

Stilbenoids and Flavonoids from Cajanus cajan (L.) Millsp. and Their α-Glucosidase Inhibitory Activities
 , and
, and
Abstract
1. Introduction
2. Results and Discussion
2.1. Structural Elucidation
2.2. α-Glucosidase Inhibitory Activity of Compounds 1-27
2.3. Inhibitory Kinetics of Compounds 8-9, 11, 13, 24-26 against α-Glucosidase
2.4. Structure–Activity Relationship Analysis
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Spectral and Physical Data of Compounds 1 and 2
3.5. Assay for α-Glucosidase Inhibitory Activity of Compounds 1-27
3.6. Kinetics of Compounds 8-9, 11, 13, 24-26 Inhibiting α-Glucosidase
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Zheng, L.; Xu, T. Eyes on the progressive study of diabetes. Chin. Bull. Life Sci. 2012, 24, 606–610. [Google Scholar]
- Zhu, X.D.; Jiang, B.J.; Liu, X.Y.; Gu, Y.; Xia, T.; Han, L.Y.; Qiao, O.; Zhang, Y.; Gao, W.Y. Mechanism and research progress of flavonoids, polyphenols and alkaloids in natural products for type 2 diabetes mellitus. Mod. Chin. Med. 2019, 21, 1592–1598. [Google Scholar]
- Lv, G.Y. A review of research progress in oral medication for diabetes. Mod. Diagn. Treat. 2019, 30, 3336–3338. [Google Scholar]
- Liu, B.; Kongstad, K.T.; Wiese, S.; Jäger, A.K.; Staerk, D. Edible seaweed as future functional food: Identification of α-glucosidase inhibitors by combined use of high-resolution α-glucosidase inhibition profiling and HPLC-HRMS-SPE-NMR. Food Chem. 2016, 203, 16–22. [Google Scholar] [CrossRef]
- He, B.Q.; Zhang, Y.Y.; Zhuang, Y.B.; Wei, A.H.; Li, R.D.; Zhang, S.Y. α-Glucosidase inhibitory activity of the extracts from Tadehagi triquetrum. Nat. Prod. Res. Dev. 2020, 32, 2026–2030. [Google Scholar]
- Cai, J.Z.; Dai, W.; Zhang, N.L. Advance on chemical constituents and pharmacological activities of Cajanus cajan (L.) Millsp. Nat. Prod. Res. Dev. 2020, 32, 515–524. [Google Scholar]
- Pal, D.; Sachan, N.; Ghosh, A.; Mishra, P. Biological activities and medicinal properties of Cajanus cajan (L.) Millsp. J. Adv. Pharm. Technol. Res. 2011, 2, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.M.; Jiang, B.P.; Shen, S.N.; Guo, Z.; Li, Z.Y.; Si, J.Y.; Pan, R.L. Chemical constituents from leaves of Cajanus cajan. Chin. Tradit. Herb. Drugs 2014, 4, 466–470. [Google Scholar]
- Ezike, A.C.; Akah, P.A.; Okoli, C.C.; Okpala, C.B. Experimental evidence for the antidiabetic activity of Cajanus cajan leaves in rats. Basic Clin. Pharm. 2010, 1, 81–84. [Google Scholar]
- Lei, X.Y.; Liu, S.H.; Zhao, Y.X.; Guo, M.J.; Tao, L.; Shen, X.C.; Zhang, N.L. Lignans and flavonoids from Cajanus cajan (L.) Millsp. and their α-glucosidase inhibitory activities. Chem. Biodivers. 2022, 19, 414–424. [Google Scholar] [CrossRef]
- Giuseppe, Z.; Joan, G.M.; Imma, E.; Dagmar, S.; Mariia, S.K.; Alba, H.P.J.; Pilar, C.; Jordan, R.B.; Antonio, M.E. Enantioselective folding of enynes by gold (I) catalysts with a remote C2-chiral element. J. Am. Chem. Soc. 2019, 141, 11858–11863. [Google Scholar]
