
Synthesis of Six-Membered N-Heterocyclic Carbene Precursors Based on Camphor

 , ,
, ,  and
and 
Abstract
:
1. Introduction
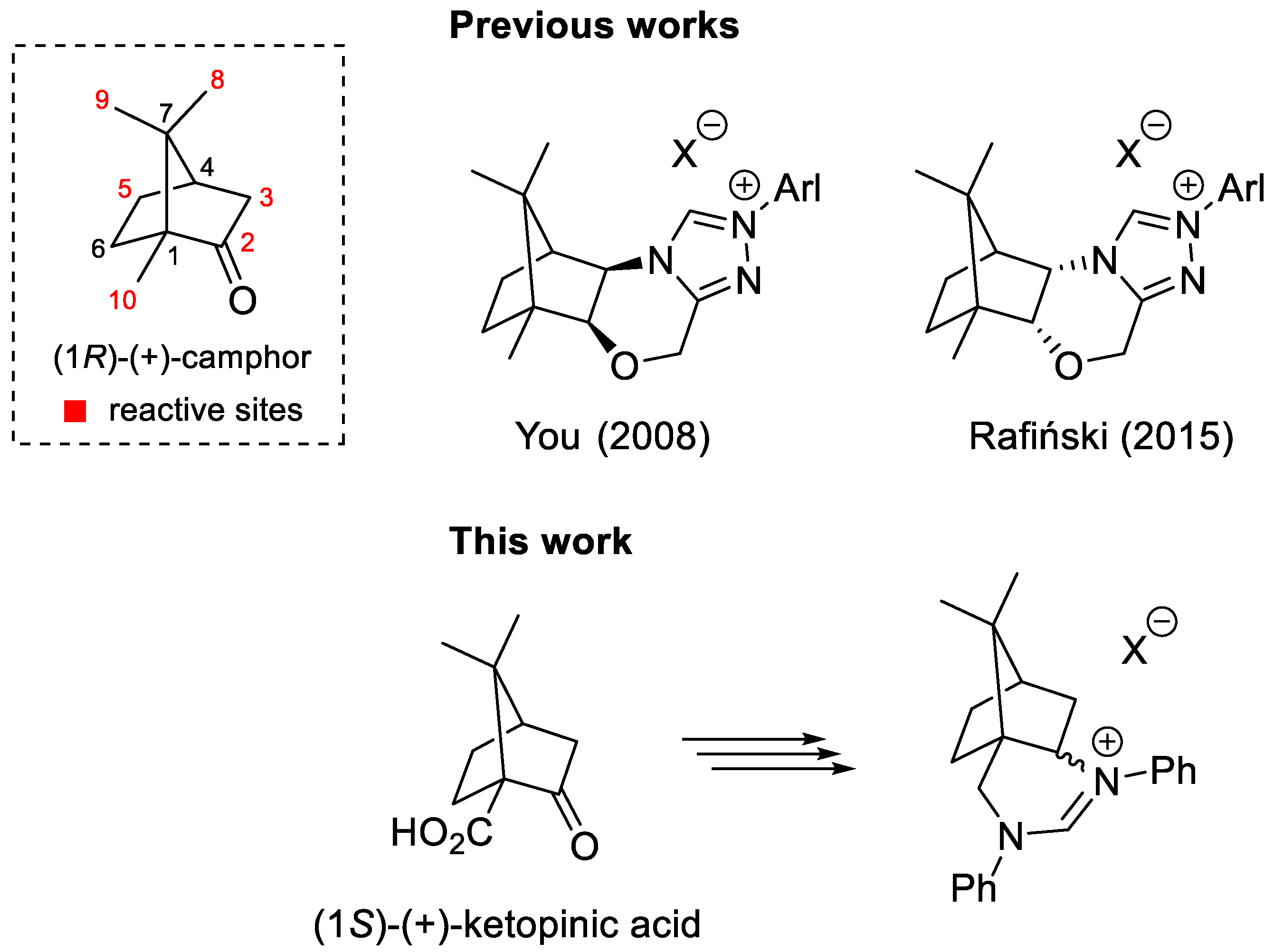
2. Results and Discussion
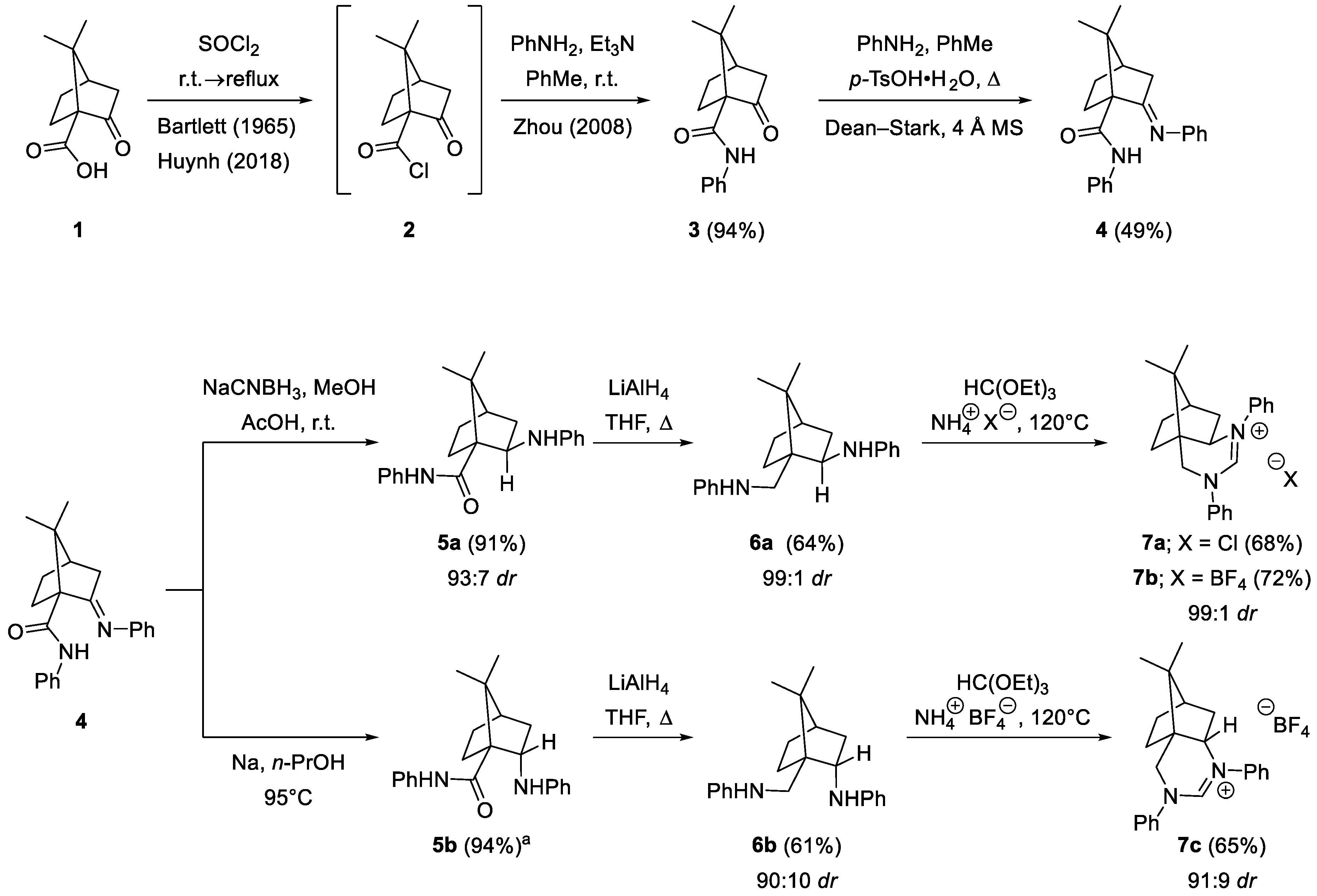
2.1. Synthesis
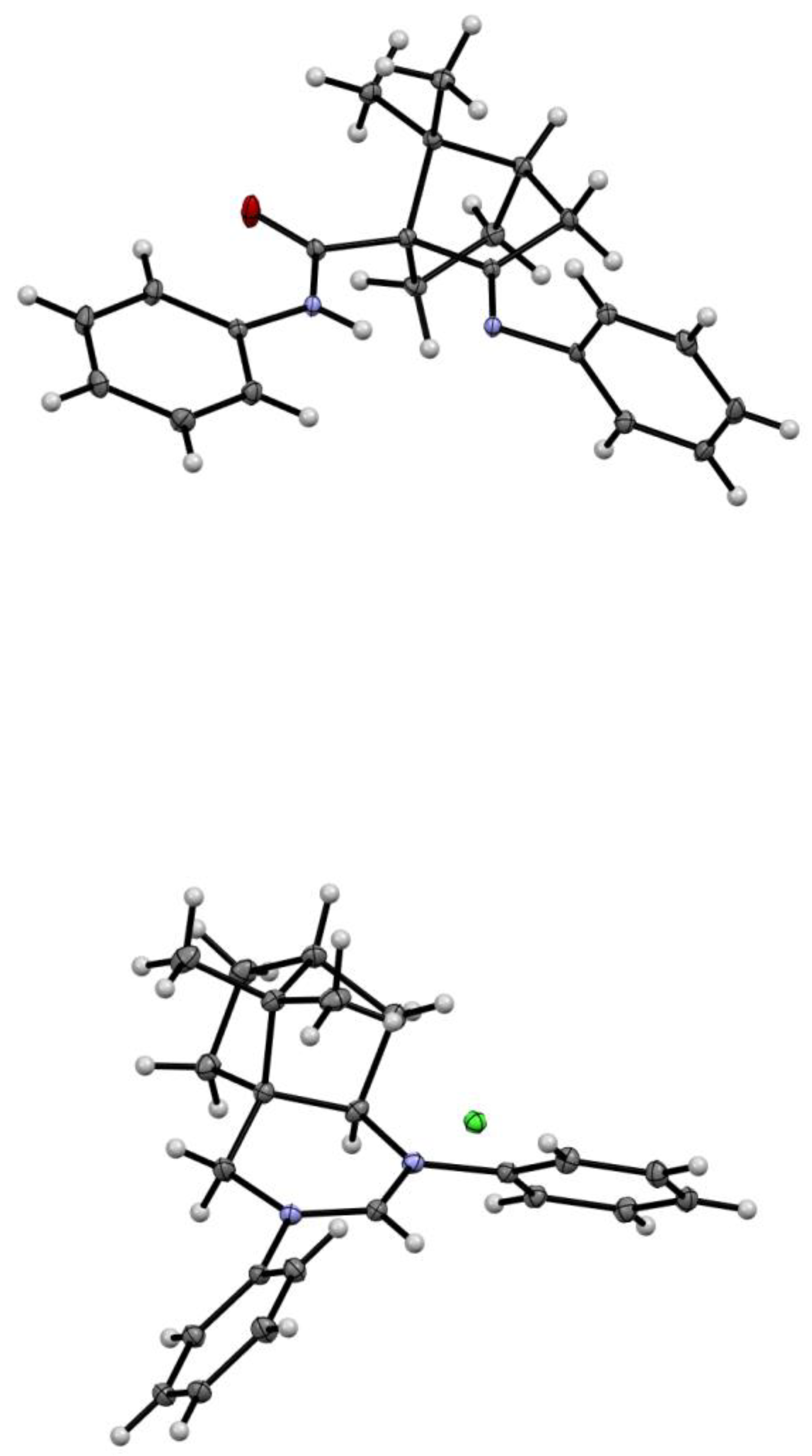
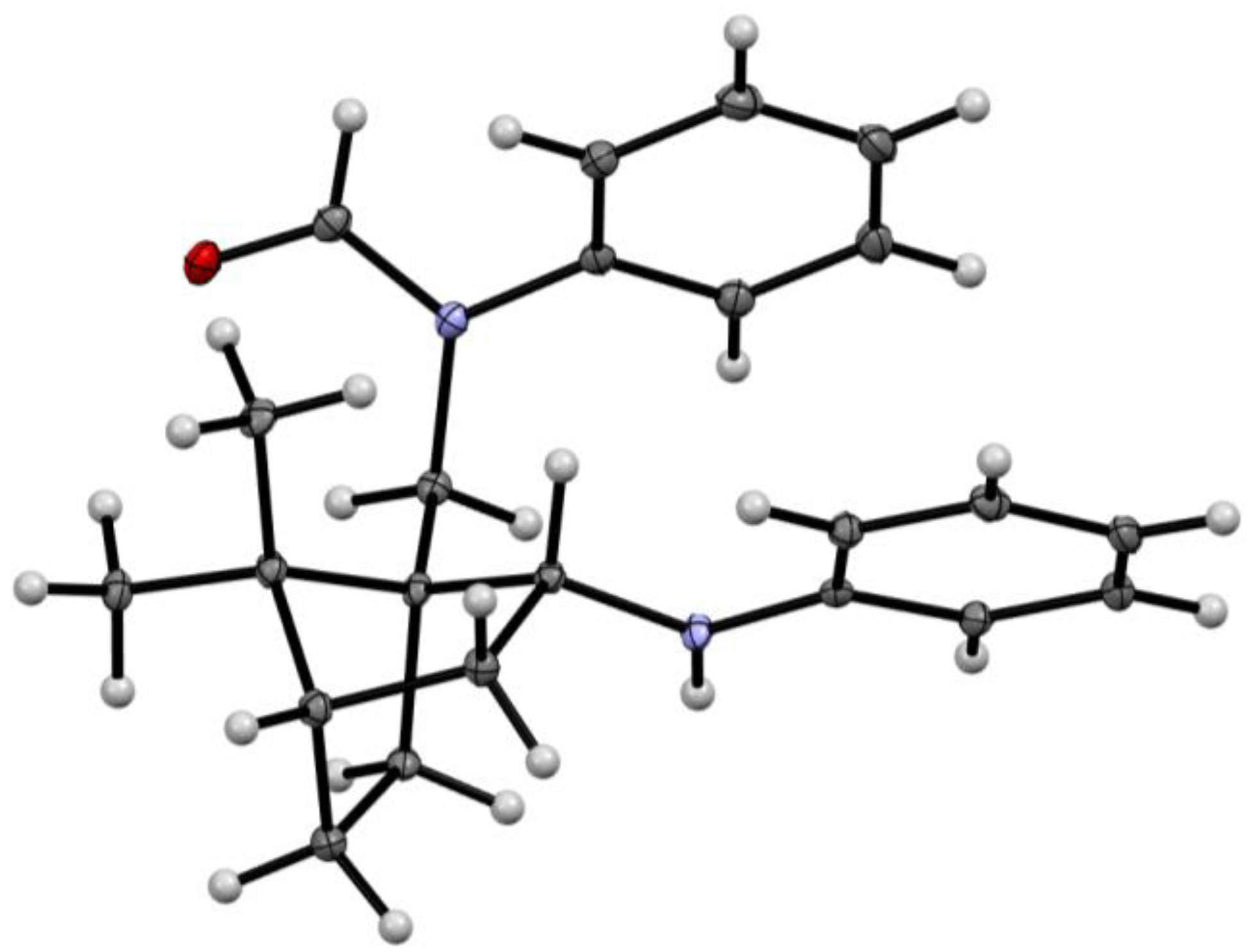
2.2. Structure Determination
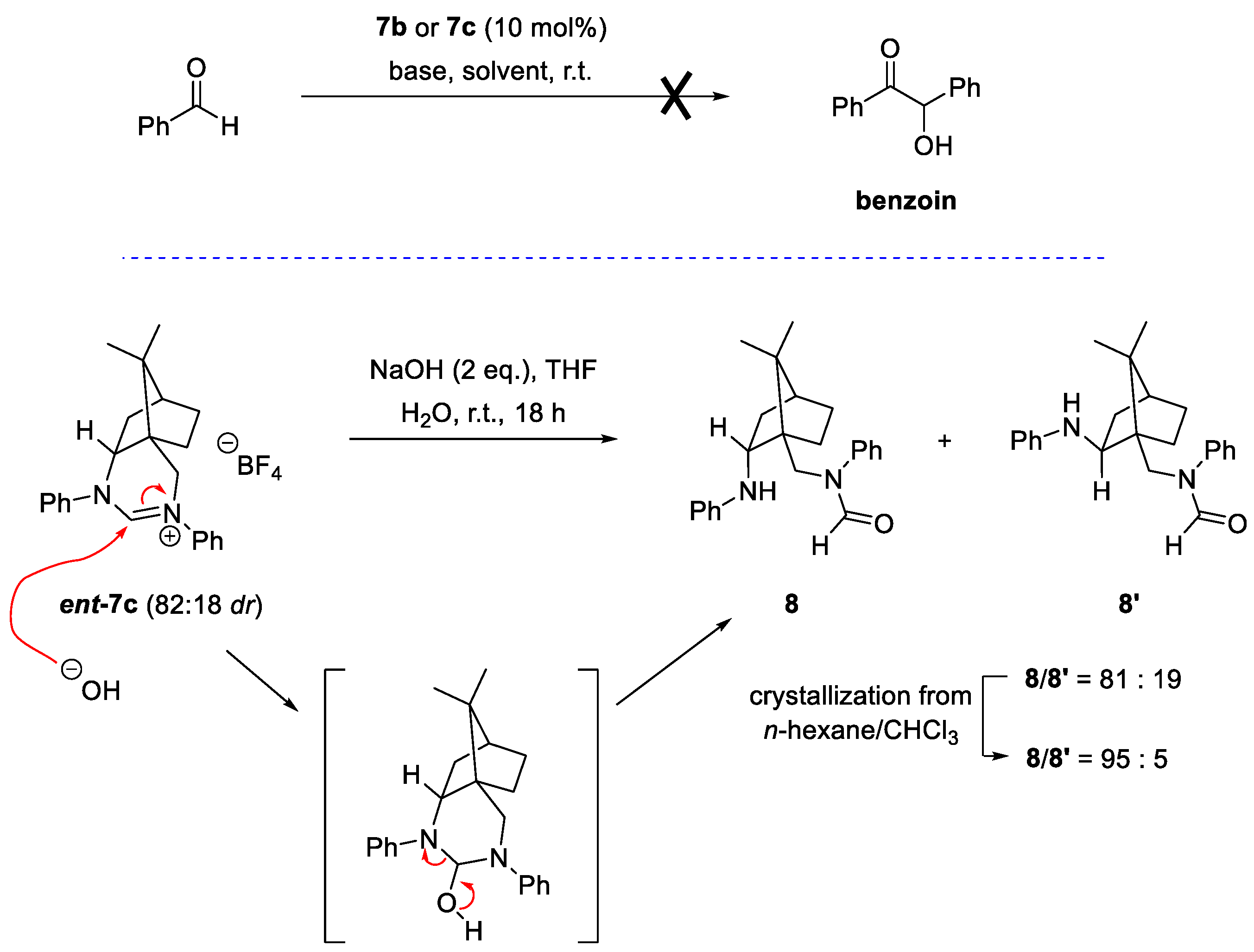
2.3. Performance of Camphor-Derived NHC Precursors in Benzoin Reaction
3. Materials and Methods
3.1. Materials and General Methods
3.1.1. Synthesis of (1R,4R)-7,7-Dimethyl-2-oxo-N-phenylbicyclo[2.2.1]heptane-1-carboxamide (3) [28]
3.1.2. Synthesis of (1R,4R,E)-7,7-Dimethyl-N-phenyl-2-(phenylimino)bicyclo[2.2.1]heptane-1-carboxamide (4)
3.1.3. Synthesis of (1R,2R,4R)-7,7-Dimethyl-N-phenyl-2-(phenylamino)bicyclo[2.2.1]heptane-1-carboxamide (5a)
3.1.4. Synthesis of (1R,2S,4R)-7,7-dimethyl-N-phenyl-2-(phenylamino)bicyclo[2.2.1]heptane-1-carboxamide (5b)
3.1.5. Synthesis of (1S,2R,4R)-7,7-Dimethyl-N-phenyl-1-((phenylamino)methyl)bicyclo[2.2.1]-heptan-2-amine (6a)
3.1.6. Synthesis of (1S,2S,4R)-7,7-Dimethyl-N-phenyl-1-((phenylamino)methyl)bicyclo[2.2.1]-heptan-2-amine (6b)
3.1.7. Synthesis of (7R,8aR)-9,9-Dimethyl-1,3-diphenyl-3,5,6,7,8,8a-hexahydro-4H-4a,7-methanoquinazolin-1-ium Chloride (7a)
