
Diffusion-Controlled Reactions: An Overview


{kind=link}
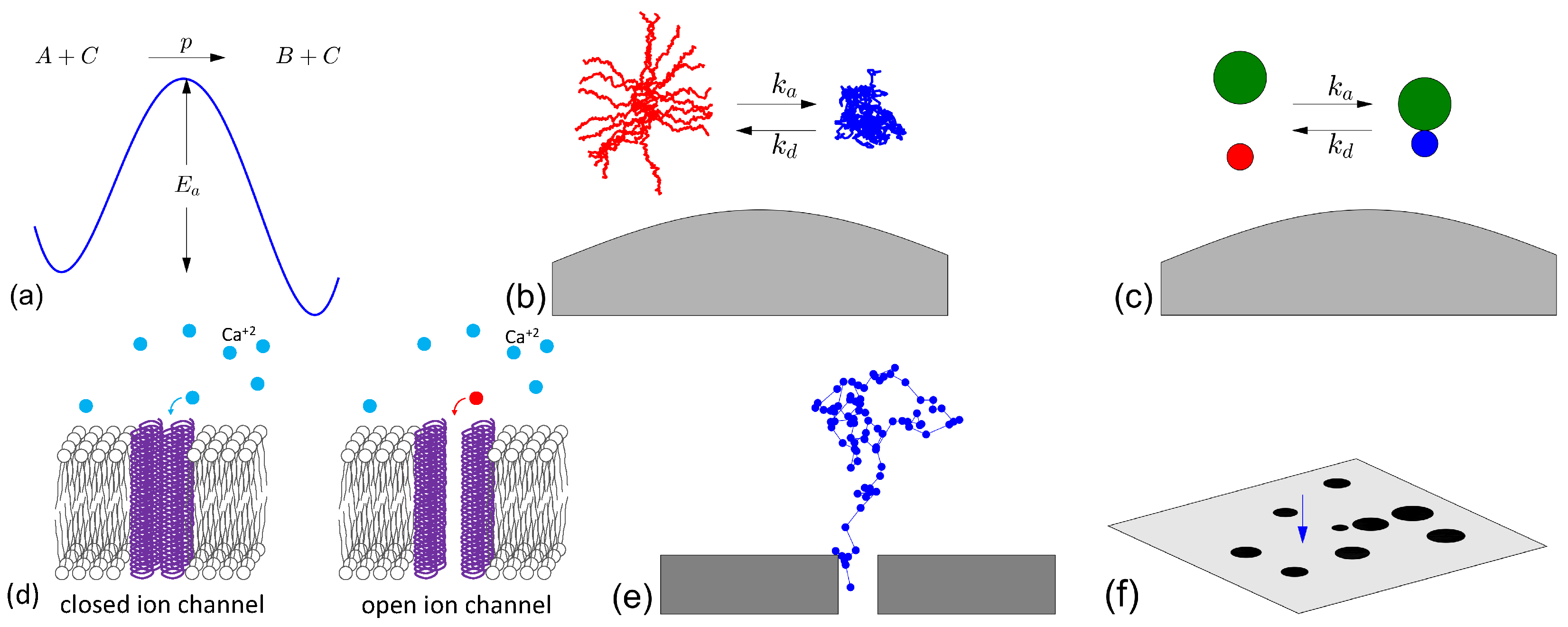{kind=link}
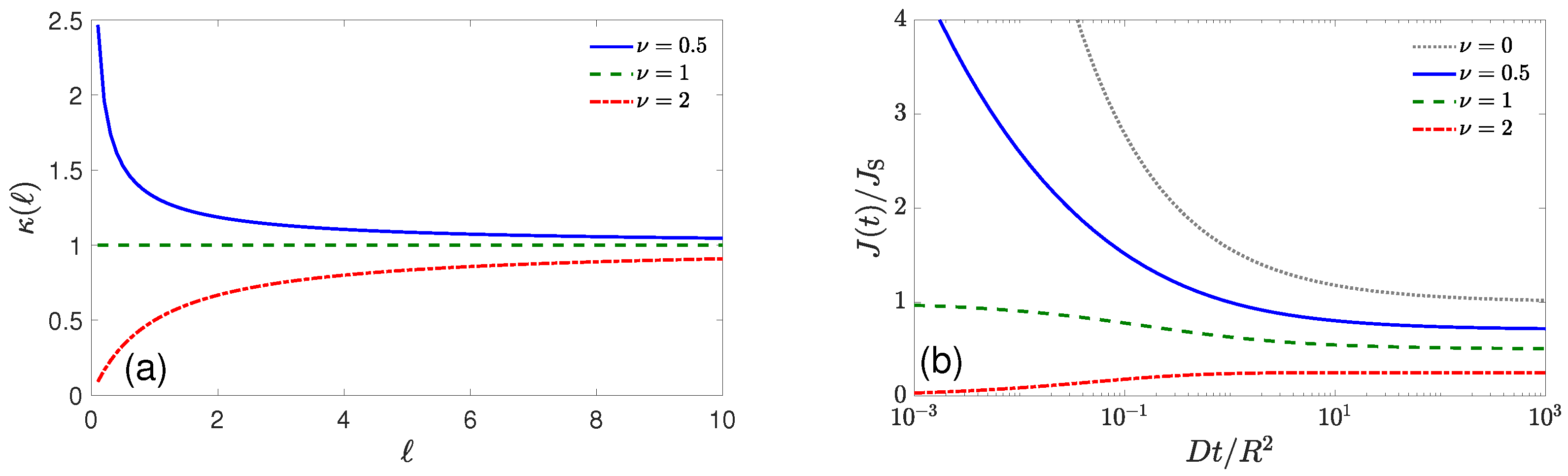{kind=link}
Abstract
1. Introduction
2. The Role of Diffusive Transport
3. Imperfect Surface Reactions
4. Various Extensions
5. Beyond the Conventional Framework
6. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Murrey, J.D. Mathematical Biology II: Spatial Models and Biomedical Applications, 3rd ed.; Springer: Berlin, Germany, 2003. [Google Scholar]
- Volpert, V. Elliptic Partial Differential Equations. Vol. 2. Reaction-Diffusion Equations; Monographs in Mathematics, 104; Springer: Basel, Switzerland, 2014. [Google Scholar]
- von Smoluchowski, M. Versuch einer Mathematischen Theorie der Koagulations Kinetic Kolloider Lousungen. Z. Phys. Chem. 1918, 92U, 129–168. [Google Scholar] [CrossRef]
- North, A.M. Diffusion-controlled reactions. Q. Rev. Chem. Soc. 1966, 20, 421–440. [Google Scholar] [CrossRef]
- Wilemski, G.; Fixman, M. General theory of diffusion-controlled reactions. J. Chem. Phys. 1973, 58, 4009–4019. [Google Scholar] [CrossRef]
- Calef, D.F.; Deutch, J.M. Diffusion-Controlled Reactions. Ann. Rev. Phys. Chem. 1983, 34, 493–524. [Google Scholar] [CrossRef]
- Berg, O.G.; von Hippel, P.H. Diffusion-Controlled Macromolecular Interactions. Ann. Rev. Biophys. Biophys. Chem. 1985, 14, 131–160. [Google Scholar] [CrossRef]
- Rice, S. Diffusion-Limited Reactions; Elsevier: Amsterdam, The Netherlands, 1985. [Google Scholar]
- Bressloff, P.C.; Newby, J.M. Stochastic models of intracellular transport. Rev. Mod. Phys. 2013, 85, 135–196. [Google Scholar] [CrossRef]
- Lindenberg, K.; Metzler, R.; Oshanin, G. (Eds.) Chemical Kinetics: Beyond the Textbook; World Scientific: Hackensack, NJ, USA, 2019. [Google Scholar]
- Witten, T.A., Jr.; Sander, L.M. Diffusion-Limited Aggregation, a Kinetic Critical Phenomenon. Phys. Rev. Lett. 1981, 47, 1400–1403. [Google Scholar] [CrossRef]
- Lauffenburger, D.A.; Linderman, J. Receptors: Models for Binding, Trafficking, and Signaling; Oxford University Press: Oxford, UK, 1993. [Google Scholar]
- Küchler, A.; Yoshimoto, M.; Luginbühl, S.; Mavelli, F.; Walde, P. Enzymatic reactions in confined environments. Nat. Nanotechnol. 2016, 11, 409–420. [Google Scholar] [CrossRef]
- Hill, T.L. Effect of rotation on the diffusion-controlled rate of ligand-protein association. Proc. Natl. Acad. Sci. USA 1975, 72, 4918–4922. [Google Scholar] [CrossRef]
- Zwanzig, R. Diffusion controlled ligand binding to spheres partially covered by receptors: An effective medium treatment. Proc. Nat. Acad. Sci. USA 1990, 87, 5856–5857. [Google Scholar] [CrossRef]
- Held, M.; Metzner, P.; Prinz, J.-H.; Noé, F. Mechanisms of Protein-Ligand Association and Its Modulation by Protein Mutations. Biophys. J. 2011, 100, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Sano, H.; Tachiya, M. Partially diffusion-controlled recombination. J. Chem. Phys. 1979, 71, 1276–1282. [Google Scholar] [CrossRef]
- Agmon, N.; Pines, E.; Huppert, D. Geminate recombination in proton-transfer reactions. II. Comparison of diffusional and kinetic schemes. J. Chem. Phys. 1988, 88, 5631–5638. [Google Scholar] [CrossRef]