- Fiorentino, A.; D’Abrosca, B.; Pacifico, S.; Izzo, A.; Uzzo, P.; Russo, A.; Di, B.B.; Monaco, P. Carexanes from Carex distachya Desf.: Revised stereochemistry and characterization of four novel polyhydroxylated prenylstilbenes. Tetrahedron 2008, 64, 7782–7786. [Google Scholar] [CrossRef]
- Loset, J.R.; Marston, A.; Gupta, M.P. Five new prenylated stilbenes from the root bark of Lonchocarpus chiricanus. J. Nat. Prod. 2001, 64, 710–715. [Google Scholar]
- De Bruijn, W.J.C.; Araya-Cloutier, C.; Bijlsma, J.; de Swart, A.; Sanders, M.G.; de Waard, P.; Gruppen, H.; Vincken, J.P. Antibacterial prenylated stilbenoids from peanut (Arachis hypogaea). Phytochem. Lett. 2018, 28, 13–18. [Google Scholar] [CrossRef]
- Geng, Z.Z.; Zhang, J.J.; Lin, J.; Huang, M.Y.; An, L.K.; Zhang, H.B.; Sun, P.H.; Ye, W.C.; Chen, W.M. Novel cajaninstilbene acid derivatives as antibacterial agents. Eur. J. Med. Chem. 2015, 100, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.H.; Zhang, Y.M.; Tan, N.H.; Chu, H.B.; Ji, C.J. Chemical constituents of Keteleeria evelyniana. Nat. Prod. Res. Dev. 2008, 2, 257–261. [Google Scholar]
- Zhang, N.L. Investigation on the Chemical Constituents of Cajanus cajan, Periploca forrestti and Their Bioactivities. Ph.D. Thesis, South China Botanical Garden, Guangzhou, China, 2013. [Google Scholar]
- Jin, X.D. Study on Chemical Constituents and Biological Activities of Ethyl Acetate Fraction of Ethanol Extract of Thuja sutchuenensis Franch. Master’s Thesis, Guangzhou University, Guangzhou, China, 2018. [Google Scholar]
- Duker-Eshun, G.; Jaroszewski, J.W.; Asomaning, W.A.; Oppong-Boachie, F.; Brøgger Christensen, S. Antiplasmodial constituents of Cajanus cajan. Phytother Res. 2004, 18, 128–130. [Google Scholar] [CrossRef]
- Chen, W.Z.; Wang, C.; Fan, L.L.; Wang, J.T.; Yue, W.Z.; Tang, L. Total synthesis of longistylin A, longistylin C and cajanotone. Chin. J. Org. Chem. 2015, 35, 1146–1151. [Google Scholar] [CrossRef]
- Han, F.; Xiao, Y.; Lee, I.S. Microbial transformation of prenylquercetins by Mucor hiemalis. Molecules 2020, 25, 528–538. [Google Scholar] [CrossRef]
- Osorio, M.; Carvajal, M.; Vergara, A.; Butassi, E.; Zacchino, S.; Mascayano, C.; Montoya, M.; Mejías, S.; Martín, M.C.; Vásquez-Martínez, Y. Prenylated flavonoids with potential antimicrobial activity: Synthesis, biological activity, and in silico study. Int. J. Mol. Sci. 2021, 22, 5472–5492. [Google Scholar] [CrossRef] [PubMed]
- Ou, L.L.; Han, S.; Ding, W.B.; Chen, Z.; Ye, Z.Q.; Yang, H.Y.; Zhang, G.L.; Lou, Y.J.; Chen, J.Z.; Yu, Y.P. Design, synthesis and 3D-QSAR study of cytotoxic flavonoid derivatives. Mol. Divers. 2011, 15, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Kamphues, C.; Kadowaki, S.; Amini, N.; van den Berg, I.; Wang, J.; Andreatos, N.; Sakamoto, Y.; Ogura, T.; Kakuta, M.; Pikouli, A.; et al. The interplay of KRAS mutational status with tumor laterality in non-metastatic colorectal cancer: An international, multi-institutional study in patients with known KRAS, BRAF, and MSI status. J. Surg. Oncol. 2021, 123, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Hai, P.; Gao, Y.; Li, R.T.; Wang, F. Chemical constituents from roots of Lindera aggregata. Chin. Tradit. Herb. Drugs 2016, 47, 872–875. [Google Scholar]
