
Molecular Docking and Simulation-Binding Analysis of Plant Phytochemicals with the Hepatocellular Carcinoma Targets Epidermal Growth Factor Receptor and Caspase-9

 ,
, 
Abstract
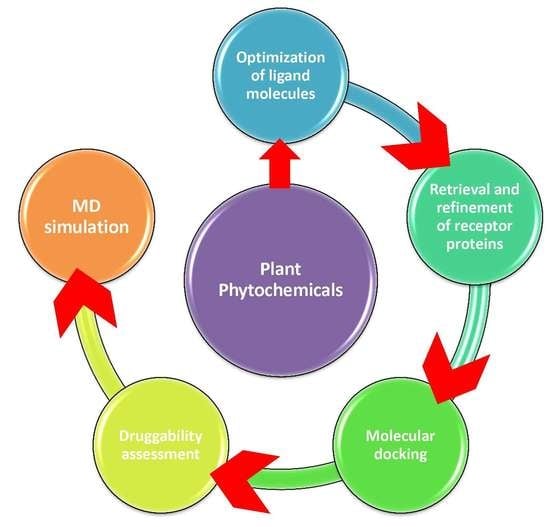
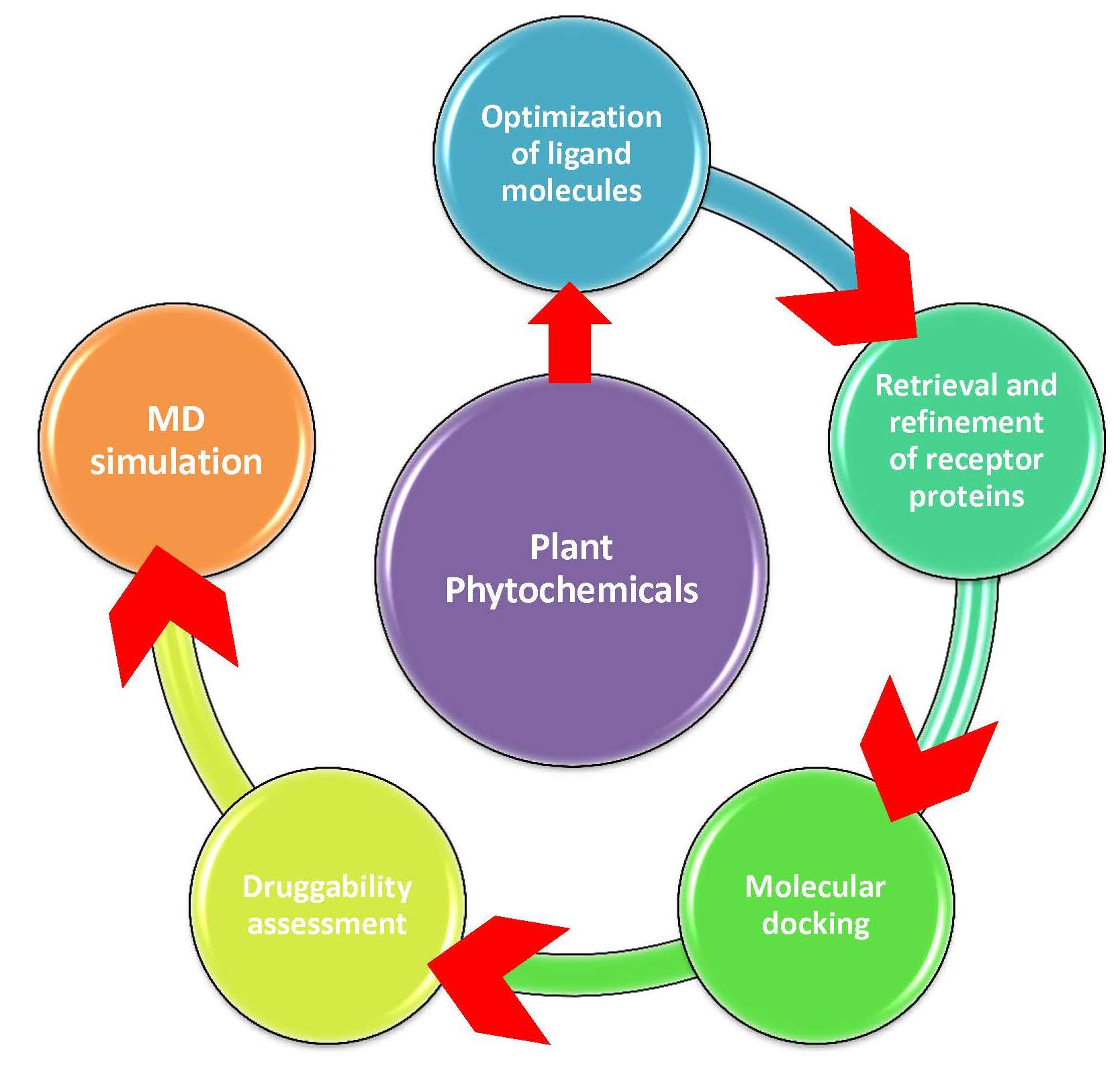
1. Introduction
2. Results
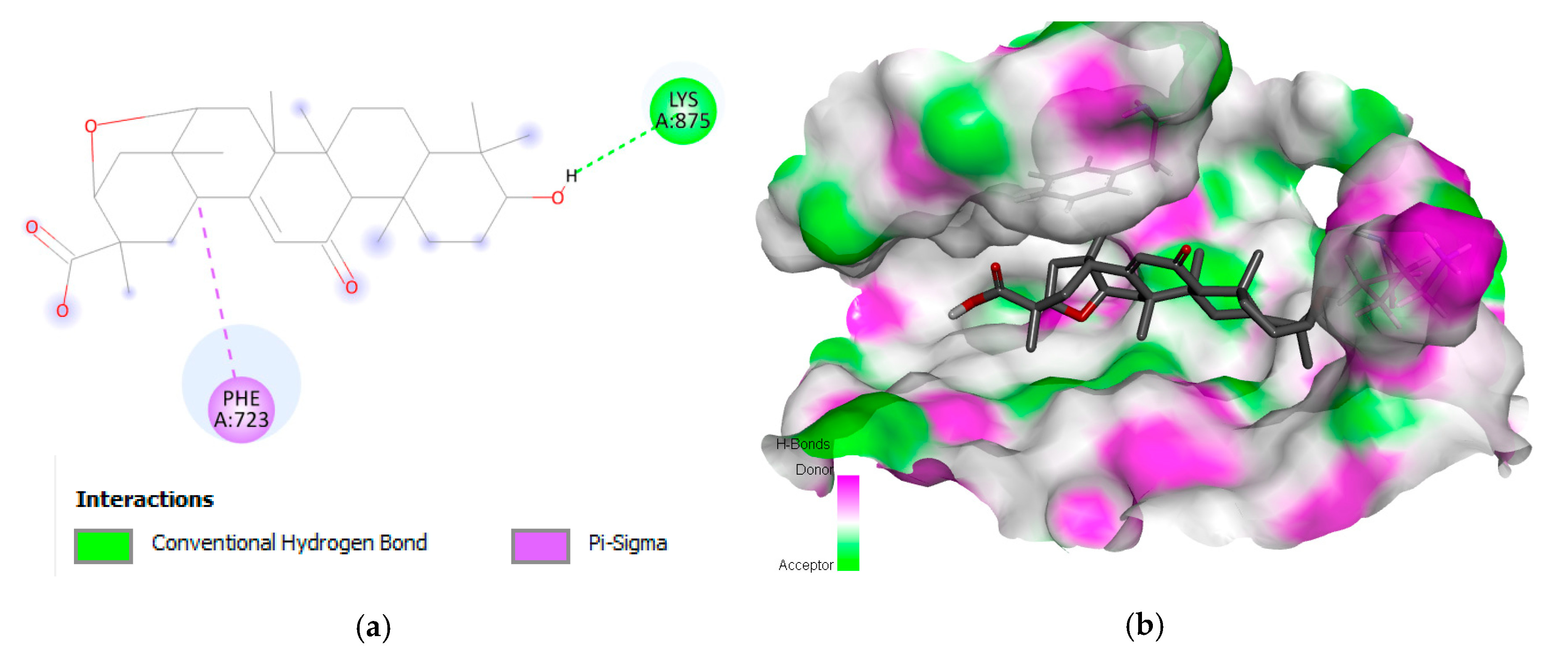
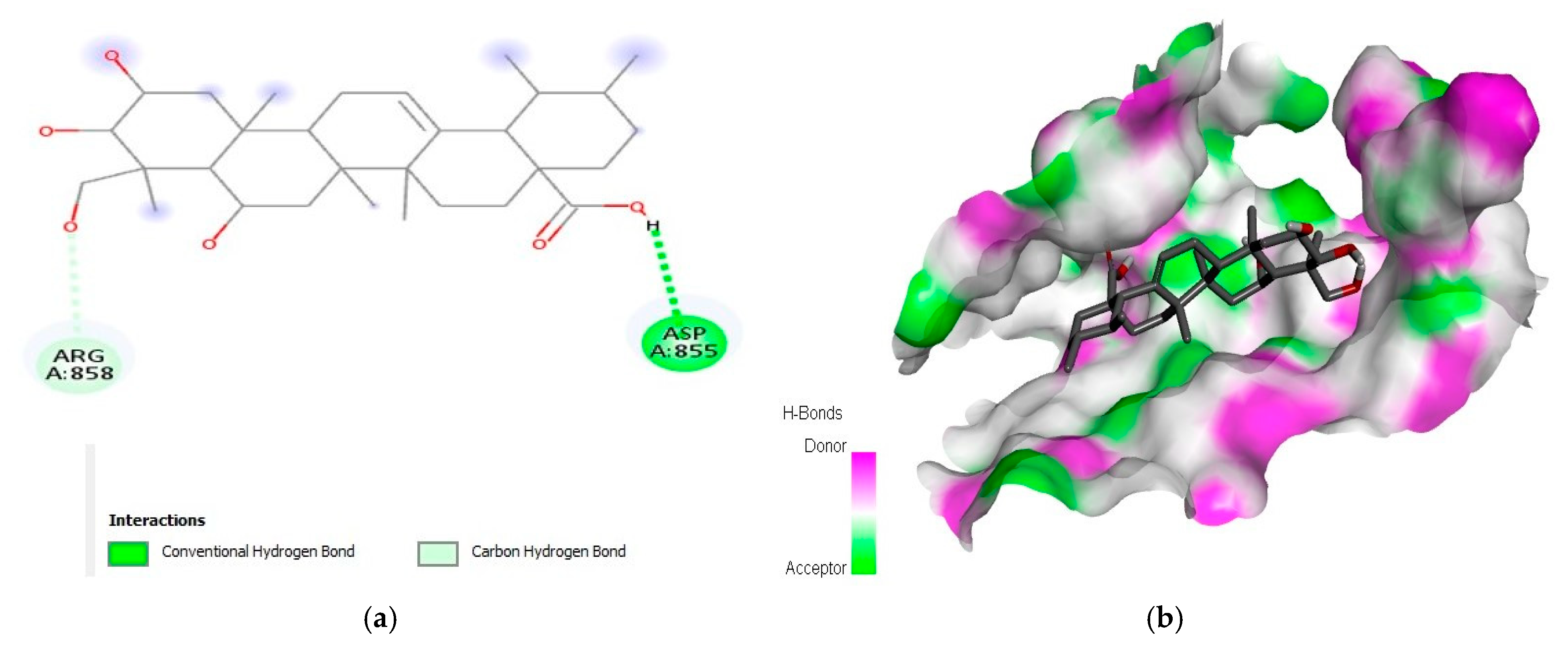
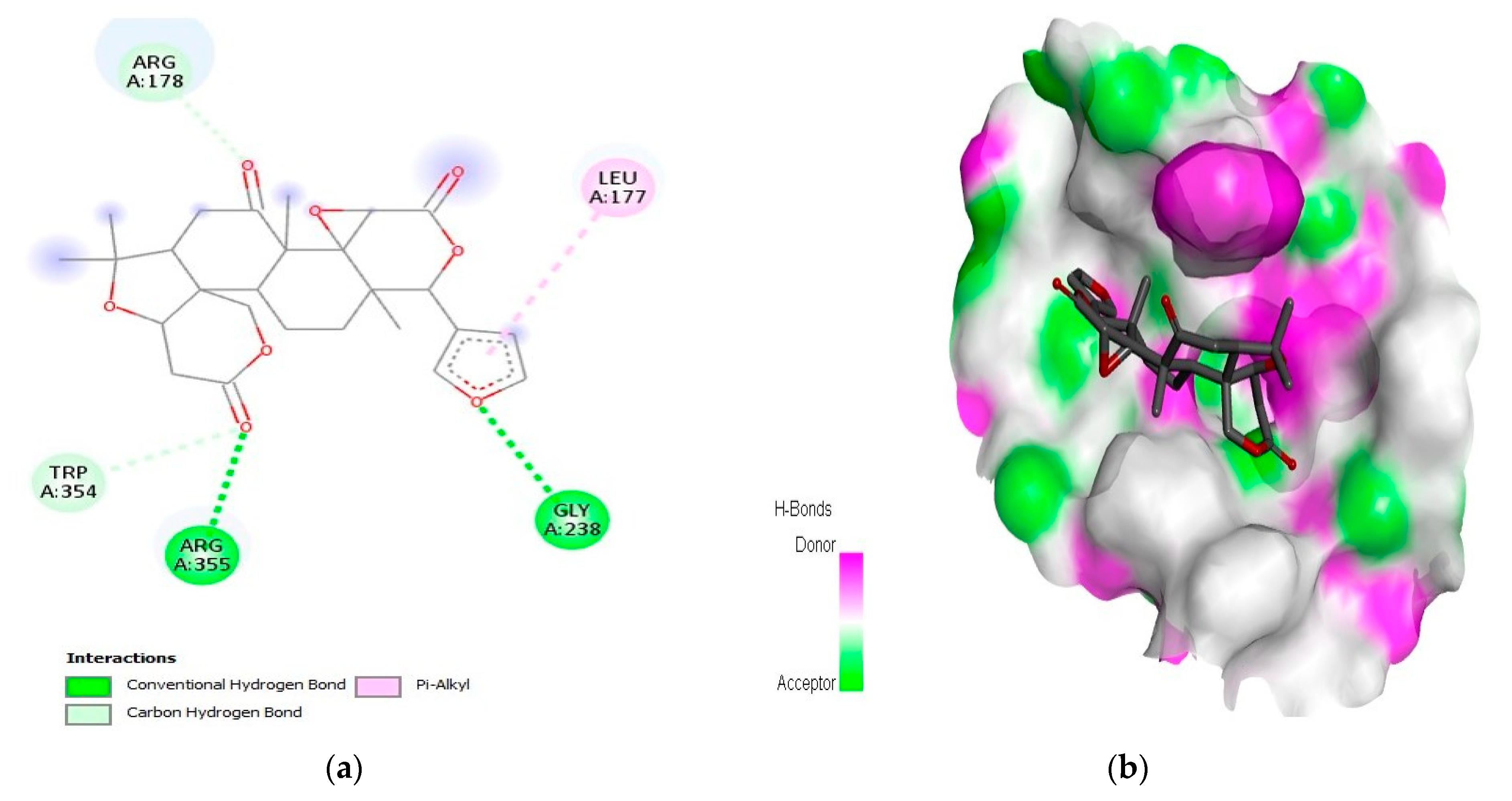
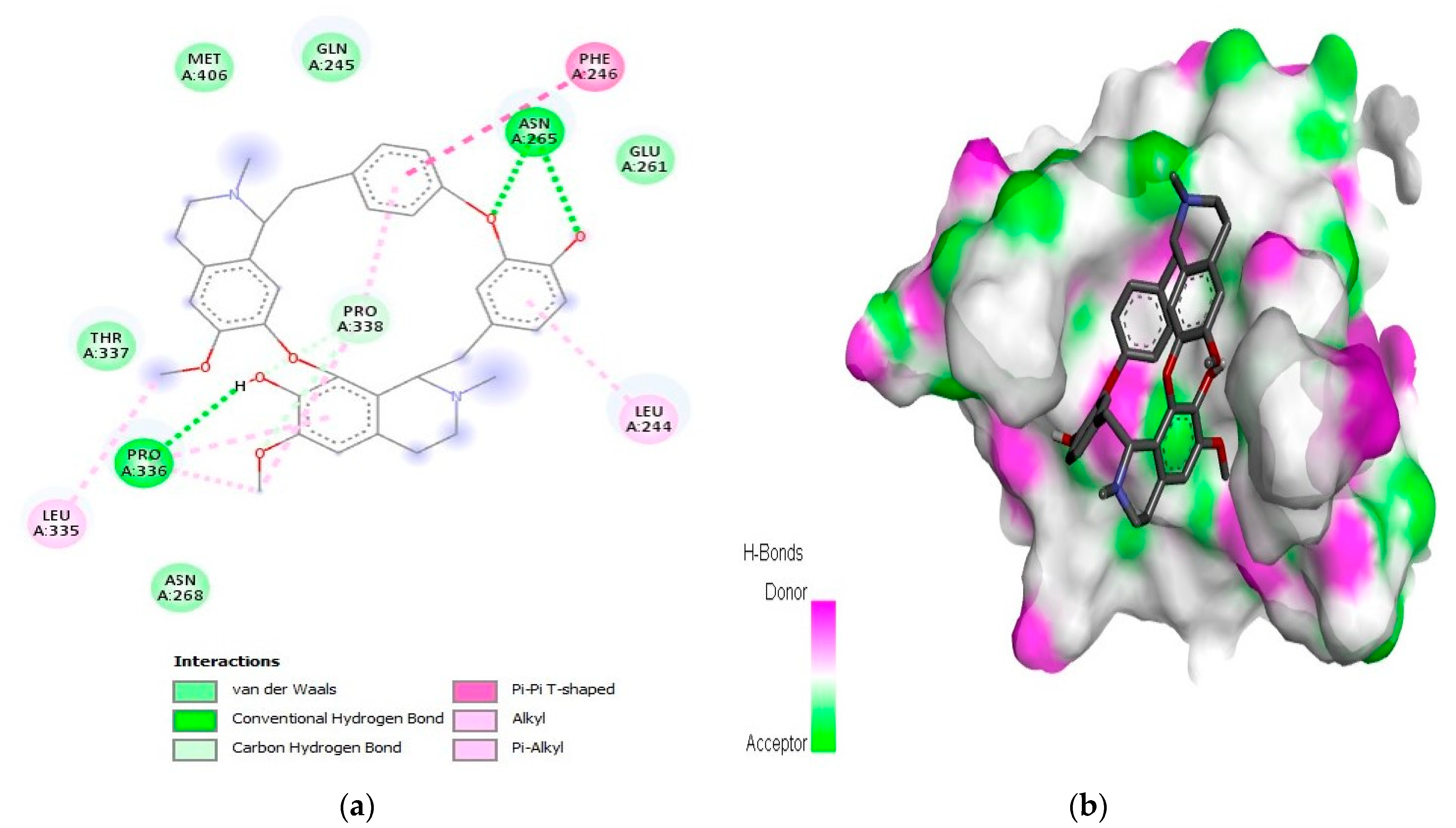
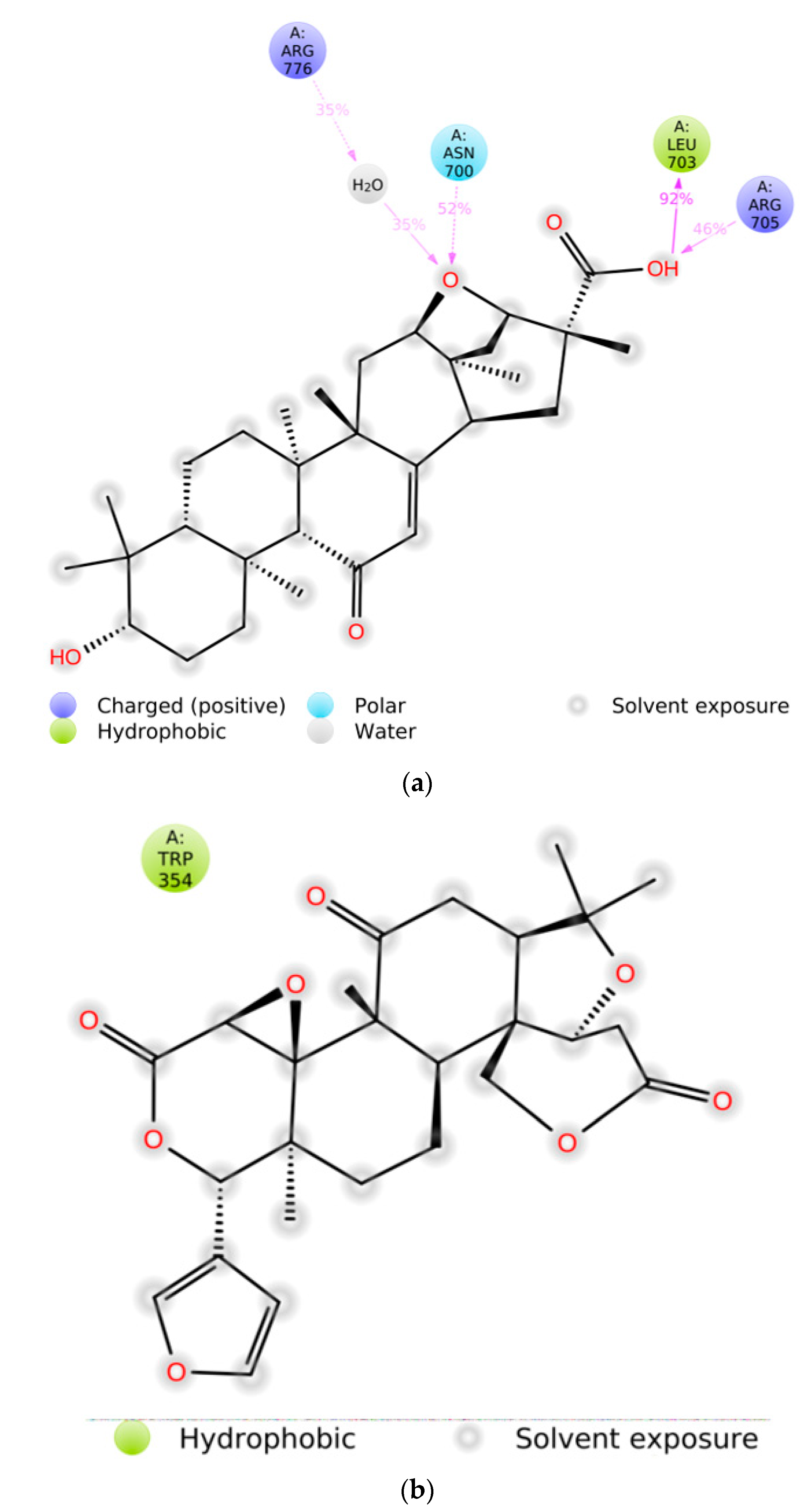
2.1. Interaction Analysis
2.2. Druggability Analyses
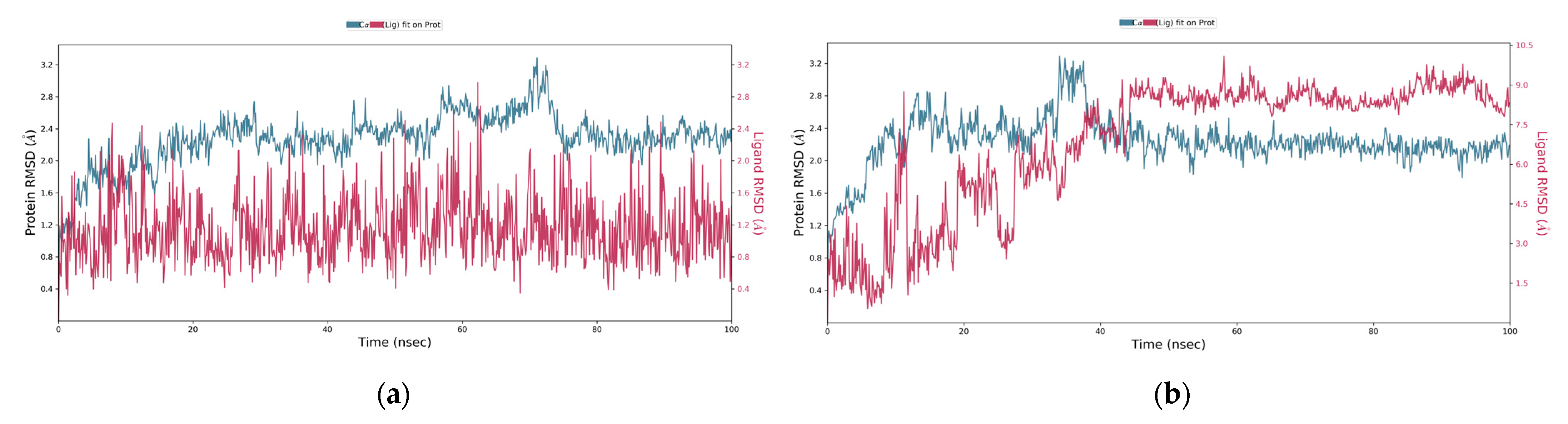
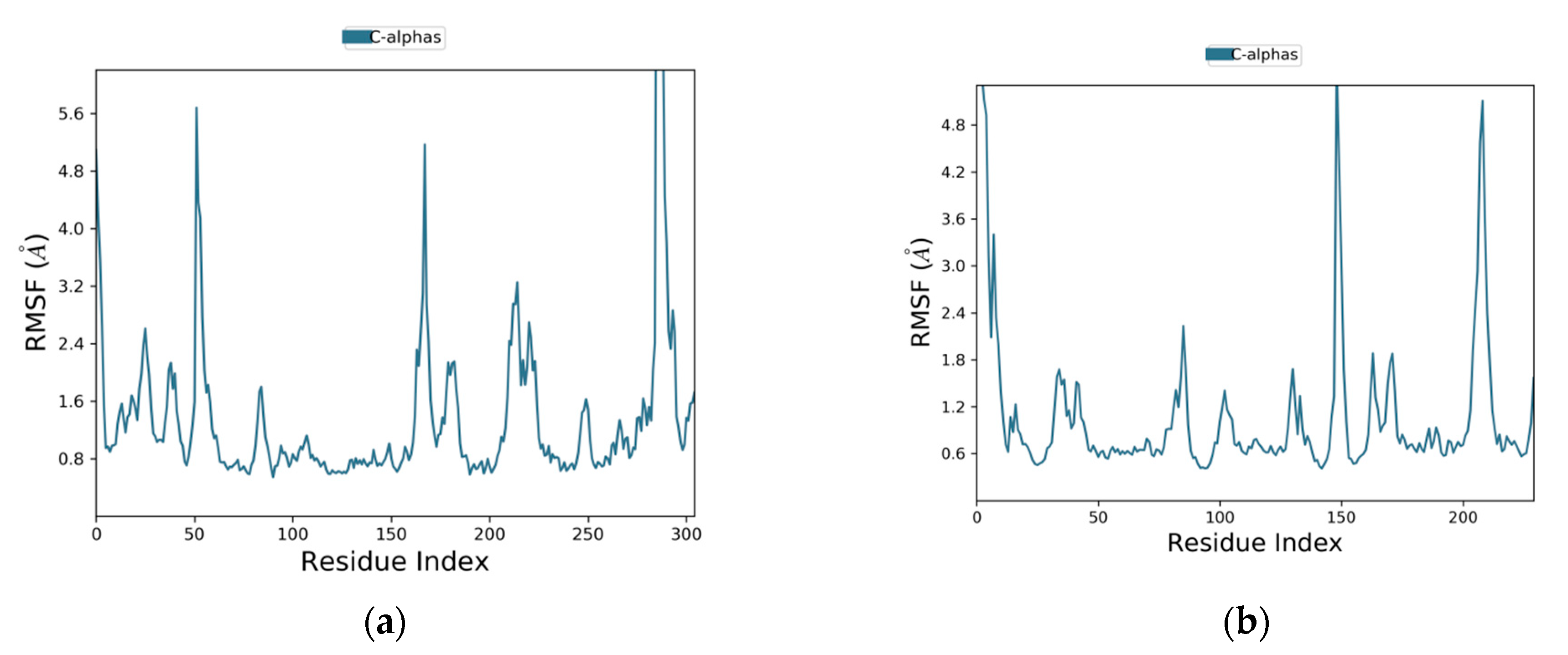
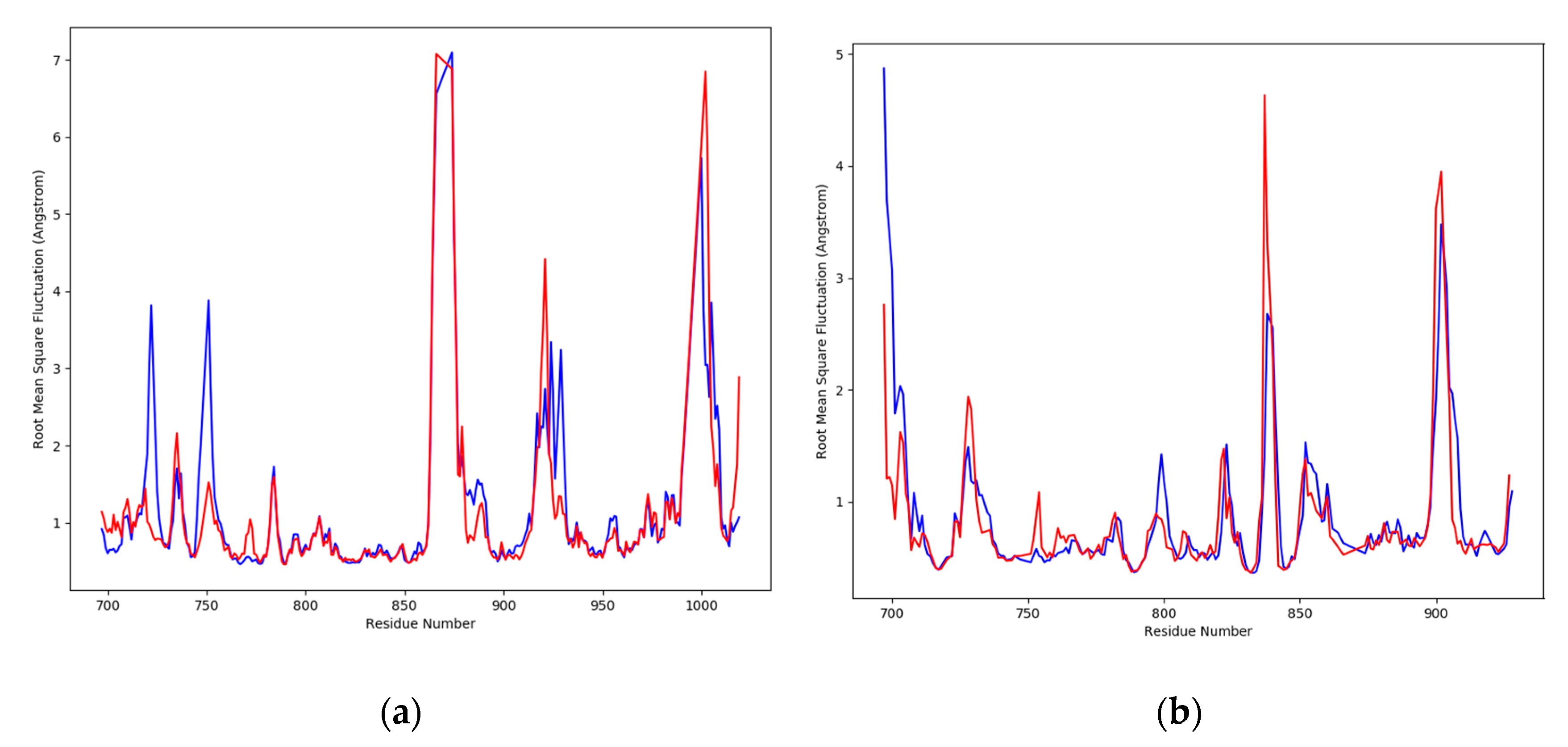
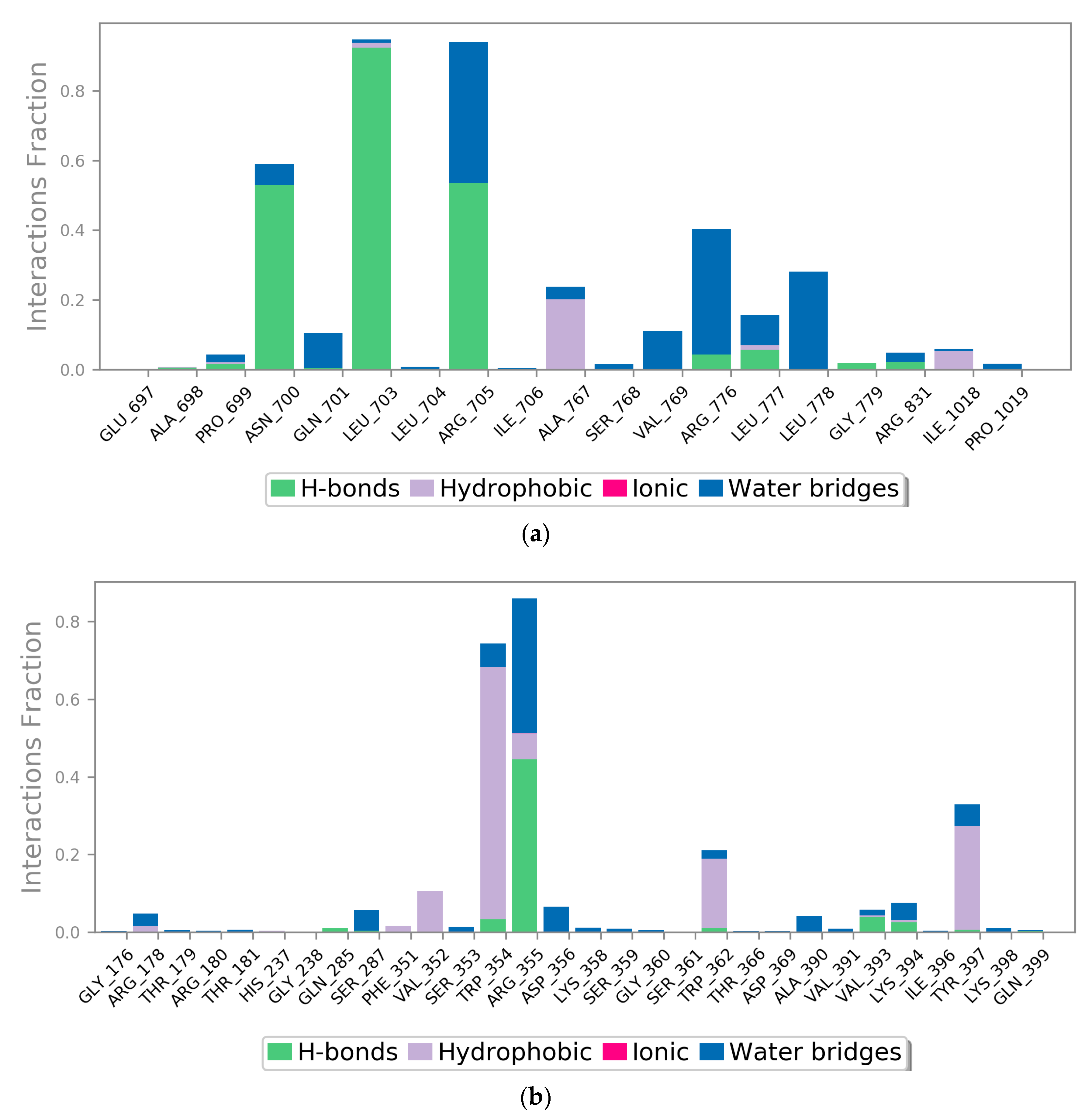
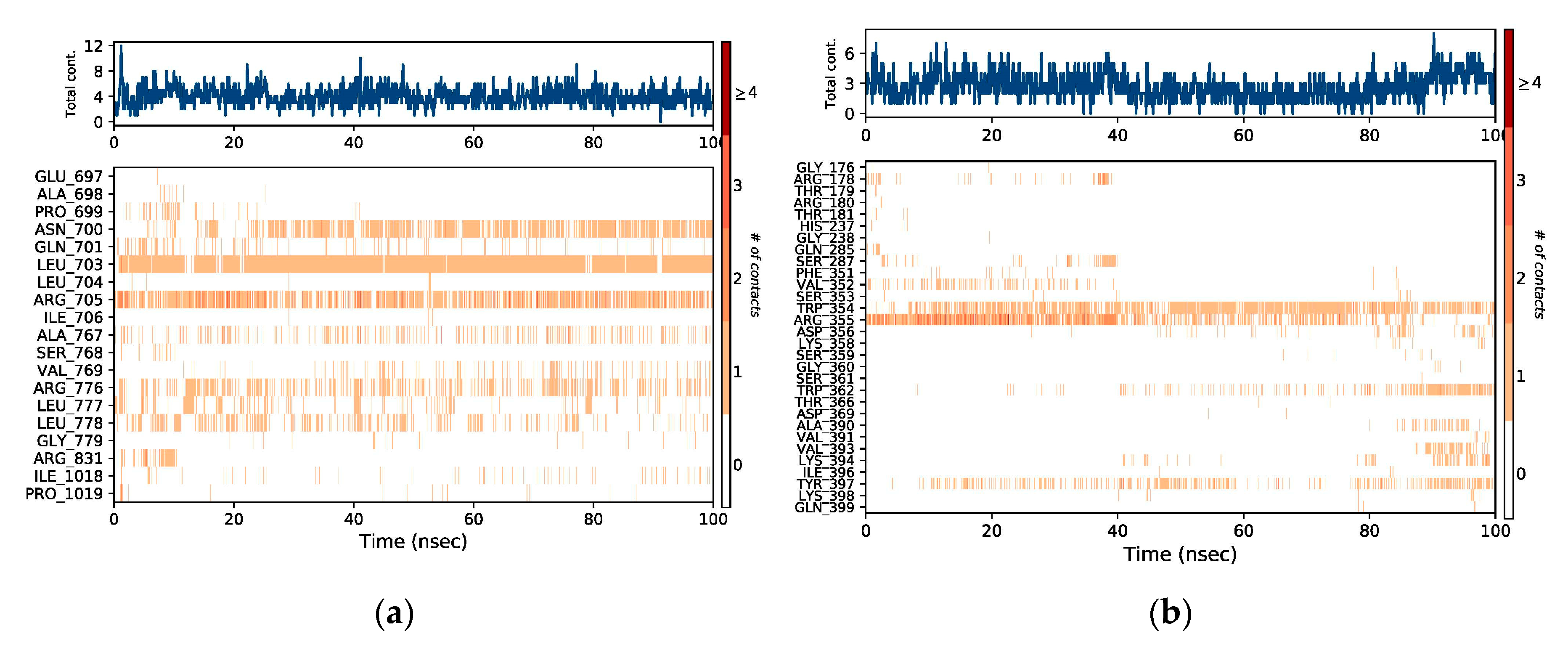
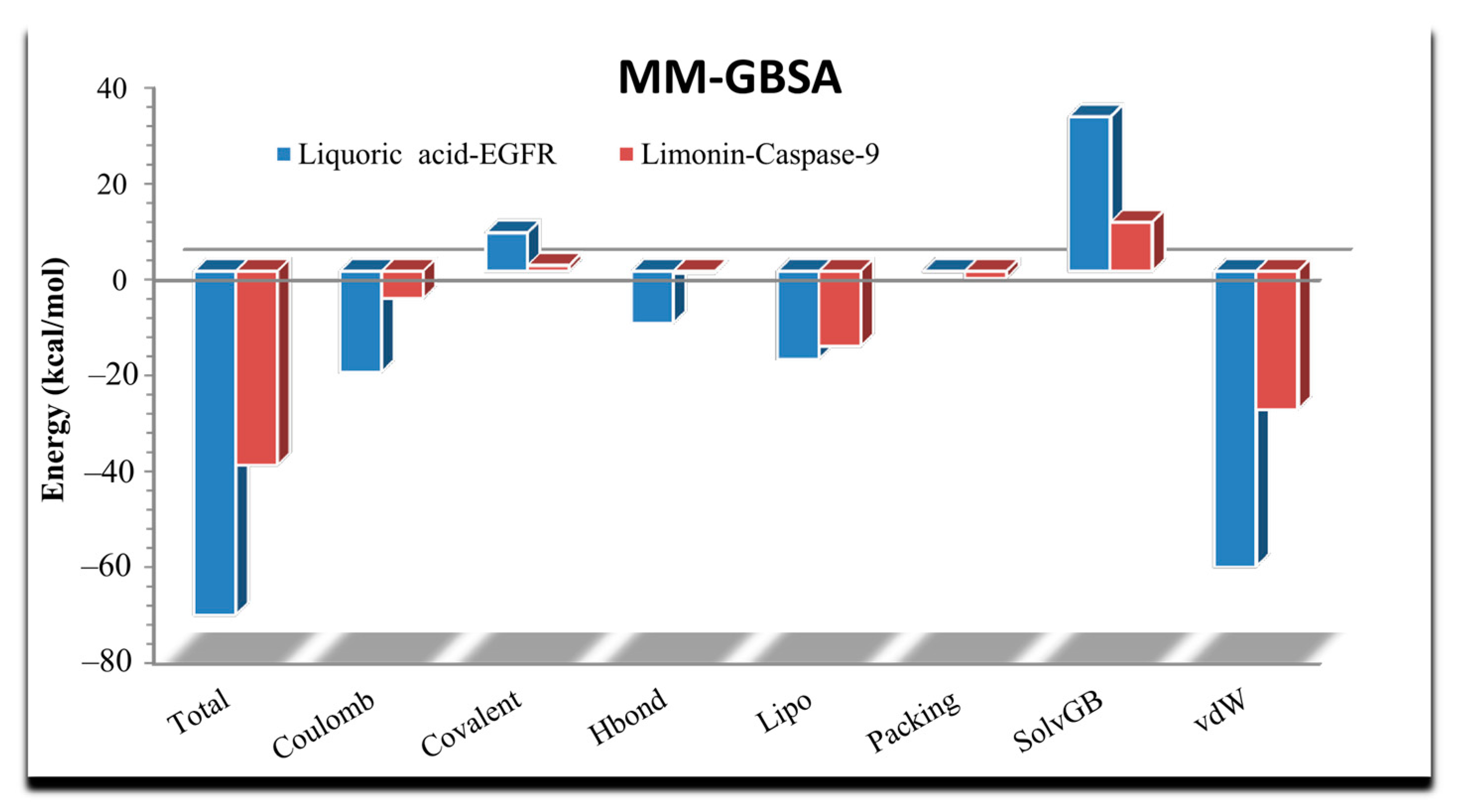
2.3. MD Simulation
3. Discussion
4. Materials and Methods
4.1. Collection and Optimization of Bioactive Compounds
4.2. Retrieval and Preparation of Receptor Proteins
4.3. Molecular Docking
4.4. Drug Scanning through Pharmacokinetics Parameters
4.5. Molecular Dynamics Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, Y.; Han, Q.-J.; Zhang, J. Hepatocellular carcinoma: Mechanisms of progression and immunotherapy. World J. Gastroenterol. 2019, 25, 3151. [Google Scholar] [CrossRef]
- Waller, L.P.; Deshpande, V.; Pyrsopoulos, N. Hepatocellular carcinoma: A comprehensive review. World J. Hepatol. 2015, 7, 2648. [Google Scholar] [CrossRef] [PubMed]