3.1.8. Synthesis of (7R,8aR)-9,9-Dimethyl-1,3-diphenyl-3,5,6,7,8,8a-hexahydro-4H-4a,7-methanoquinazolin-1-ium Tetrafluoroborate (7b)
3.1.9. Synthesis of (4aS,7R)-9,9-Dimethyl-1,3-diphenyl-3,5,6,7,8,8a-hexahydro-4H-4a,7-methanoquinazolin-1-ium Tetrafluoroborate (7c)
3.1.10. Synthesis of N-(((1S,2S,4R)-7,7-Dimethyl-2-(phenylamino)bicyclo[2.2.1]heptan-1-yl)methyl)-N-phenylformamide (8) and N-(((1S,2R,4R)-7,7-Dimethyl-2-(phenylamino)bicyclo[2.2.1]heptan-1-yl)methyl)-N-phenylformamide (8′)
3.2. General Procedure for the Catalytic Asymmetric Benzoin Condensation Reaction with Benzaldehyde
3.3. X-ray Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Money, T. Remote functionalization of camphor: Application to natural product synthesis. Org. Synth. Theory Appl. 1996, 3, 1–83. [Google Scholar]
- Money, T. Camphor: A chiral starting material in natural product synthesis. Nat. Prod. Rep. 1985, 2, 253–289. [Google Scholar] [CrossRef] [PubMed]
- Oppolzer, W. Camphor derivatives as chiral auxiliaries in asymmetric synthesis. Tetrahedron 1987, 43, 1969–2004. [Google Scholar] [CrossRef]
- Oppolzer, W. Camphor as a natural source of chirality in asymmetric synthesis. Pure Appl. Chem. 1990, 62, 1241–1250. [Google Scholar] [CrossRef]