- Yamashita, Y.; Hanaya, K.; Shoji, M.; Sugai, T. Simple synthesis of sakuranetin and selinone via a common intermediate, utilizing complementary regioselectivity in the deacetylation of naringenin triacetate. Chem. Pharm. Bull. 2016, 64, 961–965. [Google Scholar] [CrossRef]
- Tran, T.S.; Tran, T.D.; Tran, T.H.; Mai, T.T.; Nguyen, N.L.; Thai, K.M.; Le, M.T. Synthesis, in silico and in vitro evaluation of some flavone derivatives for acetylcholinesterase and BACE-1 inhibitory activity. Molecules 2020, 25, 4064–4078. [Google Scholar] [CrossRef]
- Luo, L.P.; Shen, L.M.; Sun, F.; Ma, Z.J. Immunoprecipitation coupled with HPLC-MS/MS to discover the aromatase ligands from Glycyrrhiza uralensis. Food Chem. 2013, 138, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.W.C.; Ki, N.Y.; Tan, C.Y.; Larsen, A.; Kuin, W.S.; Chan, H.T. Diosmetin and tamarixetin (methylated flavonoids): A review on their chemistry, sources, pharmacology, and anticancer properties. J. Appl. Pharm. Sci. 2021, 11, 22–28. [Google Scholar]
- Wu, Y.; Luo, Q.; Sun, C.L.; Wang, G.H.; Chen, Q.C.; Guo, Z.J.; Zou, X.H.; Chen, H.F. Chemical constituents contained in Desmodium caudatum. Chin. J. Chin. Mater. Med. 2012, 37, 1788–1792. [Google Scholar]
- Yamauchi, K.; Mitsunaga, T.; Inagaki, M.; Suzuki, T. Synthesized quercetin derivatives stimulate melanogenesis in B16 melanoma cells by influencing the expression of melanin biosynthesis proteins MITF and p38 MAPK. Bioorg. Med. Chem. 2014, 22, 3331–3340. [Google Scholar] [CrossRef]
- Yuk, H.J.; Lee, Y.S.; Ryu, H.W.; Kim, S.H.; Kim, D.S. Effects of Toona sinensis leaf extract and its chemical constituents on xanthine oxidase activity and serum uric acid levels in potassium oxonate-induced hyperuricemic rats. Molecules 2018, 23, 3254–3265. [Google Scholar] [CrossRef]
- Zou, Y.; Hong, M.; Yang, X.F. Isolation of chemical components from Thesium chinense. Chin. J. Exp. Tradit. Med. Form. 2016, 22, 74–77. [Google Scholar]
- Cheng, L.; Ning, D.S.; Xin, M.W.; Huang, S.S.; Luo, L.; Li, Z.Q.; Pan, Z.H. Chemical constituents from EtOAc fraction of Sophora dunnii. Chin. J. Chin. Mater. Med. 2015, 40, 4428–4432. [Google Scholar]
- Kwon, S.H.; Hong, S.I.; Ma, S.X.; Lee, S.Y.; Jang, C.G. 3′, 4′, 7-trihydroxyflavone prevents apoptotic cell death in neuronal cells from hydrogen peroxide-induced oxidative stress. Food Chem. Toxicol. 2015, 80, 41–51. [Google Scholar] [CrossRef]
- Urmann, C.; Bieler, L.; Priglinger, E.; Aigner, L.; Couillard-Despres, S.; Riepl, H.M. Neuroregenerative potential of prenyl- and pyranochalcones: A structure-activity study. J. Nat. Prod. 2021, 84, 2675–2682. [Google Scholar] [CrossRef] [PubMed]
- Li, L.Z.; Yang, X.S.; Zhu, H.Y.; Shi, J.S. Chemical constituents of Crotalaria ferruginea. Chin. Tradit. Herb. Drugs 2008, 39, 173–175. [Google Scholar]

 ) and 1H—1H COSY (
) and 1H—1H COSY ( ) correlations of compounds 1 and 2.
) correlations of compounds 1 and 2.