- Sano, H.; Tachiya, M. Theory of diffusion-controlled reactions on spherical surfaces and its application to reactions on micellar surfaces. J. Chem. Phys. 1981, 75, 2870–2878. [Google Scholar] [CrossRef]
- Brownstein, K.R.; Tarr, C.E. Importance of Classical Diffusion in NMR Studies of Water in Biological Cells. Phys. Rev. A 1979, 19, 2446–2453. [Google Scholar] [CrossRef]
- Grebenkov, D.S. NMR Survey of Reflected Brownian Motion. Rev. Mod. Phys. 2007, 79, 1077–1137. [Google Scholar] [CrossRef]
- Richter, P.H.; Eigen, M. Diffusion controlled reaction rates in spheroidal geometry application to repressor-operator. association and membrane bound enzymes. Biophys. Chem. 1974, 2, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Berg, O.G.; Winter, R.B.; von Hippel, P.H. Diffusion-driven mechanisms of protein translocation on nucleic acids: 1. Models and theory. Biochemistry 1981, 20, 6929–6948. [Google Scholar] [CrossRef]
- Sheinman, M.; Bénichou, O.; Kafri, Y.; Voituriez, R. Classes of fast and specific search mechanisms for proteins on DNA. Rep. Prog. Phys. 2012, 75, 026601. [Google Scholar] [CrossRef] [PubMed]
- Golestanian, R. Anomalous Diffusion of Symmetric and Asymmetric Active Colloids. Phys. Rev. Lett. 2009, 102, 188305. [Google Scholar] [CrossRef]
- Oshanin, G.; Popescu, M.N.; Dietrich, S. Active colloids in the context of chemical kinetics. J. Phys. A Math. Theor. 2017, 50, 134001. [Google Scholar] [CrossRef]
- Traytak, S.D.; Grebenkov, D.S. Diffusion-influenced reaction rates for active ’sphere-prolate spheroid’ pairs and Janus dimers. J. Chem. Phys. 2018, 148, 024107. [Google Scholar] [CrossRef] [PubMed]
- Weibel, E.R. The Pathway for Oxygen. Structure and Function in the Mammalian Respiratory System; Harvard University Press: Cambridge, MA, USA, 1984. [Google Scholar]
- Sapoval, B.; Filoche, M.; Weibel, E. Smaller is better - but not too small: A physical scale for the design of the mammalian pulmonary acinus. Proc. Natl. Acad. Sci. USA 2002, 99, 10411–10416. [Google Scholar] [CrossRef] [PubMed]
- Grebenkov, D.S.; Filoche, M.; Sapoval, B.; Felici, M. Diffusion-reaction in Branched Structures: Theory and Application to the Lung Acinus. Phys. Rev. Lett. 2005, 94, 050602. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.C.; Kimball, G.E. Diffusion-controlled reaction rates. J. Colloid Sci. 1949, 4, 425–437. [Google Scholar] [CrossRef]
- Bénichou, O.; Chevalier, C.; Klafter, J.; Meyer, B.; Voituriez, R. Geometry-controlled kinetics. Nat. Chem. 2010, 2, 472–477. [Google Scholar] [CrossRef]
- Bénichou, O.; Voituriez, R. From first-passage times of random walks in confinement to geometry-controlled kinetics. Phys. Rep. 2014, 539, 225–284. [Google Scholar] [CrossRef]
- Grebenkov, D.S. Imperfect Diffusion-Controlled Reactions. In Chemical Kinetics: Beyond the Textbook; Lindenberg, K., Metzler, R., Oshanin, G., Eds.; World Scientific: Hackensack, NJ, USA, 2019; pp. 191–219. [Google Scholar]
- Blender, R.; Dieterich, W.; Kirchhoff, T.; Sapoval, B. Impedance of Fractal Interfaces. J. Phys. A Math. Gen. 1990, 23, 1225–1232. [Google Scholar] [CrossRef]
- Coppens, M.-O. The effect of fractal surface roughness on diffusion and reaction in porous catalysts: From fundamentals to practical applications. Cat. Today 1999, 53, 225–243. [Google Scholar] [CrossRef]