- Kitamura, M.; Suga, S.; Kawai, K.; Noyori, R. Catalytic asymmetric induction. Highly enantioselective addition of dialkylzincs to aldehydes. J. Am. Chem. Soc. 1986, 108, 6071–6072. [Google Scholar] [CrossRef] [PubMed]
- Nugent, W.A. MIB: An advantageous alternative to DAIB for the addition of organozinc reagents to aldehydes. Chem. Commun. 1999, 15, 1369–1370. [Google Scholar] [CrossRef]
- Rios, R. (Ed.) Stereoselective Organocatalysis; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Jakab, G.; Schreiner, P.R. Brønsted Acids: Chiral (Thio)urea Derivatives. In Comprehensive Enantioselective Organocatalysis; Dalko, P.I., Ed.; Wiley-VCH: Weinheim, Germany, 2013; pp. 315–342. [Google Scholar]
- Rajaram, S.; Sigman, M.S. Design of Hydrogen Bond Catalysts Based on a Modular Oxazoline Template: Application to an Enantioselective Hetero Diels-Alder Reaction. Org. Lett. 2005, 7, 5473–5475. [Google Scholar] [CrossRef]
- Grošelj, U. Camphor-Derivatives in Asymmetric Organocatalysis—Synthesis and Application. Curr. Org. Chem. 2015, 19, 2048–2074. [Google Scholar] [CrossRef]
- Enders, D.; Kallfass, U. An efficient nucleophilic carbene catalyst for the asymmetric benzoin condensation. Angew. Chem. Int. Ed. 2002, 41, 1743–1745. [Google Scholar] [CrossRef]