- Balogh, J.; Victor III, D.; Asham, E.H.; Burroughs, S.G.; Boktour, M.; Saharia, A.; Li, X.; Ghobrial, R.M.; Monsour, H.P., Jr. Hepatocellular carcinoma: A review. J. Hepatocell. Carcinoma 2016, 3, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Sas, Z.; Cendrowicz, E.; Weinhäuser, I.; Rygiel, T.P. Tumor microenvironment of hepatocellular carcinoma: Challenges and opportunities for new treatment options. Int. J. Mol. Sci. 2022, 23, 3778. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Shi, X.; He, X.; Gao, Y. Macrophage phenotype and function in liver disorder. Front. Immunol. 2020, 10, 3112. [Google Scholar] [CrossRef]
- Samad, A.; Haque, F.; Nain, Z.; Alam, R.; Al Noman, M.A.; Molla, M.H.R.; Hossen, M.S.; Islam, M.R.; Khan, M.I.; Ahammad, F. Computational assessment of MCM2 transcriptional expression and identification of the prognostic biomarker for human breast cancer. Heliyon 2020, 6, e05087. [Google Scholar] [CrossRef]
- Llovet, J.M.; Fuster, J.; Bruix, J. The Barcelona approach: Diagnosis, staging, and treatment of hepatocellular carcinoma. Liver Transplant. 2004, 10, S115–S120. [Google Scholar] [CrossRef]
- Dasgupta, P.; Henshaw, C.; Youlden, D.R.; Clark, P.J.; Aitken, J.F.; Baade, P.D. Global trends in incidence rates of primary adult liver cancers: A systematic review and meta-analysis. Front. Oncol. 2020, 10, 171. [Google Scholar] [CrossRef]
- Jha, V.; Bhosale, A.; Kapadia, P.; Bhargava, A.; Marick, A.; Charania, Z.; Parulekar, O.; Shaikh, M.; Madaye, B. Multitargeted molecular docking study of phytochemicals on hepatocellular carcinoma. J. Appl. Biol. Biotechnol. 2022, 11, 116–130. [Google Scholar] [CrossRef]
- Rafi, J.H.; Jafar, T.; Pathan, M.T.; Reza, R.; Islam, S.; Sourna, I.J.; Alam, R.; Samad, A.; Ahammad, F. High expression of bone morphogenetic protein 1 (BMP1) is associated with a poor survival rate in human gastric cancer, a dataset approaches. Genomics 2021, 113, 1141–1154. [Google Scholar] [CrossRef]