- Filoche, M.; Sapoval, B. Transfer Across Random versus Deterministic Fractal Interfaces. Phys. Rev. Lett. 2000, 84, 5776–5779. [Google Scholar] [CrossRef]
- Andrade, J.S., Jr.; Araujo, S.D.; Filoche, M.; Sapoval, B. Screening Effects in Flow through Rough Channels. Phys. Rev. Lett. 2007, 98, 194101. [Google Scholar] [CrossRef]
- Serov, A.S.; Salafia, C.; Grebenkov, D.S.; Filoche, M. The Role of Morphology in Mathematical Models of Placental Gas Exchange. J. Appl. Physiol. 2016, 120, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Galanti, M.; Fanelli, D.; Traytak, S.D.; Piazza, F. Theory of diffusion-influenced reactions in complex geometries. Phys. Chem. Chem. Phys. 2016, 18, 15950–15954. [Google Scholar] [CrossRef] [PubMed]
- Grebenkov, D.S.; Traytak, S.D. Semi-analytical computation of Laplacian Green functions in three-dimensional domains with disconnected spherical boundaries. J. Comput. Phys. 2019, 379, 91–117. [Google Scholar] [CrossRef]
- Grebenkov, D.S. Diffusion toward non-overlapping partially reactive spherical traps: Fresh insights onto classic problems. J. Chem. Phys. 2020, 152, 244108. [Google Scholar] [CrossRef]
- Nguyen, B.-T.; Grebenkov, D.S. A Spectral Approach to Survival Probability in Porous Media. J. Stat. Phys. 2010, 141, 532–554. [Google Scholar] [CrossRef]
- Piazza, F. The physics of boundary conditions in reaction-diffusion problems. J. Chem. Phys. 2022, 157, 234110. [Google Scholar] [CrossRef]
- Weiss, G.H. Overview of theoretical models for reaction rates. J. Stat. Phys. 1986, 42, 3–36. [Google Scholar] [CrossRef]
- Hänggi, P.; Talkner, P.; Borkovec, M. Reaction-rate theory: Fifty years after Kramers. Rev. Mod. Phys. 1990, 62, 251–341. [Google Scholar] [CrossRef]
- Grebenkov, D.S.; Filoche, M.; Sapoval, B. Spectral Properties of the Brownian Self-Transport Operator. Eur. Phys. J. B 2003, 36, 221–231. [Google Scholar] [CrossRef]
- Bressloff, P.C. Stochastic switching in biology: From genotype to phenotype. J. Phys. A Math. Theor. 2017, 50, 133001. [Google Scholar] [CrossRef]
- Cortés, J.; Le, D.T.; Iehl, R.; Siméon, T. Simulating ligand-induced conformational changes in proteins using a mechanical disassembly method. Phys. Chem. Chem. Phys. 2010, 12, 8268–8276. [Google Scholar] [CrossRef]
- Lüking, M.; Elf, J.; Levy, Y. Conformational Change of Transcription Factors from Search to Specific Binding: A lac Repressor Case Study. J. Phys. Chem. B 2022, 126, 9971–9984. [Google Scholar] [CrossRef]
- Galanti, M.; Fanelli, D.; Piazza, F. Conformation-controlled binding kinetics of antibodies. Sci. Rep. 2016, 6, 18976. [Google Scholar] [CrossRef]
- Reva, M.; DiGregorio, D.A.; Grebenkov, D.S. A first-passage approach to diffusion-influenced reversible binding: Insights into nanoscale signaling at the presynapse. Sci. Rep. 2021, 11, 5377. [Google Scholar] [CrossRef]
- Bénichou, O.; Moreau, M.; Oshanin, G. Kinetics of stochastically gated diffusion-limited reactions and geometry of random walk trajectories. Phys. Rev. E 2000, 61, 3388–3406. [Google Scholar] [CrossRef] [PubMed]