- Reddy, P.V.G.; Tabassum, S.; Blanrue, A.; Wilhelm, R. New enantiopure NHCs derived from camphor. Chem. Commun. 2009, 39, 5910–5912. [Google Scholar] [CrossRef]
- Li, Y.; Feng, Z.; You, S.-L. D-Camphor-derived triazolium salts for catalytic intramolecular crossed aldehyde–ketone benzoin reactions. Chem. Commun. 2008, 19, 2263–2265. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, X.-Q.; Zheng, C.; You, S.-L. Highly enantioselective intramolecular Michael reactions by D-camphor-derived triazolium salts. Chem. Commun. 2009, 39, 5823–5825. [Google Scholar] [CrossRef] [PubMed]
- Rong, Z.-Q.; Jia, M.-Q.; You, S.-L. Enantioselective N-Heterocyclic Carbene-Catalyzed Michael Addition to α,β-Unsaturated Aldehydes by Redox Oxidation. Org. Lett. 2011, 13, 4080–4083. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.-Q.; You, S.-L. N-Heterocyclic Carbene-Catalyzed Enantioselective Intramolecular N-Tethered Aldehyde–Ketone Benzoin Reactions. ACS Catal. 2013, 3, 622–624. [Google Scholar] [CrossRef]
- Jia, M.-Q.; You, S.-L. Desymmetrization of Cyclohexadienones via Intramolecular Stetter Reaction to Construct Tricyclic Carbocycles. Synlett 2013, 24, 1201–1204. [Google Scholar] [CrossRef]
- Rong, Z.-Q.; Li, Y.; Yang, G.-Q.; You, S.-L. D-Camphor-Derived Triazolium Salts for Enantioselective Intramolecular Stetter Reactions. Synlett 2011, 2011, 1033–1037. [Google Scholar] [CrossRef]