- Kattan, S.W.; Nafie, M.S.; Elmgeed, G.A.; Alelwani, W.; Badar, M.; Tantawy, M.A. Molecular docking, anti-proliferative activity and induction of apoptosis in human liver cancer cells treated with androstane derivatives: Implication of PI3K/AKT/mTOR pathway. J. Steroid Biochem. Mol. Biol. 2020, 198, 105604. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Mustafa, G.; Mahrosh, H.S.; Zafar, M.; Attique, S.A.; Arif, R. Exploring the antihyperglycemic potential of tetrapeptides devised from AdMc1 via different receptor proteins inhibition using in silico approaches. Int. J. Immunopathol. Pharmacol. 2022, 36, 1–15. [Google Scholar] [CrossRef]
- Maiorov, V.N.; Crippen, G.M. Significance of root-mean-square deviation in comparing three-dimensional structures of globular proteins. J. Mol. Biol. 1994, 235, 625–634. [Google Scholar] [CrossRef]
- Singh, D.; Singh, M.; Yadav, E.; Falls, N.; Dangi, D.S.; Kumar, V.; Ramteke, P.W.; Verma, A. Attenuation of diethylnitrosamine (DEN)–Induced hepatic cancer in experimental model of Wistar rats by Carissa carandas embedded silver nanoparticles. Biomed. Pharmacother. 2018, 108, 757–765. [Google Scholar] [CrossRef]
- Ebrahimzadeh, M.A.; Tafazoli, A.; Akhtari, J.; Biparva, P.; Eslami, S. Engineered silver nanoparticles, a new nanoweapon against cancer. Anti-Cancer Agents Med. Chem. 2018, 18, 1962–1969. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, W.; Jiang, L.; Chen, Y. Recent advances in systemic therapy for hepatocellular carcinoma. Biomark. Res. 2022, 10, 1–21. [Google Scholar] [CrossRef]
