
Boron Lewis Acid Catalysis Enables the Direct Cyanation of Benzyl Alcohols by Means of Isonitrile as Cyanide Source

 , ,
, ,
Abstract
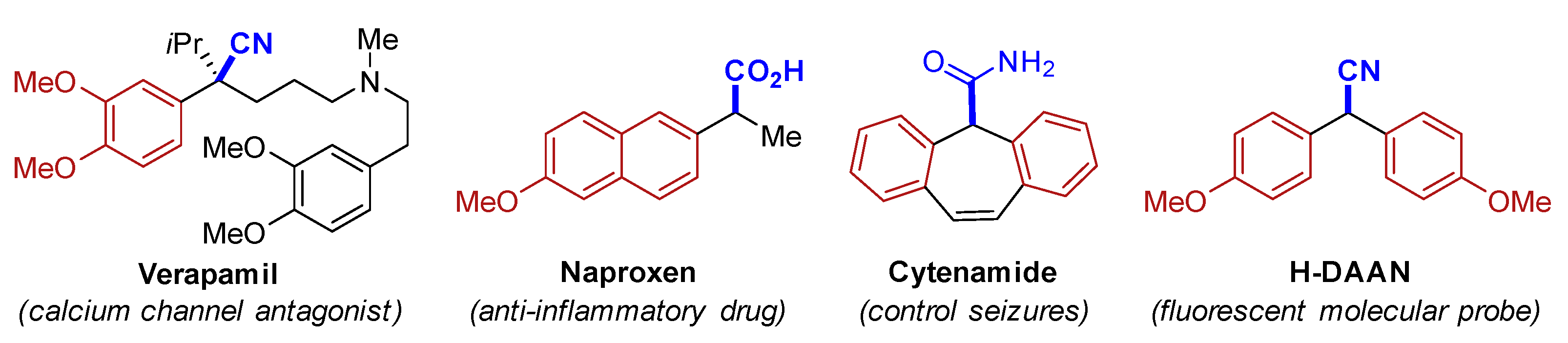
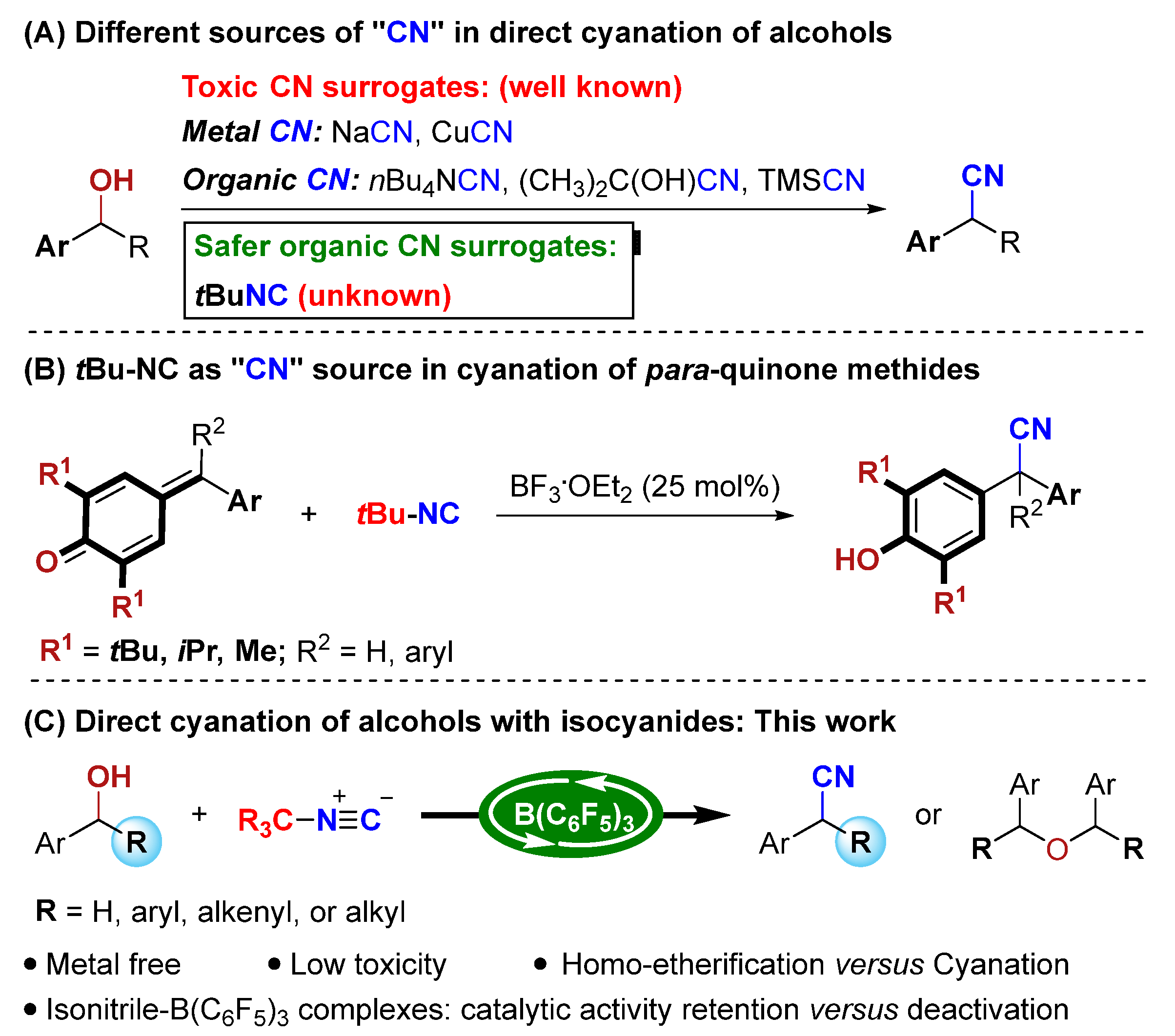
1. Introduction
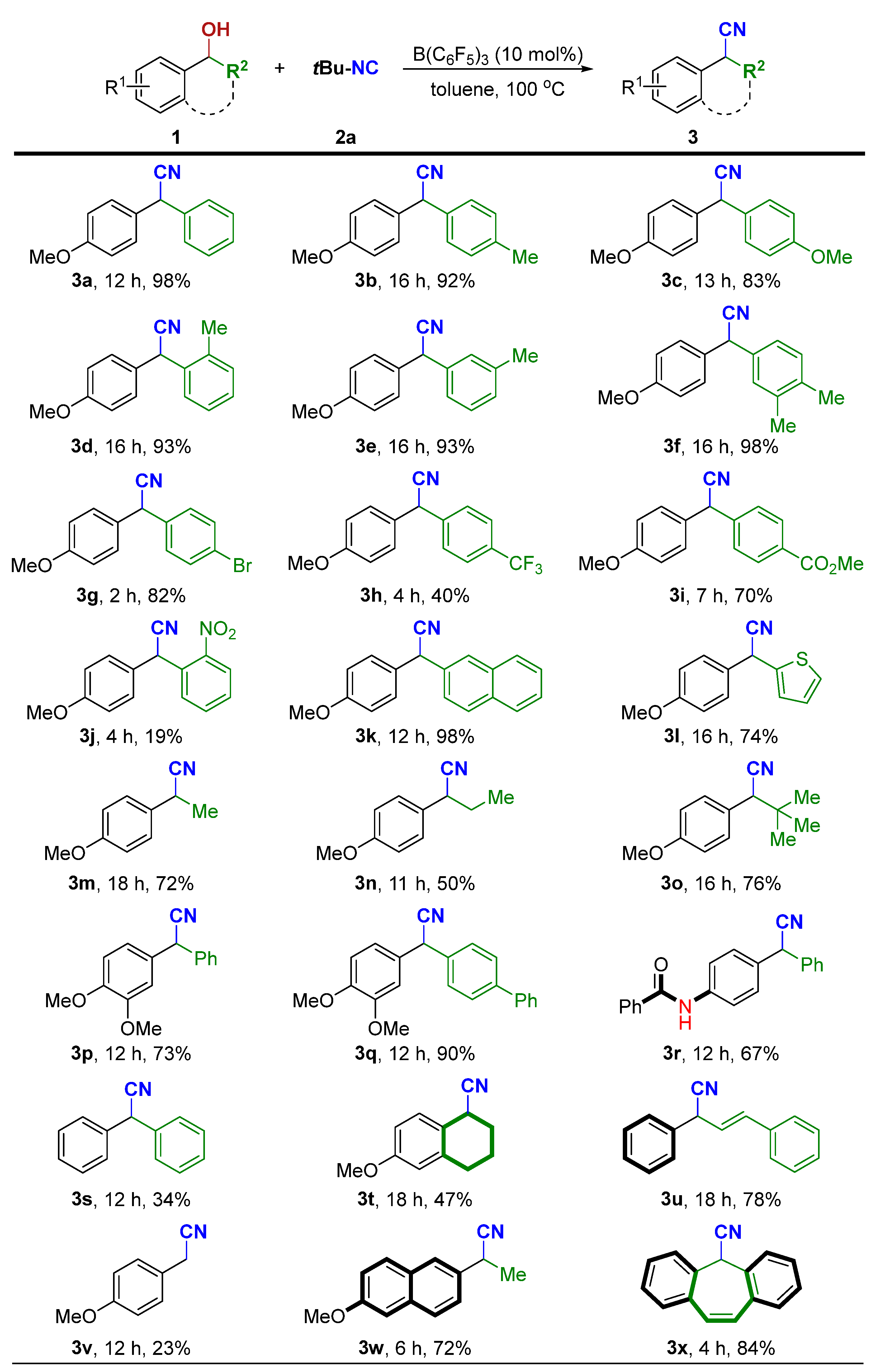
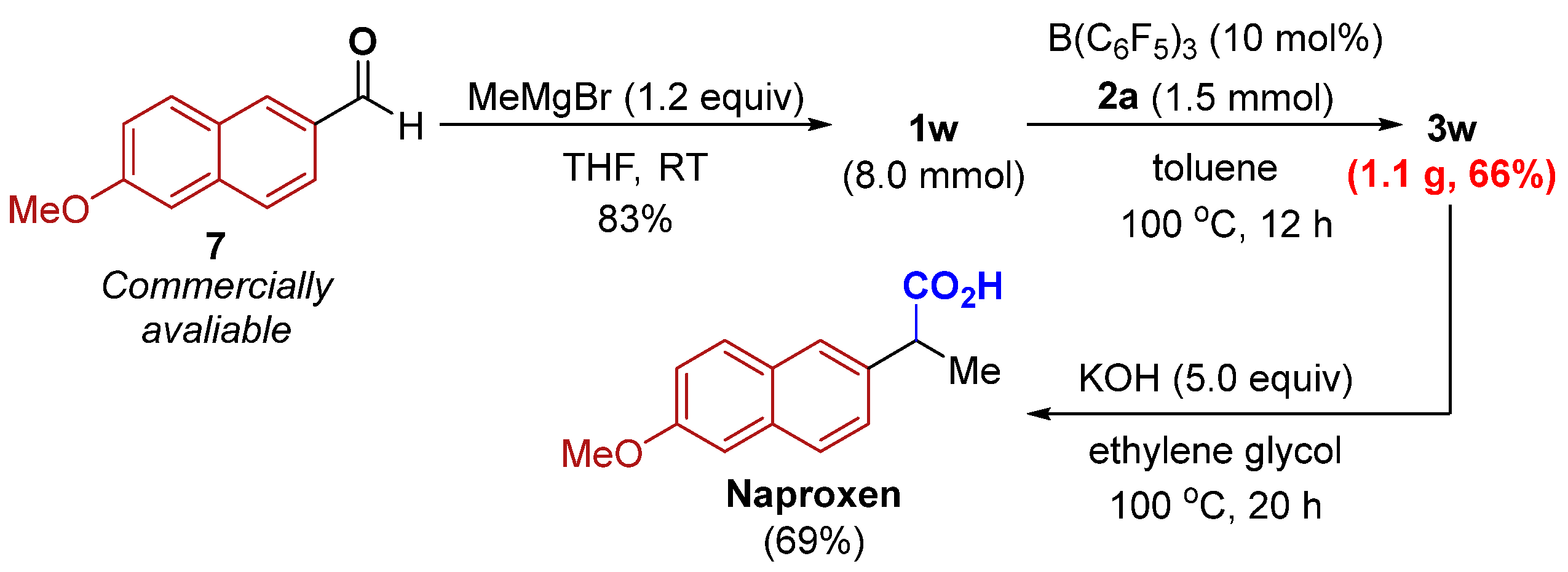
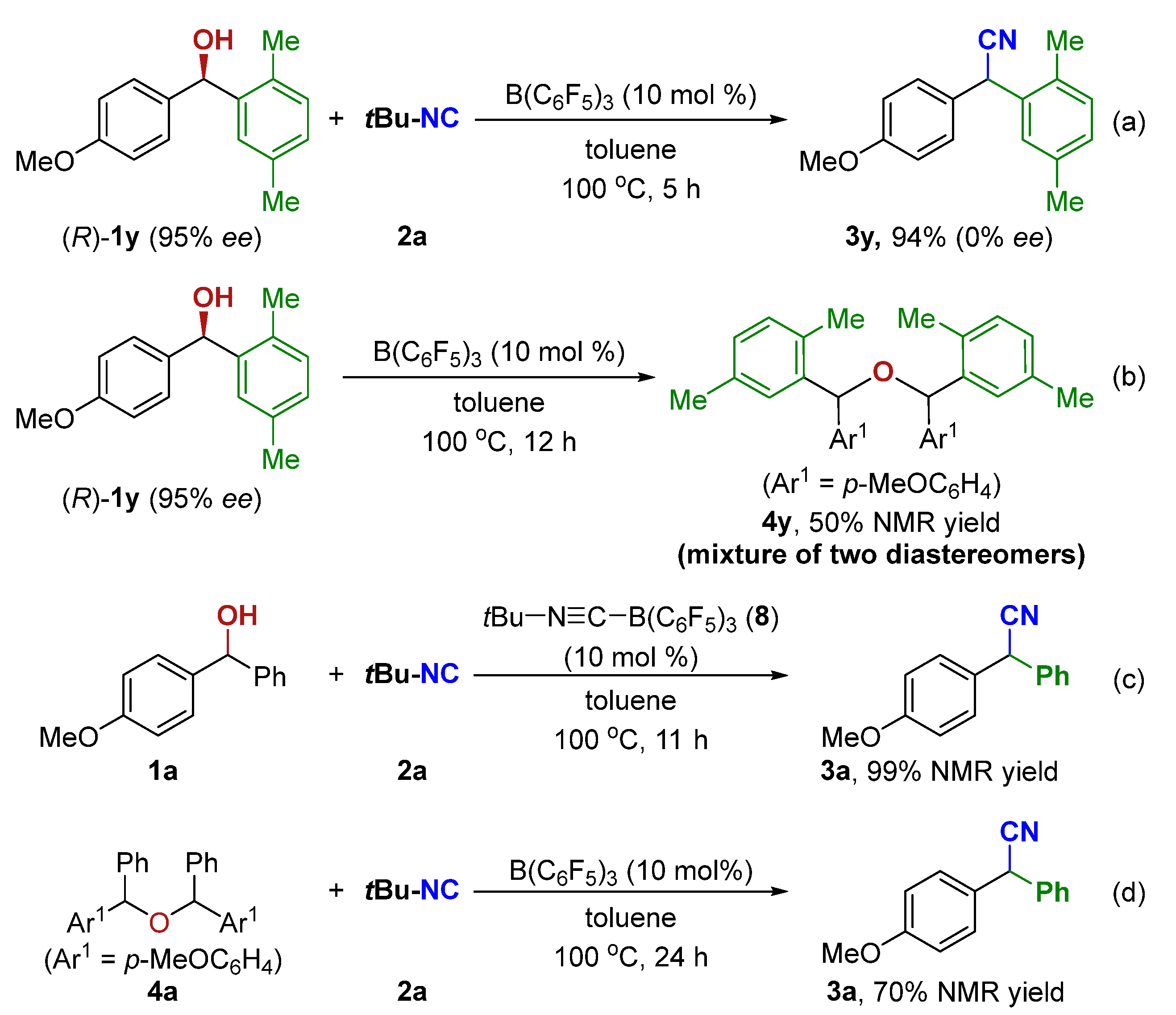
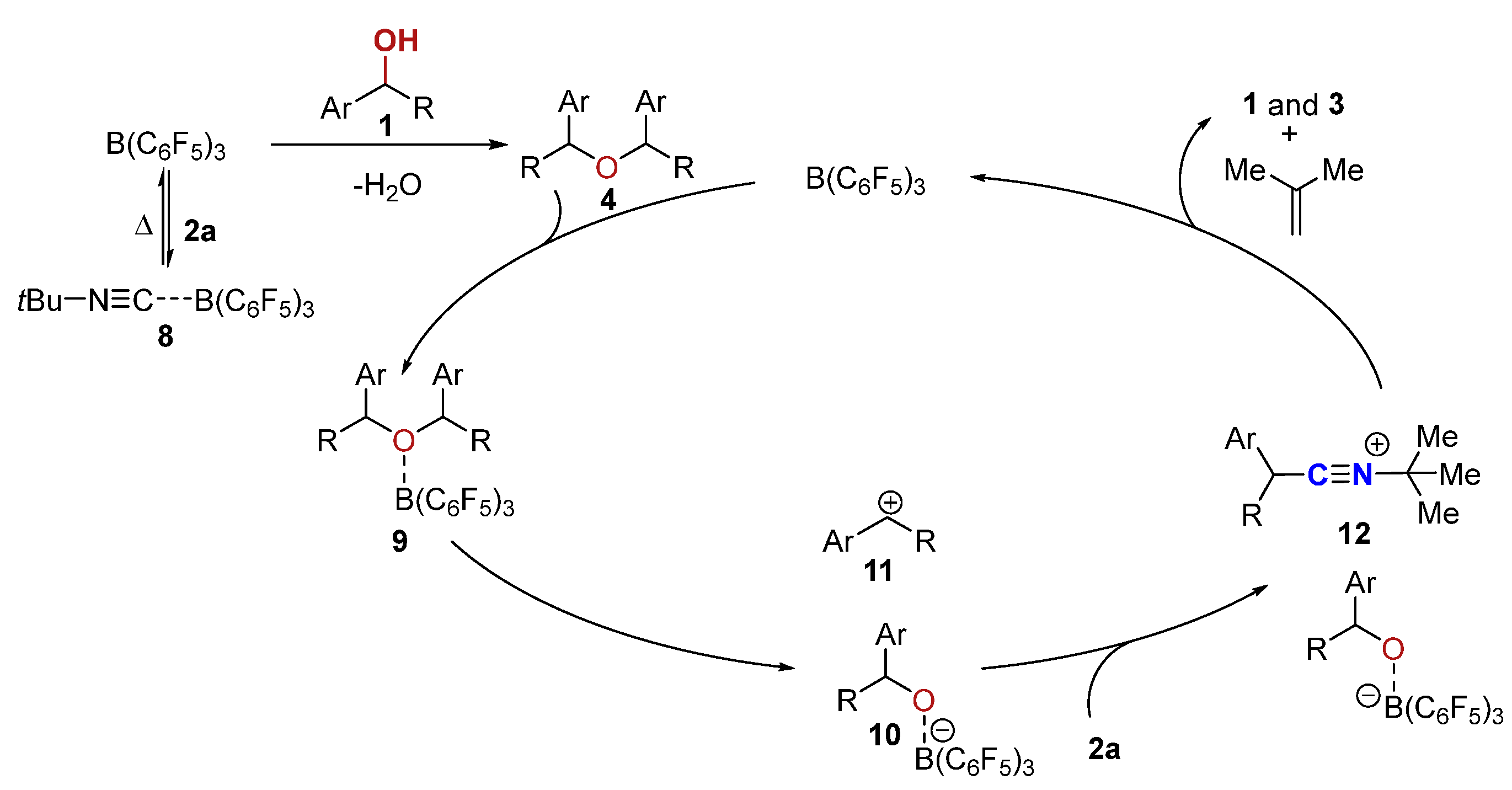
2. Results
3. Materials and Methods
3.1. General Information
3.2. Typical Procedure for Direct Cyanation of Alcohols
3.3. Procedure for the Preparation of Naproxen
3.4. Characterization Data of the Products
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Trost, B.M. Atom Economy—A Challenge for Organic Synthesis: Homogeneous Catalysis Leads the Way. Angew. Chem. Int. Ed. Engl. 1995, 34, 259–281. [Google Scholar] [CrossRef]
- Estopina-Duran, S.; Taylor, J.E. Bronsted Acid-Catalysed Dehydrative Substitution Reactions of Alcohols. Chem. Eur. J. 2021, 27, 106–120. [Google Scholar] [CrossRef]
- Huy, P.H. Lewis Base Catalysis Promoted Nucleophilic Substitutions—Recent Advances and Future Directions. Eur. J. Org. Chem. 2020, 10–27. [Google Scholar] [CrossRef]
- Moran, J.; Dryzhakov, M.; Richmond, E. Recent Advances in Direct Catalytic Dehydrative Substitution of Alcohols. Synthesis 2016, 48, 935–959. [Google Scholar] [CrossRef]
- Emer, E.; Sinisi, R.; Capdevila, M.G.; Petruzziello, D.; De Vincentiis, F.; Cozzi, P.G. Direct Nucleophilic SN1-Type Reactions of Alcohols. Eur. J. Org. Chem. 2011, 647–666. [Google Scholar] [CrossRef]
- Bryan, M.C.; Dunn, P.J.; Entwistle, D.; Gallou, F.; Koenig, S.G.; Hayler, J.D.; Hickey, M.R.; Hughes, S.; Kopach, M.E.; Moine, G.; et al. Key Green Chemistry Research Areas from a Pharmaceutical Manufacturers’ Perspective Revisited. Green Chem. 2018, 20, 5082–5103. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, F.; McCann, S.D.; Wang, D.; Chen, P.; Stahl, S.S.; Liu, G. Enantioselective Cyanation of Benzylic C–H Bonds Via Copper-Catalyzed Radical Relay. Science 2016, 353, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhu, N.; Chen, P.; Lin, Z.; Liu, G. Enantioselective Decarboxylative Cyanation Employing Cooperative Photoredox Catalysis and Copper Catalysis. J. Am. Chem. Soc. 2017, 139, 15632–15635. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Yang, J.; Zhang, J. Cu-Catalyzed Enantioselective Decarboxylative Cyanation Via the Synergistic Merger of Photocatalysis and Electrochemistry. Chem. Sci. 2023, 14, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Culkin, D.A.; Hartwig, J.F. Palladium-Catalyzed α-Arylation of Carbonyl Compounds and Nitriles. Acc. Chem. Res. 2003, 36, 234–245. [Google Scholar] [CrossRef]