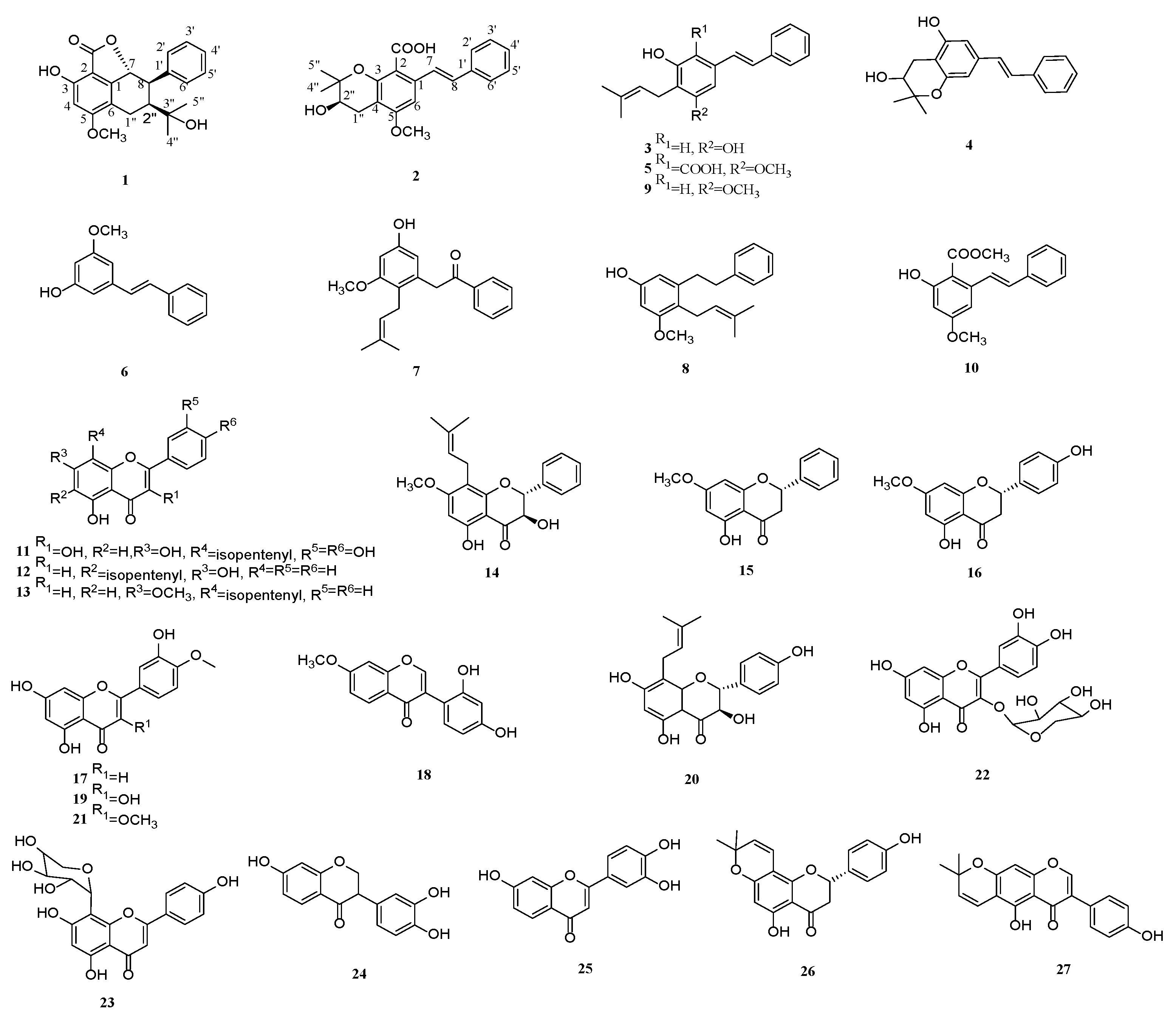{kind=link}
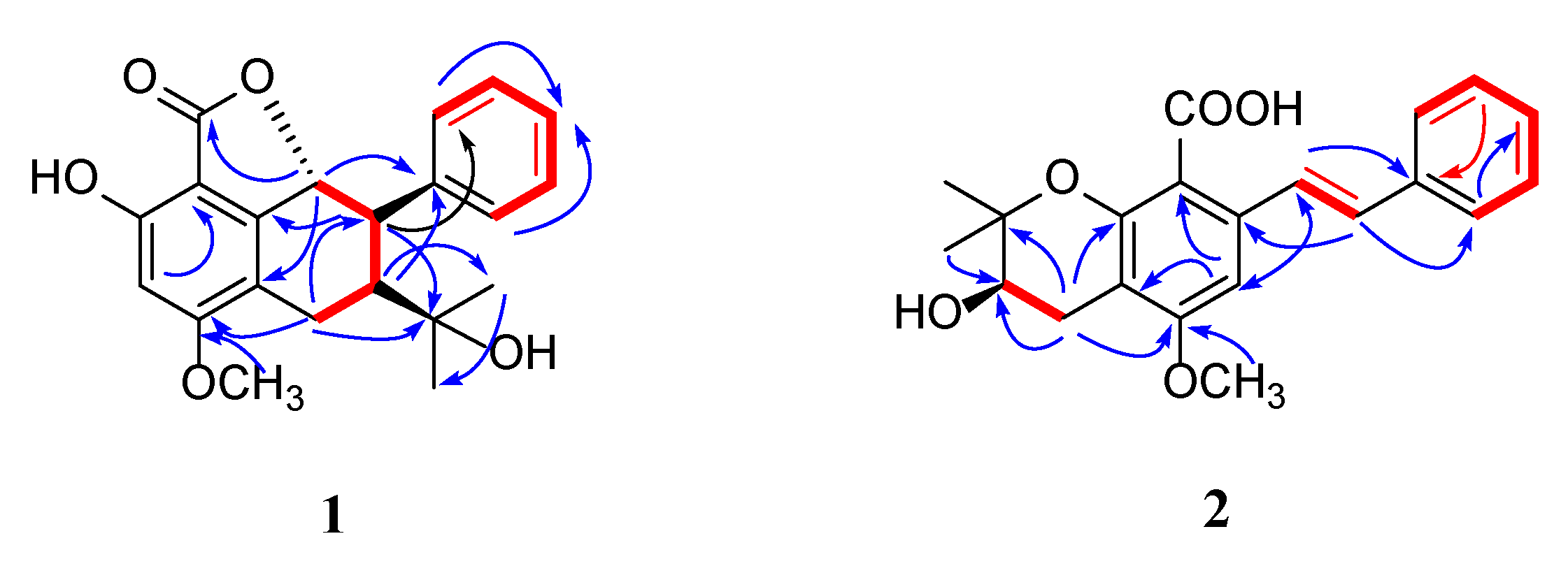{kind=link}
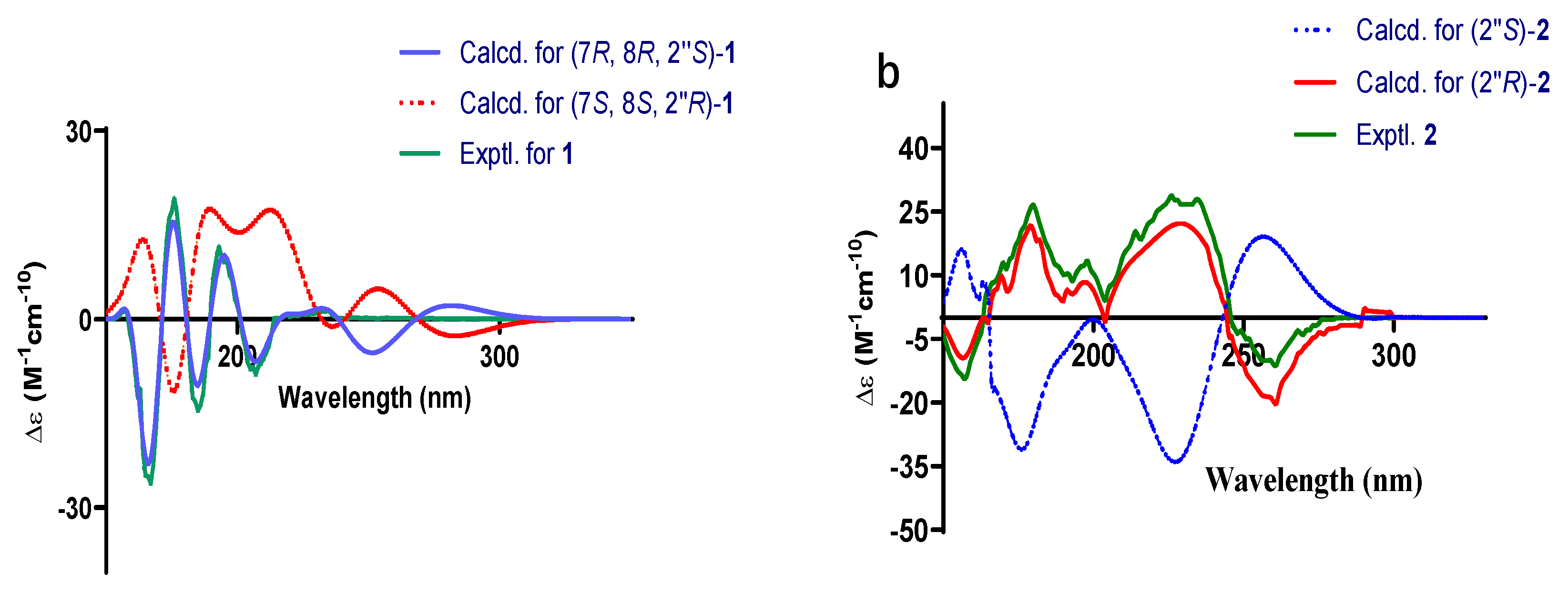{kind=link}
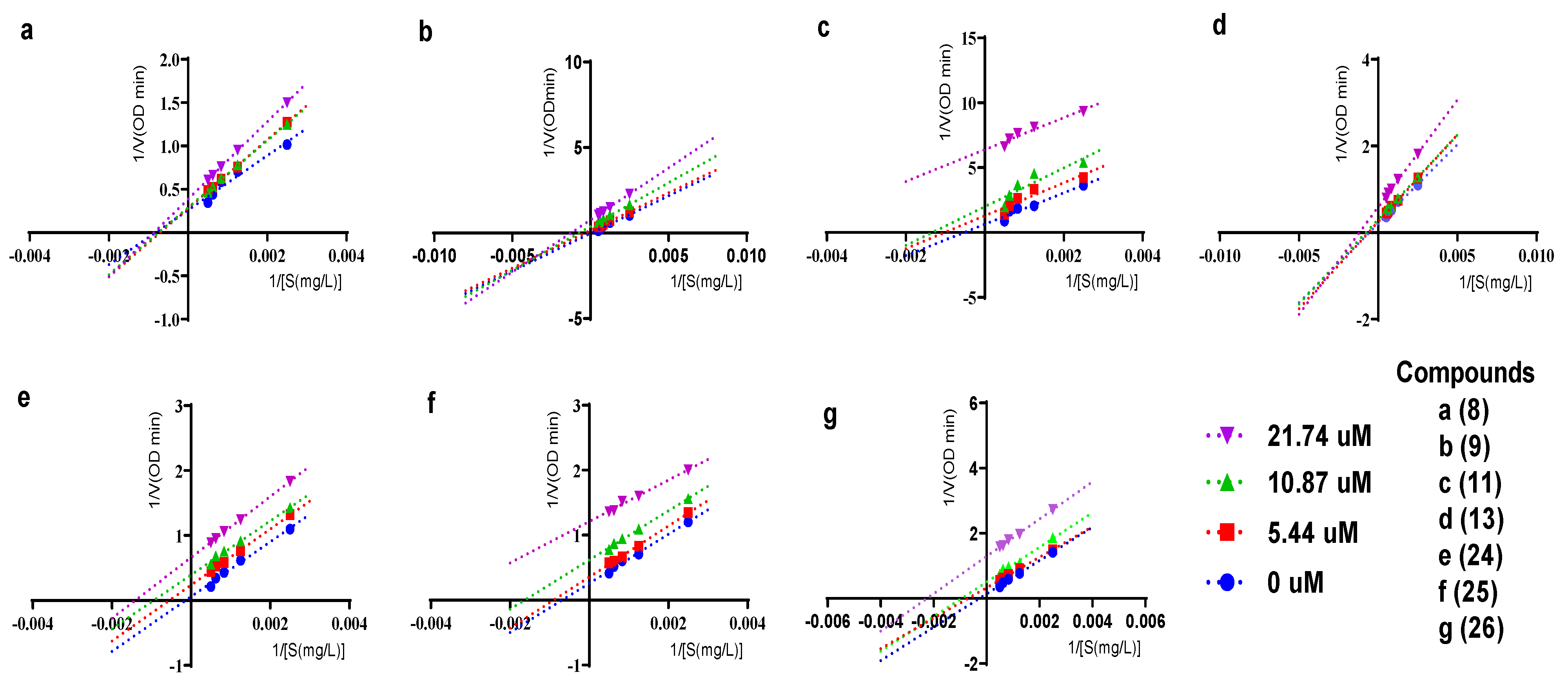{kind=link}
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δH | δC | δH | δC | |
| 1 | - | 152.0 | - | 132.9 |
| 2 | - | 101.8 | - | 109.4 |
| 3 | - | 156.9 | - | 150.4 |
| 4 | 6.45 (s, 1H) | 99.4 | - | 119.2 |
| 5 | - | 162.1 | - | 157.9 |
| 6 | - | 112.8 | 6.90 (s, 1H) | 98.3 |
| 7 | 5.22 (d, J = 10.5 Hz, 1H) | 82.0 | 7.29 (d, 1H, J = 15.1 Hz, overlapped) | 128.4 |
| 8 | 2.30 (dd, J = 10.5, 7.7 Hz, 1H) | 49.2 | 7.08 (d, J = 16.3 Hz, 1H) | 125.9 |
| 1′ | - | 145.5 | - | 137.3 |
| 2′, 6′ | 7.43 (d, J = 6.9 Hz, 2H) | 129.3 | 7.50 (d, J = 7.3 Hz, 2H) | 126.8 |