- Reingruber, J.; Holcman, D. Gated Narrow Escape Time for Molecular Signaling. Phys. Rev. Lett. 2009, 103, 148102. [Google Scholar] [CrossRef] [PubMed]
- Lawley, S.D.; Keener, J.P. A new derivation of Robin boundary conditions through homogenization of a stochastically switching boundary. SIAM J. Appl. Dyn. Syst. 2015, 14, 1845–1867. [Google Scholar] [CrossRef]
- Zhou, H.-X.; Zwanzig, R. A rate process with an entropy barrier. J. Chem. Phys. 1991, 94, 6147–6152. [Google Scholar] [CrossRef]
- Reguera, D.; Schmid, G.; Burada, P.S.; Rubí, J.-M.; Reimann, P.; Hänggi, P. Entropic Transport: Kinetics, Scaling, and Control Mechanisms. Phys. Rev. Lett. 2006, 96, 130603. [Google Scholar] [CrossRef]
- Chapman, S.J.; Erban, R.; Isaacson, S. Reactive boundary conditions as limits of interaction potentials for Brownian and Langevin dynamics. SIAM J. Appl. Math. 2016, 76, 368–390. [Google Scholar] [CrossRef]
- Berg, H.C.; Purcell, E.M. Physics of chemoreception. Biophys. J. 1977, 20, 193–219. [Google Scholar] [CrossRef]
- Berezhkovskii, A.M.; Makhnovskii, Y.A.; Monine, M.I.; Zitserman, V.Y.; Shvartsman, S.Y. Boundary homogenization for trapping by patchy surfaces. J. Chem. Phys. 2004, 121, 11390–11394. [Google Scholar] [CrossRef]
- Berezhkovskii, A.M.; Monine, M.I.; Muratov, C.B.; Shvartsman, S.Y. Homogenization of boundary conditions for surfaces with regular arrays of traps. J. Chem. Phys. 2006, 124, 036103. [Google Scholar] [CrossRef]
- Muratov, C.B.; Shvartsman, S.Y. Boundary homogenization for periodic arrays of absorbers. Multiscale Model. Simul. 2008, 7, 44–61. [Google Scholar] [CrossRef]
- Bernoff, A.; Lindsay, A.; Schmidt, D. Boundary homogenization and capture time distributions of semipermeable membranes with periodic patterns of reactive sites. Multiscale Model. Simul. 2018, 16, 1411–1447. [Google Scholar] [CrossRef]
- Punia, B.; Chaudhury, S.; Kolomeisky, A.B. Understanding the Reaction Dynamics on Heterogeneous Catalysts Using a Simple Stochastic Approach. J. Phys. Chem. Lett. 2021, 12, 11802–11810. [Google Scholar] [CrossRef]
- Sapoval, B. General Formulation of Laplacian Transfer Across Irregular Surfaces. Phys. Rev. Lett. 1994, 73, 3314–3316. [Google Scholar] [CrossRef] [PubMed]
- Sapoval, B. Transport Across Irregular Interfaces: Fractal Electrodes, Membranes and Catalysts. In Fractals and Disordered Systems; Bunde, A., Havlin, S., Eds.; Springer: Berlin, Germany, 1996; pp. 233–261. [Google Scholar]
- Grebenkov, D.S.; Filoche, M.; Sapoval, B. Mathematical Basis for a General Theory of Laplacian Transport towards Irregular Interfaces. Phys. Rev. E 2006, 73, 021103. [Google Scholar] [CrossRef] [PubMed]
- Powles, J.G.; Mallett, M.J.D.; Rickayzen, G.; Evans, W.A.B. Exact analytic solutions for diffusion impeded by an infinite array of partially permeable barriers. Proc. R. Soc. Lond. A 1992, 436, 391–403. [Google Scholar]
- Bressloff, P.C.; Earnshaw, B.A.; Ward, M.J. Diffusion of protein receptors on a cylindrical dendritic membrane with partially absorbing traps. SIAM J. Appl. Math. 2008, 68, 1223–1246. [Google Scholar] [CrossRef][Green Version]
- Grebenkov, D.S.; Oshanin, G. Diffusive escape through a narrow opening: New insights into a classic problem. Phys. Chem. Chem. Phys. 2017, 19, 2723–2739. [Google Scholar] [CrossRef]