- Wu, G.; Deng, Y.; Wu, C.; Zhang, Y.; Wang, J. Synthesis of α-Aryl Esters and Nitriles: Deaminative Coupling of α-Aminoesters and α-Aminoacetonitriles with Arylboronic Acids. Angew. Chem. Int. Ed. 2014, 53, 10510–10514. [Google Scholar] [CrossRef]
- Falk, A.; Goderz, A.L.; Schmalz, H.G. Enantioselective Nickel-Catalyzed Hydrocyanation of Vinylarenes Using Chiral Phosphine-Phosphite Ligands and TMS-CN as a Source of HCN. Angew. Chem. Int. Ed. 2013, 52, 1576–1580. [Google Scholar] [CrossRef]
- Singh, D.K.; Prasad, S.S.; Kim, J.; Kim, I. One-Pot, Three-Component Approach to Diarylacetonitriles. Org. Chem. Front. 2019, 6, 669–673. [Google Scholar] [CrossRef]
- Tan, J.-P.; Chen, Y.; Ren, X.; Guo, Y.; Yi, B.; Zhang, H.; Gao, G.; Wang, T. In Situ Phosphonium-Containing Lewis Base-Catalyzed 1,6-Cyanation Reaction: A Facile Way to Obtain α-Diaryl and α-Triaryl Acetonitriles. Org. Chem. Front. 2022, 9, 156–162. [Google Scholar] [CrossRef]
- Kadunce, N.T.; Reisman, S.E. Nickel-Catalyzed Asymmetric Reductive Cross-Coupling between Heteroaryl Iodides and α-Chloronitriles. J. Am. Chem. Soc. 2015, 137, 10480–10483. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Z.; Chee, K.W.; Zhou, J.S. Palladium-Catalyzed Asymmetric α-Arylation of Alkylnitriles. J. Am. Chem. Soc. 2016, 138, 16240–16243. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.Y.; Bae, M.; Buchwald, S.L. Mechanistic Insight Facilitates Discovery of a Mild and Efficient Copper-Catalyzed Dehydration of Primary Amides to Nitriles Using Hydrosilanes. J. Am. Chem. Soc. 2018, 140, 1627–1631. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Yu, P.; Morandi, B. Catalytic Reversible Alkene-Nitrile Interconversion through Controllable Transfer Hydrocyanation. Science 2016, 351, 832–836. [Google Scholar] [CrossRef]
- Goswami, P.; Singh, G.; Vijaya Anand, R. N-Heterocyclic Carbene Catalyzed 1,6-Conjugate Addition of Me3Si-CN to para-Quinone Methides and Fuchsones: Access to α-Arylated Nitriles. Org. Lett. 2017, 19, 1982–1985. [Google Scholar] [CrossRef] [PubMed]
- Michel, N.W.M.; Jeanneret, A.D.M.; Kim, H.; Rousseaux, S.A.L. Nickel-Catalyzed Cyanation of Benzylic and Allylic Pivalate Esters. J. Org. Chem. 2018, 83, 11860–11872. [Google Scholar] [CrossRef]
- Fleming, F.F.; Yao, L.; Ravikumar, P.C.; Funk, L.; Shook, B.C. Nitrile-Containing Pharmaceuticals: Efficacious Roles of the Nitrile Pharmacophore. J. Med. Chem. 2010, 53, 7902–7917. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Kato, S.; Aoki, D.; Otsuka, H. A Diarylacetonitrile as a Molecular Probe for the Detection of Polymeric Mechanoradicals in the Bulk State through a Radical Chain-Transfer Mechanism. Angew. Chem. Int. Ed. 2021, 60, 2680–2683. [Google Scholar] [CrossRef] [PubMed]
- Sica, D.A.; Prisant, L.M. Pharmacologic and Therapeutic Considerationsin Hypertension Therapy with Calcium Channel Blockers: Focus on Verapamil. J. Clin. Hypertens. 2007, 9, 1–22. [Google Scholar] [CrossRef]
