Abstract
The dynamic scenario of di-aryls-pyrano-chromenes was investigated using DFT calculations. The symmetry of the chromene scaffold and the presence of two ortho-substituted aryls substituents can generate two syn/anti diastereoisomers and conformational enantiomers with different rotational barriers. The relative conformations and configurations were derived using NOESY-1D experiments. Depending on the energies related to the conformational exchange, the experimental energy barriers were determined through Dynamic NMR, Dynamic HPLC or kinetic studies. The atropisomeric pairs were resolved in the latter scenario, and their absolute configuration was assigned using the ECD/TD-DFT method.
1. Introduction
In recent years, atropisomeric compounds have received significant attention due to their numerous applications, and the synthesis of new entities is a rapidly growing research area [1,2,3]. They are often involved in pharmaceuticals, are present in bioactive natural products [4,5,6] and are frequently used as chiral ligands in asymmetric synthesis [7,8,9,10,11,12,13]. The most well-known and perhaps explored atropisomers are biaryl systems, spiranes and compounds with a Csp2-N [14,15,16,17] or Csp2-B [18,19] axis bearing at least one chiral axis. Interestingly, when two stereogenic axes and sufficiently hindered substituents around the chiral axis are present, molecules with a potential “cleft” structure could be prepared [20,21,22,23,24]. In these cases, a deeper investigation concerning the rotational energy barriers of each axis could provide valuable support in developing rigid atropisomeric clefts with potential applications in molecular recognition, such as chiral catalysts or molecular machines [25,26].
Recently, Fochetti et al. reported the synthesis of 4,6-diphenyl-2H,8H-pyrano [3,2-g]chromene (hereafter referred to as 4,6-diaryls-pyrano-chromene 1) and 4,10-diphenyl-2H,8H-pyrano[2,3-f]chromene (hereafter referred to as 4,10-diaryls-pyrano-chromene 2) through a gold (I)-catalyzed intramolecular hydroarylation reaction (IMHA), which is compatible with different functional groups on the aryls [27]. In this paper, we reported the possibility of introducing two chiral axes by increasing the steric hindrance of the substituent in the ortho positions of the aryls (Scheme 1). The aryls are indeed skewed with respect to the dynamically planar scaffold of chromene, and they could be driven to be almost perpendicular to it by raising the steric hindrance in the ortho position. In this conformation, we formed two stereogenic axes that, in principle, generate two diastereoisomers (syn/anti) with four conformations for the 4,10-diaryls-pyrano-chromene derivatives 2 and three conformations in the case of 4,6-diaryls-pyrano-chromene derivative 1, due to the Cs symmetry of the syn stereoisomer.
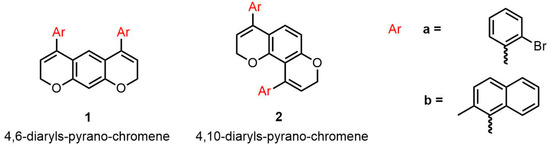
Scheme 1.
Chemical Structures of the two classes of compounds 1 and 2.
The resulting diastereomeric conformations can be either stereolabile or configurationally stable. Herein, we explored the dynamic scenario of di-aryls-pyrano-chromenes using DFT calculations, and depending on the syn/anti conformational exchange, we were able to determine the experimental free energy barriers through Dynamic NMR, Dynamic HPLC or Kinetic studies. Moreover, when the interconversion barriers were high enough, we isolated the atropisomeric compounds and assigned their absolute configuration using the ECD/TD-DFT method.
2. Results and Discussion
2.1. Synthesis
The synthesis of compounds 1a and 2a was obtained, as reported by some of the authors [27]. 1,3-bis((3-bromo-prop-2-yn-1-yl)oxy)benzene was achieved from resorcinol and propargyl bromide, followed by Sonogashira’s cross-coupling with 2-bromoiodobenzene. Intramolecular hydroarylation in the presence of a gold catalyst yielded the two regioisomers (“linear” 1a and “bent” 2a) in a 70:30 ratio. For compounds 1b/2b, the developed protocol required a slight modification, introducing the triple bond into the aryl moiety (in this case, 2-methyl naphthalene), followed by the reaction with resorcinol, as reported in Scheme 2. 1-(3-bromoprop-1-yn-1-yl)-2-methylnaphthalene 4 was obtained in four steps using 1-bromo-2-methylnaphthalene 3 as the starting material. Subsequent alkylation of resorcinol, performed with K2CO3 in DMF at room temperature, gave the intermediate 1,3-bis((3-(2-methylnaphthalen-1-yl)prop-2-yn-1-yl)oxy)benzene, 5 [27,28]. The intramolecular hydroarylation (IMHA) of compound 5 in the presence of a gold catalyst yielded compounds 1b and 2b with an 87/13 ratio (Scheme 2).