- Guérin, T.; Dolgushev, M.; Bénichou, O.; Voituriez, R. Universal kinetics of imperfect reactions in confinement. Commun. Chem. 2021, 4, 157. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, C.W. Handbook of Stochastic Methods for Physics, Chemistry and the Natural Sciences; Springer: Berlin, Germany, 1985. [Google Scholar]
- Risken, H. The Fokker-Planck Equation: Methods of Solution and Applications, 3rd ed.; Springer: Berlin, Germany, 1996. [Google Scholar]
- Van Kampen, N.G. Stochastic Processes in Physics and Chemistry; Elsevier: Amsterdam, The Netherlands, 1992. [Google Scholar]
- Schuss, Z. Brownian Dynamics at Boundaries and Interfaces in Physics, Chemistry and Biology; Springer: New York, NY, USA, 2013. [Google Scholar]
- Metzler, R.; Klafter, J. The random walk’s guide to anomalous diffusion: A fractional dynamics approach. Phys. Rep. 2000, 339, 1–77. [Google Scholar] [CrossRef]
- Metzler, R.; Klafter, J. The restaurant at the end of the random walk: Recent developments in the description of anomalous transport by fractional dynamics. J. Phys. A Math. Gen. 2004, 37, R161–R208. [Google Scholar] [CrossRef]
- Sokolov, I.M. Models of anomalous diffusion in crowded environments. Soft Matter 2012, 8, 9043–9052. [Google Scholar] [CrossRef]
- Krapf, D.; Metzler, R. Strange interfacial molecular dynamics. Phys. Today 2019, 72, 48–54. [Google Scholar] [CrossRef]
- Chubynsky, M.V.; Slater, G.W. Diffusing Diffusivity: A Model for Anomalous, yet Brownian, Diffusion. Phys. Rev. Lett. 2014, 113, 098302. [Google Scholar] [CrossRef]
- Chechkin, A.V.; Seno, F.; Metzler, R.; Sokolov, I.M. Brownian yet Non-Gaussian Diffusion: From Superstatistics to Subordination of Diffusing Diffusivities. Phys. Rev. X 2017, 7, 021002. [Google Scholar] [CrossRef]
- Lanoiselée, Y.; Moutal, N.; Grebenkov, D.S. Diffusion-limited reactions in dynamic heterogeneous media. Nat. Commun. 2018, 9, 4398. [Google Scholar] [CrossRef]
- Grebenkov, D.S. A unifying approach to first-passage time distributions in diffusing diffusivity and switching diffusion models. J. Phys. A Math. Theor. 2019, 52, 174001. [Google Scholar] [CrossRef]
- Yuste, S.B.; Abad, E.; Lindenberg, K. Exploration and Trapping of Mortal Random Walkers. Phys. Rev. Lett. 2013, 110, 220603. [Google Scholar] [CrossRef] [PubMed]
- Meerson, B.; Redner, S. Mortality, Redundancy, and Diversity in Stochastic Search. Phys. Rev. Lett. 2015, 114, 198101. [Google Scholar] [CrossRef] [PubMed]
- Grebenkov, D.S.; Rupprecht, J.-F. The escape problem for mortal walkers. J. Chem. Phys. 2017, 146, 084106. [Google Scholar] [CrossRef]
- Turing, A. The Chemical Basis of Morphogenesis. Phil. Trans. R. Soc. Lond. B 1952, 237, 37–72. [Google Scholar]
- Milo, R.; Rhillips, R. Cell Biology by the Numbers; Garland Science: New York, NY, USA, 2015. [Google Scholar]
- Redner, S. A Guide to First Passage Processes; Cambridge University Press: Cambridge, UK, 2001. [Google Scholar]
- Metzler, R.; Oshanin, G.; Redner, S. (Eds.) First-Passage Phenomena and Their Applications; World Scientific: Singapore, 2014. [Google Scholar]