- Harrington, P.J.; Lodewijk, E. Twenty Years of Naproxen Technology. Org. Process Res. Dev. 1997, 1, 72–76. [Google Scholar] [CrossRef]
- Bedford, C.T. An Efficient, Large-Scale Synthesis of Cytenamide. J. Chem. Res. 2018, 42, 153–155. [Google Scholar] [CrossRef]
- Brett, D.; Downie, I.M.; Lee, J.B. Sugars with Potential Antiviral Activity. Conversion of Hydroxy Compounds to Nitriles. J. Org. Chem. 1967, 32, 855–856. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.; Untch, K.G. Direct One-Step Conversion of Alcohols into Nitriles. J. Org. Chem. 1981, 46, 2985–2987. [Google Scholar] [CrossRef]
- Iranpoor, N.; Firouzabadi, H.; Akhlaghinia, B.; Nowrouzi, N. Conversion of Alcohols, Thiols, and Trimethysilyl Ethers to Alkyl Cyanides Using Triphenylphosphine/2,3-Dichloro-5,6-Dicyanobenzoquinone/n-Bu4NCN. J. Org. Chem. 2004, 69, 2562–2564. [Google Scholar] [CrossRef]
- Soltani Rad, M.N.; Khalafi-Nezhad, A.; Behrouz, S.; Faghihi, M.A. A Simple One-Pot Procedure for the Direct Conversion of Alcohols into Alkyl Nitriles Using TsIm. Tetrahedron Lett. 2007, 48, 6779–6784. [Google Scholar] [CrossRef]
- Aesa, M.C.; Baán, G.; Novák, L.; Szántay, C. Preparation of Unsaturated Nitriles by the Modification of Mitsunobu-Wilk Procedure. Synth. Commun. 1995, 25, 1545–1550. [Google Scholar] [CrossRef]
- Chen, G.; Wang, Z.; Wu, J.; Ding, K. Facile Preparation of α-Aryl Nitriles by Direct Cyanation of Alcohols with TMSCN under the Catalysis of InX3. Org. Lett. 2008, 10, 4573–4576. [Google Scholar] [CrossRef]
- Trillo, P.; Baeza, A.; Nájera, C. Direct Nucleophilic Substitution of Free Allylic Alcohols in Water Catalyzed by FeCl3⋅6 H2O: Which Is the Real Catalyst? ChemCatChem 2013, 5, 1538–1542. [Google Scholar] [CrossRef]
- Theerthagiri, P.; Lalitha, A. Zn(OTf)2—Catalyzed Direct Cyanation of Benzylic Alcohols—a Novel Synthesis of α-Aryl Nitriles. Tetrahedron Lett. 2012, 53, 5535–5538. [Google Scholar] [CrossRef]
- Wang, J.; Masui, Y.; Onaka, M. Direct Synthesis of Nitriles from Alcohols with Trialkylsilyl Cyanide Using Brønsted Acid Montmorillonite Catalysts. ACS Catal. 2011, 1, 446–454. [Google Scholar] [CrossRef]
- Rajagopal, G.; Kim, S.S. Synthesis of α-Aryl Nitriles through B(C6F5)3-Catalyzed Direct Cyanation of α-Aryl Alcohols and Thiols. Tetrahedron 2009, 65, 4351–4355. [Google Scholar] [CrossRef]
- Bhor, M.D.; Panda, A.G.; Nandurkar, N.S.; Bhanage, B.M. Synthesis of Alkyl Iodides/Nitriles from Carbonyl Compounds Using Novel Ruthenium Tris(2,2,6,6-Tetramethyl-3,5-Heptanedionate) as Catalyst. Tetrahedron Lett. 2008, 49, 6475–6479. [Google Scholar] [CrossRef]
- Yu, R.; Rajasekar, S.; Fang, X. Enantioselective Nickel-Catalyzed Migratory Hydrocyanation of Nonconjugated Dienes. Angew. Chem. Int. Ed. 2020, 59, 21436–21441. [Google Scholar] [CrossRef]