- Rafiński, Z.; Kozakiewicz, A. Enantioselective Synthesis of Chromanones Bearing Quaternary Substituted Stereocenters Catalyzed by (1R)-Camphor-Derived N-Heterocyclic Carbenes. J. Org. Chem. 2015, 80, 7468–7476. [Google Scholar] [CrossRef]
- Rafiński, Z.; Kozakiewicz, A.; Rafińska, K. Highly efficient synthesis of spirocyclic (1R)-camphor-derived triazolium salts: Application in the catalytic asymmetric benzoin condensation. Tetrahedron 2014, 70, 5739–5745. [Google Scholar] [CrossRef]
- Ričko, S.; Požgan, F.; Štefane, B.; Svete, J.; Golobič, A.; Grošelj, U. Stereodivergent Synthesis of Camphor-Derived Diamines and Their Application as Thiourea Organocatalysts. Molecules 2020, 25, 2978. [Google Scholar] [CrossRef]
- Ričko, S.; Svete, J.; Štefane, B.; Perdih, A.; Golobič, A.; Meden, A.; Grošelj, U. 1,3-Diamine-Derived Bifunctional Organocatalyst Prepared from Camphor. Adv. Synth. Catal. 2016, 358, 3786–3796. [Google Scholar] [CrossRef]
- Ciber, L.; Požgan, F.; Brodnik, H.; Štefane, B.; Svete, J.; Waser, M.; Grošelj, U. Synthesis and Catalytic Activity of Bifunctional Phase-Transfer Organocatalysts Based on Camphor. Molecules 2023, 28, 1515. [Google Scholar] [CrossRef] [PubMed]