- Yu, J.; Xiao, J.; Ren, X.; Lao, K.; Xie, X.S. Probing Gene Expression in Live Cells, One Protein Molecule at a Time. Science 2006, 311, 1600–1603. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; van Oudenaarden, A. Nature, Nurture, or Chance: Stochastic Gene Expression and Its Consequences. Cell 2008, 135, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.S.; Choi, P.J.; Li, G.-W.; Lee, N.K.; Lia, G. Single-Molecule Approach to Molecular Biology in Living Bacterial Cells. Annu. Rev. Biophys. 2008, 37, 417–444. [Google Scholar] [CrossRef]
- Li, G.-W.; Xie, X.S. Central dogma at the single-molecule level in living cells. Nature 2011, 475, 308–315. [Google Scholar] [CrossRef]
- Kastantin, M.; Walder, R.; Schwartz, D.K. Identifying Mechanisms of Interfacial Dynamics Using Single-Molecule Tracking. Langmuir 2012, 28, 12443–12456. [Google Scholar] [CrossRef]
- Wang, D.; Wu, H.; Schwartz, D.K. Three-Dimensional Tracking of Interfacial Hopping Diffusion. Phys. Rev. Lett. 2017, 119, 268001. [Google Scholar] [CrossRef] [PubMed]
- Norregaard, K.; Metzler, R.; Ritter, C.M.; Berg-Sørensen, K.; Oddershede, L.B. Manipulation and Motion of Organelles and Single Molecules in Living Cells. Chem. Rev. 2017, 117, 4342–4374. [Google Scholar] [CrossRef]
- Sungkaworn, T.; Jobin, M.-L.; Burnecki, K.; Weron, A.; Lohse, M.-J.; Calebiro, D. Single-molecule imaging reveals receptor-G protein interactions at cell surface hot spots. Nature 2017, 550, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Grebenkov, D.S.; Metzler, R.; Oshanin, G. Strong defocusing of molecular reaction times results from an interplay of geometry and reaction control. Commun. Chem. 2018, 1, 96. [Google Scholar] [CrossRef]
- Grebenkov, D.S.; Metzler, R.; Oshanin, G. Towards a full quantitative description of single-molecule reaction kinetics in biological cells. Phys. Chem. Chem. Phys. 2018, 20, 16393–16401. [Google Scholar] [CrossRef]
- Elf, J.; Barkefors, I. Single-molecule kinetics in living cells. Ann. Rev. Biochem. 2019, 88, 635–659. [Google Scholar] [CrossRef]
- Grebenkov, D.S. Spectral theory of imperfect diffusion-controlled reactions on heterogeneous catalytic surfaces. J. Chem. Phys. 2019, 151, 104108. [Google Scholar] [CrossRef]
- Goodrich, F.C. Random walk with semi-adsorbing barrier. J. Chem. Phys. 1954, 22, 588–594. [Google Scholar] [CrossRef]
- Mysels, K.J. Diffusion-controlled adsorption kinetics. General solution and some applications. J. Phys. Chem. 1982, 86, 4648–4651. [Google Scholar] [CrossRef]
- Agmon, N. Diffusion with back reaction. J. Chem. Phys. 1984, 81, 2811–2817. [Google Scholar] [CrossRef]
- Agmon, N.; Weiss, G.H. Theory of non-Markovian reversible dissociation reactions. J. Chem. Phys. 1989, 91, 6937–6942. [Google Scholar] [CrossRef]
- Agmon, N.; Szabo, A. Theory of reversible diffusion-influenced reactions. J. Chem. Phys. 1990, 92, 5270–5284. [Google Scholar] [CrossRef]
- Kim, H.; Shin, K.J. Exact solution of the reversible diffusion-influenced reaction for an isolated pair in three dimensions. Phys. Rev. Lett. 1999, 82, 1578–1581. [Google Scholar] [CrossRef]
- Prüstel, T.; Tachiya, M. Reversible diffusion-influenced reactions of an isolated pair on some two dimensional surfaces. J. Chem. Phys. 2013, 139, 194103. [Google Scholar] [CrossRef] [PubMed]