- Gao, J.; Jiao, M.; Ni, J.; Yu, R.; Cheng, G.J.; Fang, X. Nickel-Catalyzed Migratory Hydrocyanation of Internal Alkenes: Unexpected Diastereomeric-Ligand-Controlled Regiodivergence. Angew. Chem. Int. Ed. 2021, 60, 1883–1890. [Google Scholar] [CrossRef]
- Sun, F.; Wang, T.; Cheng, G.-J.; Fang, X. Enantioselective Nickel-Catalyzed Hydrocyanative Desymmetrization of Norbornene Derivatives. ACS Catal. 2021, 11, 7578–7583. [Google Scholar] [CrossRef]
- Ding, S.; Jiao, N. Direct Transformation of N,N-Dimethylformamide to −CN: Pd-Catalyzed Cyanation of Heteroarenes Via C–H Functionalization. J. Am. Chem. Soc. 2011, 133, 12374–12377. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Chen, J.; Chen, F.; Cheng, J. The Palladium-Catalyzed Cyanation of Indole C-H Bonds with the Combination of NH4HCO3 and DMSO as a Safe Cyanide Source. Chem. Commun. 2011, 47, 6725–6727. [Google Scholar] [CrossRef]
- Fan, C.; Zhou, Q.-L. Nickel-Catalyzed Group Transfer of Radicals Enables Hydrocyanation of Alkenes and Alkynes. Chem. Catal. 2021, 1, 117–128. [Google Scholar] [CrossRef]
- Cui, J.; Song, J.; Liu, Q.; Liu, H.; Dong, Y. Transition-Metal-Catalyzed Cyanation by Using an Electrophilic Cyanating Agent, N-Cyano-N-Phenyl-p-Toluenesulfonamide (NCTS). Chem. Asian J. 2018, 13, 482–495. [Google Scholar] [CrossRef]
- Ito, Y.; Kato, H.; Imai, H.; Saegusa, T. A Novel Conjugate Hydrocyanation with TiCl4-tert-Butyl Isocyanide. J. Am. Chem. Soc. 1982, 104, 6449–6450. [Google Scholar] [CrossRef]
- Xu, S.; Huang, X.; Hong, X.; Xu, B. Palladium-Assisted Regioselective C–H Cyanation of Heteroarenes Using Isonitrile as Cyanide Source. Org. Lett. 2012, 14, 4614–4617. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zhao, J.; Hu, Z.; Liang, D.; Huang, J.; Zhu, Q. Palladium-Catalyzed C(Sp2)–H Cyanation Using Tertiary Amine Derived Isocyanide as a Cyano Source. Org. Lett. 2012, 14, 4966–4969. [Google Scholar] [CrossRef]
- Dewanji, A.; van Dalsen, L.; Rossi-Ashton, J.A.; Gasson, E.; Crisenza, G.E.M.; Procter, D.J. A General Arene C-H Functionalization Strategy Via Electron Donor-Acceptor Complex Photoactivation. Nat. Chem. 2022, 15, 43–52. [Google Scholar] [CrossRef]
- Tang, S.; Guillot, R.; Grimaud, L.; Vitale, M.R.; Vincent, G. Electrochemical Benzylic C-H Functionalization with Isocyanides. Org. Lett. 2022, 24, 2125–2130. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, J.-M.; Zhang, Y.; Chen, Z.; Zhu, Y.-M.; Ji, S.-J. Synthesis of Aryl Nitriles by Palladium-Assisted Cyanation of Aryl Iodides Using tert-Butyl Isocyanide as Cyano Source. Tetrahedron 2015, 71, 4883–4887. [Google Scholar] [CrossRef]