- Mousa, A.B. Sorafenib in the treatment of advanced hepatocellular carcinoma. Saudi J. Gastroenterol. 2008, 14, 40. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Mahrosh, H.S.; Mustafa, G. An in silico approach to target RNA-dependent RNA polymerase of COVID-19 with naturally occurring phytochemicals. Environ. Dev. Sustain. 2021, 23, 16674–16687. [Google Scholar] [CrossRef]
- Mustafa, G.; Majid, M.; Ghaffar, A.; Yameen, M.; Samad, H.A.; Mahrosh, H.S. Screening and molecular docking of selected phytochemicals against NS5B polymerase of hepatitis C virus. Pak. J. Pharm. Sci. 2020, 33, 2317–2322. [Google Scholar] [PubMed]
- Hsu, P.-C.; Jablons, D.M.; Yang, C.-T.; You, L. Epidermal growth factor receptor (EGFR) pathway, yes-associated protein (YAP) and the regulation of programmed death-ligand 1 (PD-L1) in non-small cell lung cancer (NSCLC). Int. J. Mol. Sci. 2019, 20, 3821. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Srivastava, S.K.; Kim, S.-H. Caspase-9 as a therapeutic target for treating cancer. Expert Opin. Ther. Targets 2015, 19, 113–127. [Google Scholar] [CrossRef]
- Boston, S.R.; Deshmukh, R.; Strome, S.; Priyakumar, U.D.; MacKerell, A.D.; Shapiro, P. Characterization of ERK docking domain inhibitors that induce apoptosis by targeting Rsk-1 and caspase-9. BMC Cancer 2011, 11, 7. [Google Scholar] [CrossRef]
- Ghorab, W.M.; El-Sebaey, S.A.; Ghorab, M.M. Design, synthesis and Molecular modeling study of certain EGFR inhibitors with a quinazolinone scaffold as anti-hepatocellular carcinoma and Radio-sensitizers. Bioorganic Chem. 2023, 131, 106310. [Google Scholar] [CrossRef]