- Scher, Y.; Lauber Bonomo, O.; Pal, A.; Reuveni, S. Microscopic theory of adsorption kinetics. J. Chem. Phys. 2023, 158, 094107. [Google Scholar] [CrossRef] [PubMed]
- Grebenkov, D.S. Diffusion-controlled reactions with non-Markovian binding/unbinding kinetics. J. Chem. Phys. 2023, 158, 214111. [Google Scholar] [CrossRef]
- Chechkin, A.V.; Zaid, I.M.; Lomholt, M.; Sokolov, I.M.; Metzler, R. Bulk-mediated surface diffusion along a cylinder: Propagators and crossovers. Phys. Rev. E 2009, 79, 040105. [Google Scholar] [CrossRef]
- Bénichou, O.; Grebenkov, D.S.; Levitz, P.E.; Loverdo, C.; Voituriez, R. Optimal Reaction Time for Surface-Mediated Diffusion. Phys. Rev. Lett. 2010, 105, 150606. [Google Scholar] [CrossRef]
- Bénichou, O.; Grebenkov, D.S.; Levitz, P.E.; Loverdo, C.; Voituriez, R. Mean first-passage time of surface-mediated diffusion in spherical domains. J. Stat. Phys. 2011, 142, 657–685. [Google Scholar] [CrossRef][Green Version]
- Rojo, F.; Budde, C.E. Enhanced diffusion through surface excursion: A master-equation approach to the narrow-escape-time problem. Phys. Rev. E 2011, 84, 021117. [Google Scholar] [CrossRef]
- Chechkin, A.V.; Zaid, I.M.; Lomholt, M.A.; Sokolov, I.M.; Metzler, R. Effective surface motion on a reactive cylinder of particles that perform intermittent bulk diffusion. J. Chem. Phys. 2011, 134, 204116. [Google Scholar] [CrossRef]
- Chechkin, A.V.; Zaid, I.M.; Lomholt, M.; Sokolov, I.M.; Metzler, R. Bulk-mediated diffusion on a planar surface: Full solution. Phys. Rev. E 2012, 86, 041101. [Google Scholar] [CrossRef]
- Rupprecht, J.-F.; Bénichou, O.; Grebenkov, D.S.; Voituriez, R. Exact mean exit time for surface-mediated diffusion. Phys. Rev. E 2012, 86, 041135. [Google Scholar] [CrossRef]
- Rupprecht, J.-F.; Bénichou, O.; Grebenkov, D.S.; Voituriez, R. Kinetics of active surface-mediated diffusion in spherically symmetric domains. J. Stat. Phys. 2012, 147, 891–918. [Google Scholar] [CrossRef][Green Version]
- Berezhkovskii, A.M.; Dagdug, L.; Bezrukov, S.M. A new approach to the problem of bulk-mediated surface diffusion. J. Chem. Phys. 2015, 143, 084103. [Google Scholar] [CrossRef]
- Berezhkovskii, A.M.; Dagdug, L.; Bezrukov, S.M. Bulk-mediated surface transport in the presence of bias. J. Chem. Phys. 2017, 147, 014103. [Google Scholar] [CrossRef] [PubMed]
- Bénichou, O.; Loverdo, C.; Moreau, M.; Voituriez, R. Intermittent search strategies. Rev. Mod. Phys. 2011, 83, 81–129. [Google Scholar] [CrossRef]
- Filoche, M.; Sapoval, B.; Andrade, J.S., Jr. Deactivation Dynamics of Rough Catalytic Surfaces. AIChE 2005, 51, 998–1008. [Google Scholar] [CrossRef]
- Filoche, M.; Grebenkov, D.S.; Andrade, A.S., Jr.; Sapoval, B. Passivation of irregular surfaces accessed by diffusion. Proc. Natl. Acad. Sci. USA 2008, 105, 7636–7640. [Google Scholar] [CrossRef]
- Grebenkov, D.S. Paradigm Shift in Diffusion-Mediated Surface Phenomena. Phys. Rev. Lett. 2020, 125, 078102. [Google Scholar] [CrossRef]
- Grebenkov, D.S. Residence times and other functionals of reflected Brownian motion. Phys. Rev. E 2007, 76, 041139. [Google Scholar] [CrossRef]
- Grebenkov, D.S. Probability distribution of the boundary local time of reflected Brownian motion in Euclidean domains. Phys. Rev. E 2019, 100, 062110. [Google Scholar] [CrossRef]