- Higashimae, S.; Kurata, D.; Kawaguchi, S.I.; Kodama, S.; Sonoda, M.; Nomoto, A.; Ogawa, A. Palladium-Catalyzed Cyanothiolation of Internal Alkynes Using Organic Disulfides and tert-Butyl Isocyanide. J. Org. Chem. 2018, 83, 5267–5273. [Google Scholar] [CrossRef] [PubMed]
- Shirsath, S.R.; Shinde, G.H.; Shaikh, A.C.; Muthukrishnan, M. Accessing α-Arylated Nitriles Via BF3.OEt2 Catalyzed Cyanation of para-Quinone Methides Using tert-Butyl Isocyanide as a Cyanide Source. J. Org. Chem. 2018, 83, 12305–12314. [Google Scholar] [CrossRef]
- Nauth, A.M.; Opatz, T. Non-Toxic Cyanide Sources and Cyanating Agents. Org. Biomol. Chem. 2019, 17, 11–23. [Google Scholar] [CrossRef]
- Kim, J.; Kim, H.J.; Chang, S. Synthesis of Aromatic Nitriles Using Nonmetallic Cyano-Group Sources. Angew. Chem. Int. Ed. 2012, 51, 11948–11959. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, T.; Ding, C.-H.; Xu, B. C(Sp3)–H Functionalization with Isocyanides. Org. Chem. Front. 2021, 8, 3525–3542. [Google Scholar] [CrossRef]
- Wang, J.; Li, D.; Li, J.; Zhu, Q. Advances in Palladium-Catalysed Imidoylative Cyclization of Functionalized Isocyanides for the Construction of N-Heterocycles. Org. Biomol. Chem. 2021, 19, 6730–6745. [Google Scholar] [CrossRef]
- Giustiniano, M.; Basso, A.; Mercalli, V.; Massarotti, A.; Novellino, E.; Tron, G.C.; Zhu, J. To Each His Own: Isonitriles for All Flavors. Functionalized Isocyanides as Valuable Tools in Organic Synthesis. Chem. Soc. Rev. 2017, 46, 1295–1357. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Xu, B. Metal-Catalyzed C-H Functionalization Involving Isocyanides. Chem. Soc. Rev. 2017, 46, 1103–1123. [Google Scholar] [CrossRef]
- Zhang, B.; Studer, A. Recent Advances in the Synthesis of Nitrogen Heterocycles via Radical Cascade Reactions Using Isonitriles as Radical Acceptors. Chem. Soc. Rev. 2015, 44, 3505–3521. [Google Scholar] [CrossRef]
- Boyarskiy, V.P.; Bokach, N.A.; Luzyanin, K.V.; Kukushkin, V.Y. Metal-Mediated and Metal-Catalyzed Reactions of Isocyanides. Chem. Rev. 2015, 115, 2698–2779. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.; Ding, Q.; Wu, J. Recent Advances in Isocyanide Insertion Chemistry. Chem. Soc. Rev. 2013, 42, 5257–5269. [Google Scholar] [CrossRef]
- Dömling, A. Recent Developments in Isocyanide Based Multicomponent Reactions in Applied Chemistry. Chem. Rev. 2006, 106, 17–89. [Google Scholar] [CrossRef]
- Fang, H.; Oestreich, M. Defunctionalisation Catalysed by Boron Lewis Acids. Chem. Sci. 2020, 11, 12604–12615. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Lou, S.J.; Hou, Z. Electron-Deficient Boron-Based Catalysts for C-H Bond Functionalisation. Chem. Soc. Rev. 2021, 50, 1945–1967. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Winfrey, L.; Kustiana, B.A.; Melen, R.L.; Morrill, L.C.; Pulis, A.P. Electron Deficient Borane-Mediated Hydride Abstraction in Amines: Stoichiometric and Catalytic Processes. Chem. Soc. Rev. 2021, 50, 3720–3737. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Roy, S.; Chatterjee, I. Tris(Pentafluorophenyl)Borane Catalyzed C-C and C-Heteroatom Bond Formation. Org. Biomol. Chem. 2021, 19, 1230–1267. [Google Scholar] [CrossRef]