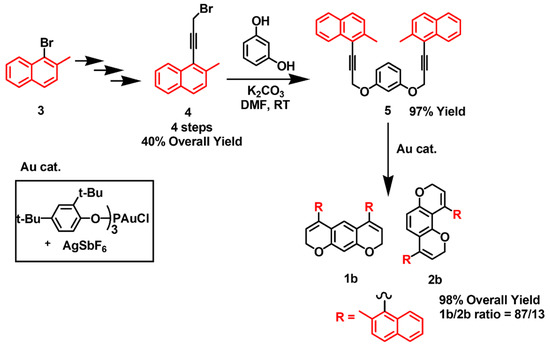
Scheme 2.
Synthesis of 1b and 2b were shown.
2.2. DFT Calculations
The investigation of atropisomer stability was carried out by DFT calculations, and the results were supported by HPLC and/or NMR at variable-temperature (dynamic-HPLC and dynamic-NMR). As a general remark, the DFT calculations assume that two diastereoisomers for each compound, syn/anti, are generated by the restricted Csp2-aryl rotation. In terms of stereoisomers, compound 1 yields only three stereoisomers due to the two anti-enantiomeric conformations (C2 point group) and the meso/syn conformation (Cs point group), while compounds 2, with an asymmetric pyrano-chromene scaffold, yield four stereoisomers (as shown in Scheme 3).
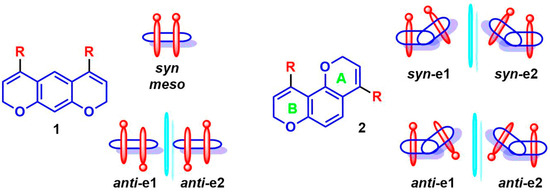
Scheme 3.
Schematic representation of the syn/anti conformations of compounds 1 and 2.
The DFT study began with the ortho-bromide phenyl substituted pyrano-chromenes (1a and 2a), then moved on to the more sterically hindered compounds (1b and 2b). As in similar systems, the activation entropy was determined to be negligible [23,24], and the ZPE-corrected enthalpy was considered in the calculation of rotational barriers and was consistently in agreement with the experimental barriers. The free energy term as is, or after frequency cut-off at 100 cm−1 (Table S6 in SI) [29] and also including empirical dispersion as B3LYP-D3 (Table S7 in SI), did not fit with the experimental data. All the ground states (GS) and transition states (TS) geometries for compounds 1b, 2a and 2b are reported in the Supporting Information. The syn/anti calculated conformations of compound 1a at the B3LYP/6-311++G(d,p) level [30], including chloroform as solvent (IEF-PCM approach) [31,32], are displayed in Figure 1, and Table 1 reports all the descriptors identifying the different conformations.
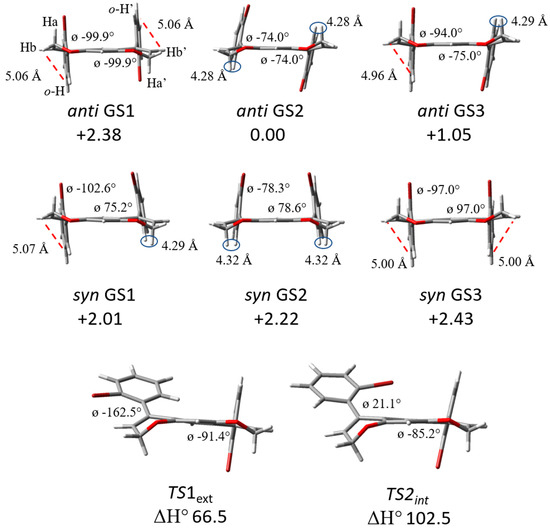
Figure 1.
Predicted DFT conformations of syn/anti for compounds 1a are shown. The relative energies are reported in kJ/mol.

Table 1.
Descriptors are shown r = atom distance; ø = dihedral angle.