- Grebenkov, D.S. Joint distribution of multiple boundary local times and related first-passage time problems with multiple targets. J. Stat. Mech. 2020, 103205. [Google Scholar] [CrossRef]
- Grebenkov, D.S. Surface Hopping Propagator: An Alternative Approach to Diffusion-Influenced Reactions. Phys. Rev. E 2020, 102, 032125. [Google Scholar] [CrossRef]
- Grebenkov, D.S. Statistics of boundary encounters by a particle diffusing outside a compact planar domain. J. Phys. A Math. Theor. 2021, 54, 015003. [Google Scholar] [CrossRef]
- Bressloff, P.C. Narrow capture problem: An encounter-based approach to partially reactive targets. Phys. Rev. E 2022, 105, 034141. [Google Scholar] [CrossRef] [PubMed]
- Grebenkov, D.S. Statistics of diffusive encounters with a small target: Three complementary approaches. J. Stat. Mech. 2022, 083205. [Google Scholar] [CrossRef]
- Levitin, M.; Mangoubi, D.; Polterovich, I. Topics in Spectral Geometry. Preliminary Version. Available online: https://bookstore.ams.org/view?ProductCode=GSM/237 (accessed on 29 May 2023).
- Grebenkov, D.S. An encounter-based approach for restricted diffusion with a gradient drift. J. Phys. A Math. Theor. 2022, 55, 045203. [Google Scholar] [CrossRef]
- Evans, M.R.; Majumdar, S.N. Diffusion with Stochastic Resetting. Phys. Rev. Lett. 2011, 106, 160601. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.R.; Majumdar, S.N.; Schehr, G. Stochastic resetting and applications. J. Phys. A Math. Theor. 2020, 53, 193001. [Google Scholar] [CrossRef]
- Bressloff, P.C. Diffusion-mediated surface reactions and stochastic resetting. J. Phys. A Math. Theor. 2022, 55, 275002. [Google Scholar] [CrossRef]
- Benkhadaj, Z.; Grebenkov, D.S. Encounter-based approach to diffusion with resetting. Phys. Rev. E 2022, 106, 044121. [Google Scholar] [CrossRef]
- Grebenkov, D.S. Depletion of Resources by a Population of Diffusing Species. Phys. Rev. E 2022, 105, 054402. [Google Scholar] [CrossRef] [PubMed]
- Grebenkov, D.S. Encounter-based approach to the escape problem. Phys. Rev. E 2023, 107, 044105. [Google Scholar] [CrossRef] [PubMed]
- Bressloff, P.C. A probabilistic model of diffusion through a semipermeable barrier. Proc. R. Soc. A 2022, 478, 20220615. [Google Scholar] [CrossRef]
- Bressloff, P.C. Renewal equation for single-particle diffusion through a semipermeable interface. Phys. Rev. E. 2023, 107, 014110. [Google Scholar] [CrossRef]
- Bressloff, P.C. Renewal equations for single-particle diffusion in multilayered media. SIAM J. Appl. Math. 2023, 83, 1518–1545. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grebenkov, D.S. Diffusion-Controlled Reactions: An Overview. Molecules 2023, 28, 7570. https://doi.org/10.3390/molecules28227570
Grebenkov DS. Diffusion-Controlled Reactions: An Overview. Molecules. 2023; 28(22):7570. https://doi.org/10.3390/molecules28227570
Chicago/Turabian StyleGrebenkov, Denis S. 2023. "Diffusion-Controlled Reactions: An Overview" Molecules 28, no. 22: 7570. https://doi.org/10.3390/molecules28227570
APA StyleGrebenkov, D. S. (2023). Diffusion-Controlled Reactions: An Overview. Molecules, 28(22), 7570. https://doi.org/10.3390/molecules28227570