- Oestreich, M.; Hermeke, J.; Mohr, J. A Unified Survey of Si-H and H-H Bond Activation Catalysed by Electron-Deficient Boranes. Chem. Soc. Rev. 2015, 44, 2202–2220. [Google Scholar] [CrossRef]
- Melen, R.L. Applications of Pentafluorophenyl Boron Reagents in the Synthesis of Heterocyclic and Aromatic Compounds. Chem. Commun. 2014, 50, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Erker, G. Tris(Pentafluorophenyl)Borane: A Special Boron Lewis Acid for Special Reactions. Dalton Trans. 2005, 1883–1890. [Google Scholar] [CrossRef]
- Meng, S.-S.; Tang, X.; Luo, X.; Wu, R.; Zhao, J.-L.; Chan, A.S.C. Borane-Catalyzed Chemoselectivity-Controllable N-Alkylation and ortho C-Alkylation of Unprotected Arylamines Using Benzylic Alcohols. ACS Catal. 2019, 9, 8397–8403. [Google Scholar] [CrossRef]
- Meng, S.S.; Wang, Q.; Huang, G.B.; Lin, L.R.; Zhao, J.L.; Chan, A.S.C. B(C6F5)3 Catalyzed Direct Nucleophilic Substitution of Benzylic Alcohols: An Effective Method of Constructing C-O, C-S and C-C Bonds from Benzylic Alcohols. RSC Adv. 2018, 8, 30946–30949. [Google Scholar] [CrossRef]
- Chen, X.; Patel, K.; Marek, I. Stereoselective Construction of Tertiary Homoallyl Alcohols and Ethers by Nucleophilic Substitution at Quaternary Carbon Stereocenters. Angew. Chem. Int. Ed. 2023. [Google Scholar] [CrossRef]
- San, H.H.; Huang, J.; Lei Aye, S.; Tang, X.Y. Boron-Catalyzed Dehydrative Friedel-Crafts Alkylation of Arenes Using β-Hydroxyl Ketone as MVK Precursor. Adv. Synth. Catal. 2021, 363, 2386–2391. [Google Scholar] [CrossRef]
- Guru, M.M.; Thorve, P.R.; Maji, B. Boron-Catalyzed N-Alkylation of Arylamines and Arylamides with Benzylic Alcohols. J. Org. Chem. 2020, 85, 806–819. [Google Scholar] [CrossRef]
- Rubin, M.; Gevorgyan, V. B(C6F5)3-Catalyzed Allylation of Secondary Benzyl Acetates with Allylsilanes. Org. Lett. 2001, 3, 2705–2707. [Google Scholar] [CrossRef]
- Dryzhakov, M.; Hellal, M.; Wolf, E.; Falk, F.C.; Moran, J. Nitro-Assisted Bronsted Acid Catalysis: Application to a Challenging Catalytic Azidation. J. Am. Chem. Soc. 2015, 137, 9555–9558. [Google Scholar] [CrossRef]
- Xiao, Y.; Tang, L.; Xu, T.T.; Feng, J.J. Boron Lewis Acid Catalyzed Intermolecular Trans-Hydroarylation of Ynamides with Hydroxyarenes. Org. Lett. 2022, 24, 2619–2624. [Google Scholar] [CrossRef]
- Lin, T.-Y.; Wu, H.-H.; Feng, J.-J.; Zhang, J. Design and Enantioselective Synthesis of β-Vinyl Tryptamine Building Blocks for Construction of Privileged Chiral Indole Scaffolds. ACS Catal. 2017, 7, 4047–4052. [Google Scholar] [CrossRef]