- Siddiqui, S.; Upadhyay, S.; Ahmad, I.; Hussain, A.; Ahamed, M. Cytotoxicity of Moringa oleifera fruits on human liver cancer and molecular docking analysis of bioactive constituents against caspase-3 enzyme. J. Food Biochem. 2021, 45, e13720. [Google Scholar] [CrossRef]
- Suganya, V.; Anuradha, V. In silico molecular docking of astaxanthin and sorafenib with different apoptotic proteins involved in hepatocellular carcinoma. Biocatal. Agric. Biotechnol. 2019, 19, 101076. [Google Scholar] [CrossRef]
- Khalid, H.R.; Aamir, M.; Tabassum, S.; Alghamdi, Y.S.; Alzamami, A.; Ashfaq, U.A. Integrated System Pharmacology Approaches to Elucidate Multi-Target Mechanism of Solanum surattense against Hepatocellular Carcinoma. Molecules 2022, 27, 6220. [Google Scholar] [CrossRef]
- Mohanraj, K.; Karthikeyan, B.S.; Vivek-Ananth, R.; Chand, R.B.; Aparna, S.; Mangalapandi, P.; Samal, A. IMPPAT: A curated database of I ndian M edicinal P lants, P hytochemistry A nd T herapeutics. Sci. Rep. 2018, 8, 4329. [Google Scholar] [CrossRef]
- Ru, J.; Li, P.; Wang, J.; Zhou, W.; Li, B.; Huang, C.; Li, P.; Guo, Z.; Tao, W.; Yang, Y. TCMSP: A database of systems pharmacology for drug discovery from herbal medicines. J. Cheminformatics 2014, 6, 13. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed]
- Althagafi, I.; El-Metwaly, N.; Farghaly, T.A. New series of thiazole derivatives: Synthesis, structural elucidation, antimicrobial activity, molecular modeling and MOE docking. Molecules 2019, 24, 1741. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. In Chemical Biology: Methods in Molecular Biology; Hempel, J., Williams, C., Hong, C., Eds.; Humana Press: New York, NY, USA, 2015; Volume 1263, pp. 243–250. [Google Scholar]
