-Assisted Laser Desorption–Ionization Time of Flight Mass Spectra: A Case Study of Acetic Acid Bacteria. Appl. Environ. Microbiol. 2014, 80, 1528–1538. [Google Scholar] [CrossRef] [PubMed]
- Andrés-Barrao, C.; Benagli, C.; Chappuis, M.; Ortega Pérez, R.; Tonolla, M.; Barja, F. Rapid Identification of Acetic Acid Bacteria Using MALDI-TOF Mass Spectrometry Fingerprinting. Syst. Appl. Microbiol. 2013, 36, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Pérez, R.; Torres, C.; Sanz, S.; Ruiz-Larrea, F. Strain Typing of Acetic Acid Bacteria Responsible for Vinegar Production by the Submerged Elaboration Method. Food Microbiol. 2010, 27, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Andres-Barrao, C.; Weber, A.; Chappuis, M.L. Acetic Acid Bacteria Population Dynamics and Natural Imposition of Gluconacetobacter europaeus during Submerged Vinegar Production. Arch. Sci. 2011, 64, 99–114. [Google Scholar] [CrossRef]
- Rizo, J.; Guillén, D.; Farrés, A.; Díaz-Ruiz, G.; Sánchez, S.; Wacher, C.; Rodríguez-Sanoja, R. Omics in Traditional Vegetable Fermented Foods and Beverages. Crit. Rev. Food Sci. Nutr. 2018, 60, 791–809. [Google Scholar] [CrossRef] [PubMed]
- Minden, J.S. Two-Dimensional Difference Gel Electrophoresis (2D DIGE). In Methods in Cell Biology; Elsevier: Amsterdam, The Netherlands, 2012; Volume 112, pp. 111–141. ISBN 978-0-12-405914-6. [Google Scholar]
- Porras-Agüera, J.A.; Román-Camacho, J.J.; Moreno-García, J.; Mauricio, J.C.; Moreno, J.; García-Martínez, T. Effect of Endogenous CO2 Overpressure on the Yeast “Stressome” during the “Prise de Mousse” of Sparkling Wine. Food Microbiol. 2020, 89, 103431. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.H.; Stensballe, A.; Nielsen, P.H.; Herbst, F.-A. Metaproteomics: Evaluation of Protein Extraction from Activated Sludge. Proteomics 2014, 14, 2535–2539. [Google Scholar] [CrossRef]
- Román-Camacho, J.J.; Mauricio, J.C.; Santos-Dueñas, I.M.; García-Martínez, T.; García-García, I. Recent Advances in Applying Omic Technologies for Studying Acetic Acid Bacteria in Industrial Vinegar Production: A Comprehensive Review. Biotechnol. J. 2024, 19, 2300566. [Google Scholar] [CrossRef]
- Bimmer, M.; Reimer, M.; Klingl, A.; Ludwig, C.; Zollfrank, C.; Liebl, W.; Ehrenreich, A. Analysis of Cellulose Synthesis in a High-Producing Acetic Acid Bacterium Komagataeibacter hansenii. Appl. Microbiol. Biotechnol. 2023, 107, 2947–2967. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, R.; Yin, H.; Bai, X.; Chang, Y.; Xia, M.; Wang, M. Acetobacter pasteurianus Metabolic Change Induced by Initial Acetic Acid to Adapt to Acetic Acid Fermentation Conditions. Appl. Microbiol. Biotechnol. 2017, 101, 7007–7016. [Google Scholar] [CrossRef]
- Huang, T.; Lu, Z.-M.; Peng, M.-Y.; Chai, L.-J.; Zhang, X.-J.; Shi, J.-S.; Li, Q.; Xu, Z.-H. Constructing a Defined Starter for Multispecies Vinegar Fermentation via Evaluation of the Vitality and Dominance of Functional Microbes in an Autochthonous Starter. Appl. Environ. Microbiol. 2022, 88, e02175-21. [Google Scholar] [CrossRef] [PubMed]
- Pinu, F.; De Carvalho-Silva, S.; Trovatti Uetanabaro, A.; Villas-Boas, S. Vinegar Metabolomics: An Explorative Study of Commercial Balsamic Vinegars Using Gas Chromatography-Mass Spectrometry. Metabolites 2016, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Campos-Vázquez, C.; Román-Camacho, J.J.; Consuegra-Rivera, R.; Santos-Dueñas, I.M.; García-García, I.; García-Martínez, T.; Mauricio, J.C. Exploring Microbial Diversity and Functionality in Verdejo Wine Vinegar Fermentation through Lc-Ms/Ms Analysis; Elsevier: Rochester, NY, USA, 2024. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Román-Camacho, J.J.; Mauricio, J.C.; Sánchez-León, I.; Santos-Dueñas, I.M.; Fuentes-Almagro, C.A.; Amil-Ruiz, F.; García-Martínez, T.; García-García, I. Implementation of a Novel Method for Processing Proteins from Acetic Acid Bacteria via Liquid Chromatography Coupled with Tandem Mass Spectrometry. Molecules 2024, 29, 2548. https://doi.org/10.3390/molecules29112548
Román-Camacho JJ, Mauricio JC, Sánchez-León I, Santos-Dueñas IM, Fuentes-Almagro CA, Amil-Ruiz F, García-Martínez T, García-García I. Implementation of a Novel Method for Processing Proteins from Acetic Acid Bacteria via Liquid Chromatography Coupled with Tandem Mass Spectrometry. Molecules. 2024; 29(11):2548. https://doi.org/10.3390/molecules29112548
Chicago/Turabian StyleRomán-Camacho, Juan J., Juan C. Mauricio, Irene Sánchez-León, Inés M. Santos-Dueñas, Carlos A. Fuentes-Almagro, Francisco Amil-Ruiz, Teresa García-Martínez, and Isidoro García-García. 2024. "Implementation of a Novel Method for Processing Proteins from Acetic Acid Bacteria via Liquid Chromatography Coupled with Tandem Mass Spectrometry" Molecules 29, no. 11: 2548. https://doi.org/10.3390/molecules29112548
APA StyleRomán-Camacho, J. J., Mauricio, J. C., Sánchez-León, I., Santos-Dueñas, I. M., Fuentes-Almagro, C. A., Amil-Ruiz, F., García-Martínez, T., & García-García, I. (2024). Implementation of a Novel Method for Processing Proteins from Acetic Acid Bacteria via Liquid Chromatography Coupled with Tandem Mass Spectrometry. Molecules, 29(11), 2548. https://doi.org/10.3390/molecules29112548