- Feng, J.-J.; Zhang, J. Rhodium-Catalyzed Stereoselective Intramolecular Tandem Reaction of Vinyloxiranes with Alkynes: Atom- and Step-Economical Synthesis of Multifunctional Mono-, Bi-, and Tricyclic Compounds. ACS Catal. 2017, 7, 1533–1542. [Google Scholar] [CrossRef]
- Zhu, C.Z.; Feng, J.J.; Zhang, J. Rhodium(I)-Catalyzed Intermolecular Aza-[4+3] Cycloaddition of Vinyl Aziridines and Dienes: Atom-Economical Synthesis of Enantiomerically Enriched Functionalized Azepines. Angew. Chem. Int. Ed. 2017, 56, 1351–1355. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, H.; Berke, H.; Döring, S.; Kehr, G.; Erker, G.; Fröhlich, R.; Meyer, O. Lewis Acid Properties of Tris(Pentafluorophenyl)Borane. Structure and Bonding in L−B(C6F5)3 Complexes. Organometallics 1999, 18, 1724–1735. [Google Scholar] [CrossRef]
- Tumanov, V.V.; Tishkov, A.A.; Mayr, H. Nucleophilicity Parameters for Alkyl and Aryl Isocyanides. Angew. Chem. Int. Ed. 2007, 46, 3563–3566. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Zhang, Y.Q.; Zhang, K.; Yang, M.Y.; Chen, Y.B.; Li, Y.; Peng, Q.; Zhu, S.F.; Zhou, Q.L.; Ye, L.W. Stereoselective Synthesis of Medium Lactams Enabled by Metal-Free Hydroalkoxylation/Stereospecific [1,3]-Rearrangement. Nat. Commun. 2019, 10, 3234. [Google Scholar] [CrossRef] [PubMed]
- Nambo, M.; Yar, M.; Smith, J.D.; Crudden, C.M. The Concise Synthesis of Unsymmetric Triarylacetonitriles via Pd-Catalyzed Sequential Arylation: A New Synthetic Approach to Tri- and Tetraarylmethanes. Org. Lett. 2015, 17, 50–53. [Google Scholar] [CrossRef]
- Choi, I.; Chung, H.; Park, J.W.; Chung, Y.K. Active and Recyclable Catalytic Synthesis of Indoles by Reductive Cyclization of 2-(2-Nitroaryl)Acetonitriles in the Presence of Co-Rh Heterobimetallic Nanoparticles with Atmospheric Hydrogen under Mild Conditions. Org. Lett. 2016, 18, 5508–5511. [Google Scholar] [CrossRef]
- Asai, K.; Hirano, K.; Miura, M. Divergent Synthesis of Isonitriles and Nitriles by Palladium-Catalyzed Benzylic Substitution with TMSCN. J. Org. Chem. 2020, 85, 12703–12714. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Jiang, Y.Y.; Kang, L.; Yang, L. Ni-Catalyzed hydrocyanation of alkenes with formamide as the cyano source. Green Chem. 2020, 22, 2734–2738. [Google Scholar] [CrossRef]
- Weinreb, S.; Sengupta, R. A One-Pot Umpolung Method for Preparation of α-Aryl Nitriles from α-Chloro Aldoximes via Organocuprate Additions to Transient Nitrosoalkenes. Synthesis 2012, 44, 2933–2937. [Google Scholar] [CrossRef]
- Aksenov, A.V.; Aksenov, N.A.; Dzhandigova, Z.V.; Aksenov, D.A.; Rubin, M. Nitroalkenes as surrogates for cyanomethylium species in a one-pot synthesis of non-symmetric diarylacetonitriles. RSC Adv. 2015, 5, 106492–106497. [Google Scholar] [CrossRef]