Six conformations were found for compound 1a, depending on the flexibility of the pyrano-chromene scaffold. Looking towards it, the oxygens could be considered in the plane, while the CH2 of pyrano-chromene are out of the plane and could be either towards or away from the bromine atoms, generating three conformations for both anti and syn diastereoisomers. The syn/anti interconversion occurs through the transition state TS1ext where the aryl group rotates with the bromine outside the pyrano-chromene plane. The calculated enthalpy energy barrier was 66.5 kJ/mol (15.9 kcal/mol) for compound 1a.
Similar conformations were found in compound 1b (Figure S14), where the steric hindrance of the 2-methyl-naphthyl group raised the energy barrier to 130.5 kJ/mol (31.2 kcal/mol). The TS2EXT shows a highly distorted conformation where the ortho-methyl is outside the plane while the naphthyl ring is inside. This high barrier implies that the two syn/anti configurations could be isolated at room temperature.
For compounds 2a and 2b, the corresponding anti-to-syn interconversion barrier is due to the rotation of the aryl ring with the lower barrier (Figures S6 and S15). In contrast, the higher energy barrier leads to racemization. To distinguish the two aryl groups, in Scheme 3 and Figure S6, the pyrano-chromene scaffold and the corresponding bonded aryl group were labelled as A and B, respectively. The asymmetry of the scaffold generated four transition states, of which only two are effective. Focusing on compound 2a, the aryls could rotate with the bromine inside the plane (named INT) or outside the plane (named EXT), while the other aryl group does not turn (named FIX). In detail, the transition state with the lowest energy barrier was found to be TS2A-EXT-B-FIX (65.3 kJ/mol or 15.6 kcal/mol). This barrier interconverts the syn into anti-diastereoisomers (hereafter referred to as the diastereomerization barrier). The second aryl group can rotate with a calculated energy of 70.7 kJ/mol (16.9 kcal/mol, TS3A-FIX-B-EXT). This barrier interconverts the enantiomers (hereafter referred to as the enantiomerization barrier).
Compared to compound 1b, compound 2b has diastereomerization and enantiomerization barriers higher than 125.5 kJ/mol (30 kcal/mol). Therefore, their separation should be easily achieved by chromatographic techniques.
A comprehensive comparison of calculated vs experimental data (population of conformers and rotational barriers) can be found at the bottom of the text (Table 2 and Table 3).
Table 2.
Summary of the calculated (IEF-PCM) vs experimental populations for the syn/anti diasteroisomers derived from 1H NMR.
Table 3.
Summary of the calculated (IEF-PCM) vs experimental barriers for compounds 1 and 2. The calculated barriers are reported as enthalpies (kJ/mol). For compounds 2a, 2b, the lower barrier represents the diasteromerization process, while the higher one represents the enantiomerization.
2.3. Dynamic NMR
Due to the low energy calculated for the diastereomerization process (TS1ext = 66.5 kJ/mol for 1a and TS2A-EXT-B-FIX = 65.3 kJ/mol for 2a), variable-temperature NMR (D-NMR) is the most appropriate technique to confirm these predicted values experimentally. The flexibility of the oxygen bridges in the chromene moieties makes the ring inversion very fast and averaged over the NMR time scale. The chromene scaffold can be dynamically planar at any temperature reachable by the D-NMR technique. After HPLC separation of the 1a/2a mixture, the linear 1a and bent 2a were subjected to variable-temperature 1H NMR investigation.
For compound 1a, the alkenyl 1H NMR region showed broadened signals at +26 °C (Figure 2). The 1H NMR signal of the aromatic CH between the two oxygens (H-10) splits into two singlets at −12 °C. Similarly, the alkenyl signal splits into two triplets. At this temperature, the aryl-chromene rotational barrier is frozen in the NMR time scale, showing anisochronous signals corresponding to the two syn/anti diastereoisomers.
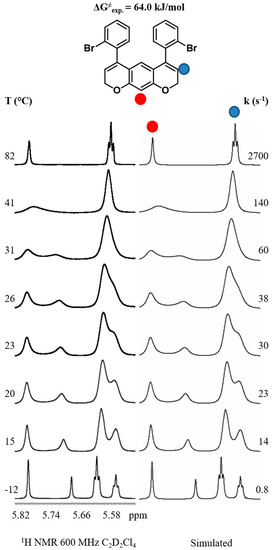
Figure 2.