- Sharma, S.; Sharma, A.; Gupta, U. Molecular Docking studies on the Anti-fungal activity of Allium sativum (Garlic) against Mucormycosis (black fungus) by BIOVIA discovery studio visualizer 21.1. 0.0. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 84-es. [Google Scholar]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Hildebrand, P.W.; Rose, A.S.; Tiemann, J.K. Bringing molecular dynamics simulation data into view. Trends Biochem. Sci. 2019, 44, 902–913. [Google Scholar] [CrossRef]
- Rasheed, M.A.; Iqbal, M.N.; Saddick, S.; Ali, I.; Khan, F.S.; Kanwal, S.; Ahmed, D.; Ibrahim, M.; Afzal, U.; Awais, M. Identification of lead compounds against Scm (fms10) in Enterococcus faecium using computer aided drug designing. Life 2021, 11, 77. [Google Scholar] [CrossRef]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No. | Ligand | Receptor | Binging Affinity (kcal/mol) | Interacting Amino Acids |
|---|---|---|---|---|
| 1 | Liquoric acid | EGFR | −9.8 | PheA:723 and LysA:875 |
| 2 | Madecassic acid | −9.3 | AspA:855 and ArgA:858 | |
| 3 | Berbamine | −8.7 | PheA:723, ValA:726, LysA:745, AspA:837, ArgA:841, AsnA:842, LeuA:844, and ThrA:854 | |
| 4 | Obamegine | −8.4 | PheA:723, HisA:835, AsnA:842, AspA:855, ArgA:858, GlyA:874, and ProA:877 | |
| 5 | Isotetrandrine | −8.2 | PheA:723, AspA:837, GlyA:857, LysA:875, ValA:876, and ProA:877 | |
| 6 | Limonin | Caspase-9 | −10.5 | LeuA:177, ArgA:178, GlyA:238, TrpA:354, and ArgA:355 |
| 7 | Obamegine | −9.3 | LeuA:244, PheA:246, AsnA:265, LeuA:335, ProA:336, and ProA:338 | |
| 8 | Liquoric acid | −9.1 | TyrA:153 and LysA:409 | |