| 3′, 5′ | 7.34 (t, J = 7.6 Hz, 2H) | 128.7 | 7.38 (t, J = 7.6 Hz, 2H) | 129.3 |
| 4′ | 7.24 (t, J = 7.4 Hz, 1H) | 126.7 | 7.29 (s, 1H, overlapped) | 130.3 |
| 1” | 2.58 (dd, J = 16.1, 8.3 Hz, 1H); | 21.2 | 2.41 (dd, J =17.4, 7.6 Hz, 1H); | 26.8 |
| 3.21 (d, J = 16.1 Hz, 1H) | 2.79 (dd, J = 17.4, 5.5 Hz, 1H) | |||
| 2” | 2.46 (t, J = 8.6 Hz, 1H) | 51.3 | 3.62–3.65 (m, 1H) | 67.9 |
| 3” | - | 73.1 | - | 77.7 |
| 4” | 0.66 (s, 3H) | 30.1 | 1.25 (s, 3H) | 25.9 |
| 5” | 0.68 (s, 3H) | 25.0 | 1.13 (s, 3H) | 20.6 |
| -OCH3 | 3.85 (s, 3H) | 56.4 | 3.88 (s, 3H) | 56.1 |
| -COO- | - | 168.4 | - | 169.4 |
| Compound | IC50 (μM) | Compound | IC50 (μM) |
|---|---|---|---|
| 1 | >2000 | 15 | >2000 |
| 2 | >2000 | 16 | >2000 |
| 3 | 30.3 ± 2.33 | 17 | >2000 |
| 4 | >2000 | 18 | >2000 |