- Ričko, S.; Meden, A.; Ivančič, A.; Perdih, A.; Štefane, B.; Svete, J.; Grošelj, U. Organocatalyzed Deracemization of Δ2-Pyrrolin-4-ones. Adv. Synth. Catal. 2017, 359, 2288–2296. [Google Scholar] [CrossRef]
- Bartlett, P.D.; Knox, L.H. DL-Ketopinic acid. Org. Synth. 1965, 45, 55–56. [Google Scholar]
- Bartlett, P.D.; Knox, L.H. D,L-10-Camphorsulfonyl chloride. Org. Synth. 1965, 45, 14–16. [Google Scholar]
- Huynh, U.; McDonald, S.L.; Lim, D.; Uddin, M.N.; Wengryniuk, S.E.; Dey, S.; Coltart, D.M. Formation, Alkylation, and Hydrolysis of Chiral Nonracemic N-Amino Cyclic Carbamate Hydrazones: An Approach to the Enantioselective α-Alkylation of Ketones. J. Org. Chem. 2018, 83, 12951–12964. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Bian, Y. Synthesis of novel chiral Schiff bases and their application in asymmetric transfer hydrogenation of prochiral ketones. Heteroat. Chem. 2008, 19, 682–687. [Google Scholar] [CrossRef]
- Grošelj, U.; Sevšek, A.; Ričko, S.; Golobič, A.; Svete, J.; Stanovnik, B. Synthesis and Structural Characterization of Novel Camphor-derived Amines. Chirality 2012, 24, 778–788. [Google Scholar] [CrossRef]
- Zhang, J.; Qin, X.; Fu, J.; Wang, X.; Su, X.; Hu, F.; Jiao, J.; Shi, M. Fine-tunable 3, 4-dihydroquinazol-2-ylidene carbenes: Synthesis, rhodium (I) complexes, and reactivity. Organometallics 2012, 31, 8275–8282. [Google Scholar] [CrossRef]