| 9 | 3-O-caffeoyloleanolic acid | −8.7 | ArgA:146, GlyA:147, IleA:154, and LeuA:155 | |
| 10 | Betulin | −8.3 | ArgA:408 andLysA:409 |
| Ligands | Molecular Properties † | ||||||
|---|---|---|---|---|---|---|---|
| Molecular Mass (≤500 Dalton) | Hydrogen Bond Donor (≤5) | Hydrogen Bond Acceptor (≤10) | Number of Rotatable Bonds (≤10) | Log P (≤5) | Molar Refractivity (40–130) | Violations | |
| Limonin | 470.51 | 0 | 8 | 1 | 2.55 | 116.17 | 0 |
| Liquoric acid | 484.67 | 2 | 5 | 1 | 4.48 | 135.82 | 1 |
| Madecassic acid | 504.70 | 5 | 6 | 2 | 3.59 | 140.40 | 2 |
| Obamegine | 594.70 | 2 | 8 | 2 | 4.84 | 177.14 | 2 |
| Betulin | 442.72 | 2 | 2 | 2 | 6.36 | 136.30 | 2 |
| Lupeol | 426.72 | 1 | 1 | 1 | 7.26 | 135.14 | 2 |
| Berbamine | 608.72 | 1 | 8 | 3 | 5.15 | 181.60 | 3 |
| Isotetrandrine | 622.75 | 0 | 8 | 4 | 5.41 | 186.07 | 3 |
| Phytochemicals | ||||||||
|---|---|---|---|---|---|---|---|---|
| Liquoric Acid | Limonin | Madecassic Acid | Berbamine | Isotetrandrine | Obamegine | Betulin | Lupeol | |
| Absorption | ||||||||
| BBB | No | No | Yes | No | Yes | Yes | Yes | No |
| HIA | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Caco-2 Permeability | No | No | No | Yes | Yes | Yes | No | No |
| PGS | No | No | No | Yes | Yes | Yes | No | No |
| PGI | No | Yes | No | Yes | Yes | Yes | No | No |
| Metabolism | ||||||||
| CYP3A4 substrate | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| CYP2C9 substrate | No | No | No | Yes | Yes | Yes | No | No |
| CYP2D6 substrate | No | No | No | Yes | Yes | Yes | No | No |
| CYP3A4 inhibition | No | Yes | No | No | No | No | No | No |
| CYP2C9 inhibition | No | No | No | No | No | No | No | No |