- Liu, S.W.; Meng, L.L.; Zeng, X.J.; Hammond, G.B.; Xu, B. Synthesis of Acrylonitriles via Mild Base Promoted Tandem Nucleophilic Substitution-Isomerization of α-Cyanohydrin Methanesulfonates. Chin. J. Chem. 2021, 39, 913–917. [Google Scholar] [CrossRef]
- Lindsay-Scott, P.J.; Clarke, A.; Richardson, J. Two-Step Cyanomethylation Protocol: Convenient Access to Functionalized Aryl- and Heteroarylacetonitriles. Org. Lett. 2015, 17, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, L.T.; Jiang, Y.W.; Ma, D.W. Assembly of α-(Hetero)aryl Nitriles via Copper-Catalyzed Coupling Reactions with (Hetero)aryl Chlorides and Bromides. Angew. Chem. Int. Ed. 2021, 60, 7082–7086. [Google Scholar] [CrossRef] [PubMed]
- Yuen, O.Y.; Chen, X.; Wu, J.; So, C.M. Palladium-Catalyzed Direct α-Arylation of Arylacetonitriles with Aryl Tosylates and Mesylates. Eur. J. Org. Chem. 2020, 1912–1916. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | T [°C] | Conv. (%) | 3a [%] [b] | 4a [%] [b] |
| 1 | FeCl3 | toluene | 100 | 10 | 0 | 0 |
| 2 | AlCl3 | toluene | 100 | 86 | 16 | 28 |
| 3 | Cu(OTf)2 | toluene | 100 | 100 | 57 | 0 |
| 4 | AgClO4 | toluene | 100 | 21 | 7 | 0 |
| 5 | BF3·Et2O | toluene | 100 | 48 | 24 | 11 |
| 6 | B(C6F5)3 | toluene | 100 | 100 | >99(98) | 0 |
| 7 | TsOH·H2O | toluene | 100 | 84 | 32 | 29 |
| 8 | Tf2NH | toluene | 100 | 100 | 72 | 0 |
| 9 | (PhO)2P(=O)OH | toluene | 100 | 100 | 0 | 45 |
| 10 | B(C6F5)3 | toluene | 80 | 70 | 20 | 30 |
| 11 | B(C6F5)3 | toluene | 50 | 0 | 0 | 0 |
| 12 | B(C6F5)3 | DCE | reflux | 100 | 93 | 0 |
| 13 | B(C6F5)3 | THF | reflux | 100 | 82 | 0 |
| 15 | B(C6F5)3 | HFIP | reflux | 50 | 20 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, T.-T.; Zhou, J.-L.; Cong, G.-Y.; Sheng, J.-Y.-H.; Wang, S.-Q.; Ma, Y.; Feng, J.-J. Boron Lewis Acid Catalysis Enables the Direct Cyanation of Benzyl Alcohols by Means of Isonitrile as Cyanide Source. Molecules 2023, 28, 2174. https://doi.org/10.3390/molecules28052174
Xu T-T, Zhou J-L, Cong G-Y, Sheng J-Y-H, Wang S-Q, Ma Y, Feng J-J. Boron Lewis Acid Catalysis Enables the Direct Cyanation of Benzyl Alcohols by Means of Isonitrile as Cyanide Source. Molecules. 2023; 28(5):2174. https://doi.org/10.3390/molecules28052174
Chicago/Turabian StyleXu, Tong-Tong, Jin-Lan Zhou, Guang-Yuan Cong, Jiang-Yi-Hui Sheng, Shi-Qi Wang, Yating Ma, and Jian-Jun Feng. 2023. "Boron Lewis Acid Catalysis Enables the Direct Cyanation of Benzyl Alcohols by Means of Isonitrile as Cyanide Source" Molecules 28, no. 5: 2174. https://doi.org/10.3390/molecules28052174
APA StyleXu, T.-T., Zhou, J.-L., Cong, G.-Y., Sheng, J.-Y.-H., Wang, S.-Q., Ma, Y., & Feng, J.-J. (2023). Boron Lewis Acid Catalysis Enables the Direct Cyanation of Benzyl Alcohols by Means of Isonitrile as Cyanide Source. Molecules, 28(5), 2174. https://doi.org/10.3390/molecules28052174