| 5 | >2000 | 19 | 8.7 ± 0.08 |
| 6 | >2000 | 20 | 4.5 ± 0.49 |
| 7 | >2000 | 21 | 19.4 ± 1.02 |
| 8 | 7.0 ± 0.45 | 22 | >2000 |
| 9 | 12.9 ± 1.31 | 23 | >2000 |
| 10 | >2000 | 24 | 6.5 ± 0.42 |
| 11 | 0.87 ± 0.05 | 25 | 3.17 ± 0.55 |
| 12 | >2000 | 26 | 2.35 ± 0.54 |
| 13 | 2.3 ± 0.51 | 27 | >2000 |
| 14 | >2000 | Acarbose | 352.87 ± 0.82 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Zhao, X.; Guo, M.; Varier, K.M.; Gajendran, B.; Liu, S.; Tao, L.; Shen, X.; Zhang, N. Stilbenoids and Flavonoids from Cajanus cajan (L.) Millsp. and Their α-Glucosidase Inhibitory Activities. Molecules 2023, 28, 3779. https://doi.org/10.3390/molecules28093779
Zhao Y, Zhao X, Guo M, Varier KM, Gajendran B, Liu S, Tao L, Shen X, Zhang N. Stilbenoids and Flavonoids from Cajanus cajan (L.) Millsp. and Their α-Glucosidase Inhibitory Activities. Molecules. 2023; 28(9):3779. https://doi.org/10.3390/molecules28093779
Chicago/Turabian StyleZhao, Yaxian, Xinman Zhao, Mengjia Guo, Krishnapriya M. Varier, Babu Gajendran, Shaohuan Liu, Ling Tao, Xiangchun Shen, and Nenling Zhang. 2023. "Stilbenoids and Flavonoids from Cajanus cajan (L.) Millsp. and Their α-Glucosidase Inhibitory Activities" Molecules 28, no. 9: 3779. https://doi.org/10.3390/molecules28093779
APA StyleZhao, Y., Zhao, X., Guo, M., Varier, K. M., Gajendran, B., Liu, S., Tao, L., Shen, X., & Zhang, N. (2023). Stilbenoids and Flavonoids from Cajanus cajan (L.) Millsp. and Their α-Glucosidase Inhibitory Activities. Molecules, 28(9), 3779. https://doi.org/10.3390/molecules28093779