| CYP2C19 inhibition | No | No | No | No | No | No | No | No |
| CYP2D6 inhibition | No | No | No | No | No | No | No | No |
| CYP1A2 inhibition | No | No | No | No | No | No | No | No |
| Toxicity | ||||||||
| AMES Toxicity | No | No | No | Yes | Yes | Yes | No | No |
| Carcinogens | No | No | No | No | No | No | No | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mustafa, G.; Younas, S.; Mahrosh, H.S.; Albeshr, M.F.; Bhat, E.A. Molecular Docking and Simulation-Binding Analysis of Plant Phytochemicals with the Hepatocellular Carcinoma Targets Epidermal Growth Factor Receptor and Caspase-9. Molecules 2023, 28, 3583. https://doi.org/10.3390/molecules28083583
Mustafa G, Younas S, Mahrosh HS, Albeshr MF, Bhat EA. Molecular Docking and Simulation-Binding Analysis of Plant Phytochemicals with the Hepatocellular Carcinoma Targets Epidermal Growth Factor Receptor and Caspase-9. Molecules. 2023; 28(8):3583. https://doi.org/10.3390/molecules28083583
Chicago/Turabian StyleMustafa, Ghulam, Shumaila Younas, Hafiza Salaha Mahrosh, Mohammed Fahad Albeshr, and Eijaz Ahmed Bhat. 2023. "Molecular Docking and Simulation-Binding Analysis of Plant Phytochemicals with the Hepatocellular Carcinoma Targets Epidermal Growth Factor Receptor and Caspase-9" Molecules 28, no. 8: 3583. https://doi.org/10.3390/molecules28083583
APA StyleMustafa, G., Younas, S., Mahrosh, H. S., Albeshr, M. F., & Bhat, E. A. (2023). Molecular Docking and Simulation-Binding Analysis of Plant Phytochemicals with the Hepatocellular Carcinoma Targets Epidermal Growth Factor Receptor and Caspase-9. Molecules, 28(8), 3583. https://doi.org/10.3390/molecules28083583