- Mincheva, R.; Narayana Murthy Chilla, S.; Todd, R.; Guillerm, B.; De Winter, J.; Gerbaux, P.; Coulembier, O.; Dubois, P.; Raquez, J.-M. Reactive Extrusion and Magnesium (II) N-Heterocyclic Carbene Catalyst in Continuous PLA Production. Polymers 2019, 11, 1987. [Google Scholar] [CrossRef]
- Yan, J.; Sun, R.; Shi, K.; Li, K.; Yang, L.; Zhong, G. N-heterocyclic carbene-catalyzed asymmetric benzoin reaction in water. J. Org. Chem. 2018, 83, 7547–7552. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, F.; Xue, X.-S.; Ji, P. Acidity Scale of N-Heterocyclic Carbene Precursors: Can We Predict the Stability of NHC–CO2 Adducts? Org. Lett. 2018, 20, 6041–6045. [Google Scholar] [CrossRef]
- Wang, Z.; Xue, X.-S.; Fu, Y.; Ji, P. Comprehensive Basicity Scales for N-Heterocyclic Carbenes in DMSO: Implications on the Stabilities of N-Heterocyclic Carbene and CO2 Adducts. Chem. Asian J. 2020, 15, 169–181. [Google Scholar] [CrossRef]
- Wang, N.; Xu, J.; Lee, J.K. The importance of N-heterocyclic carbene basicity in organocatalysis. Org. Biomol. Chem. 2018, 16, 8230–8244. [Google Scholar] [CrossRef]
- Soeta, T.; Mizuno, S.; Hatanaka, Y.; Ukaji, Y. Asymmetric cross-benzoin condensation promoted by a chiral triazolium precatalyst bearing a pyridine moiety. Tetrahedron 2017, 73, 3430–3437. [Google Scholar] [CrossRef]
- CrysAlis PRO; Agilent Technologies UK Ltd.: Yarnton, UK, 2011.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cristallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šegina, J.; Ciber, L.; Brodnik, H.; Požgan, F.; Svete, J.; Štefane, B.; Grošelj, U. Synthesis of Six-Membered N-Heterocyclic Carbene Precursors Based on Camphor. Molecules 2023, 28, 7973. https://doi.org/10.3390/molecules28247973
Šegina J, Ciber L, Brodnik H, Požgan F, Svete J, Štefane B, Grošelj U. Synthesis of Six-Membered N-Heterocyclic Carbene Precursors Based on Camphor. Molecules. 2023; 28(24):7973. https://doi.org/10.3390/molecules28247973
Chicago/Turabian StyleŠegina, Jan, Luka Ciber, Helena Brodnik, Franc Požgan, Jurij Svete, Bogdan Štefane, and Uroš Grošelj. 2023. "Synthesis of Six-Membered N-Heterocyclic Carbene Precursors Based on Camphor" Molecules 28, no. 24: 7973. https://doi.org/10.3390/molecules28247973
APA StyleŠegina, J., Ciber, L., Brodnik, H., Požgan, F., Svete, J., Štefane, B., & Grošelj, U. (2023). Synthesis of Six-Membered N-Heterocyclic Carbene Precursors Based on Camphor. Molecules, 28(24), 7973. https://doi.org/10.3390/molecules28247973