Simulated and experimental dynamic 1H NMR (600 MHz) spectra in C2D2Cl4 of compound 1a. For each temperature (left column), the kinetic constant was determined by full-line shape simulation (right column).
Albeit, for different reasons, the two methylene hydrogens for diastereoisomers have different magnetic environments and are diastereomeric. This means they do not help detect which signals belong to the chiral anti-diastereoisomer. However, the assignment of the anti-chiral conformation was achieved by acquiring the 1H NMR spectrum in a chiral environment (Figure 3), by adding (R)-2,2,2-trifluoro-1-(9-anthryl)-ethanol (R-TFAE) [33] to a solution of compound 1a in CD2Cl2 at −20 °C (18.2:1 molar ratio R-TFAE:1a). Only the signal of H-10 of the anti-chiral isomer splits into a pair of diastereotopic singlets, corresponding to the M,M and P,P enantiomers, while the same hydrogen for the Cs-symmetric syn conformation does not further split. In this way, it was possible to determine the syn/anti populations as 44.2:55.8.
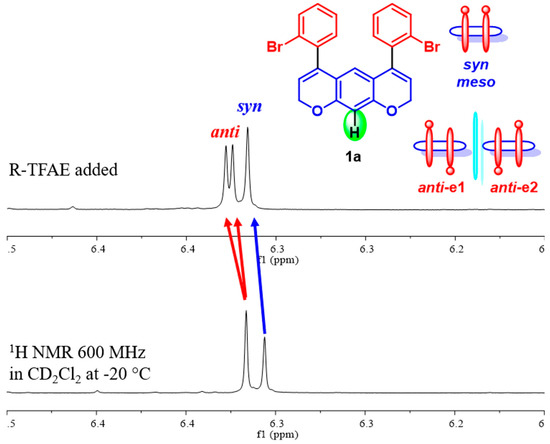
Figure 3.
Bottom: 1H NMR (600 MHz) spectra in CD2Cl2 of compound 1a at −20 °C. Top: spectrum after the addition of R-TFAE (18.2 eq).
When the temperature is raised, the alkenyl triplet and the single aromatic signals exhibit line broadening due to the syn/anti exchange. Above +82 °C, when the interconversion process becomes fast, these signals become narrow and well-defined triplet and singlet signals, respectively. By line shape simulations of the spectra at different temperatures and using the Eyring equation, a ΔG≠ value of 64.0 kJ/mol (15.3 kcal/mol) is obtained, corresponding to the barrier for the interconversion of the anti into the syn conformer. Due to the symmetrical scaffold of 1a, the syn/anti exchange can be achieved by rotating a single aryl ring. Thus, to derive the barrier corresponding to the rotation of a single aryl ring, the values of the rate constants reported must be halved, yielding a barrier of 65.7 kJ/mol (15.7 kcal/mol) [23,24]. This value is in perfect agreement with the DFT calculated enthalpy energies (see Figure 1).
The D-NMR spectra for compound 2a showed a similar trend (see Figures S7 and S8 of Supporting Information). The vinylic 1H NMR signal at 5.77 ppm in C2D2Cl4 at each temperature was simplified from ABX to AB system by homodecoupling of the CH2. In this way, at +25 °C, two doublets, at 4.83 ppm and 4.70 ppm with a 2JAB = 14 Hz, were visible, and they split into two AB systems at −10 °C, indicating the presence of both anti and syn conformations. The experimental diastereomerization syn/anti-energy barrier has been found by DNMR simulations to be 65.3 kJ/mol (15.6 kcal/mol). However, in the case of 2a, both diastereoisomers are chiral, and it was not experimentally possible to assign the anti:syn ratio. DFT calculations suggest that the 2a-syn has a larger dipole moment with respect to 2a-anti. Therefore, the syn/anti ratio was evaluated in solvents with increasing polarity (CDCl3 vs CD3CN). The calculated syn/anti ratio of 39/61 (PCM: chloroform) agrees with the experimental 1HNMR ratio 38.5/64.5 recorded in CDCl3 at −10 °C. Again, the calculated syn/anti ratio of 44/56 (PCM: acetonitrile) agrees with the experimental 1HNMR ratio of 43.4/56.6 recorded in CD3CN. As expected, the syn population slightly increases in more polar solvents (Figure S9). Both spectra were recorded at −10 °C to have a better resolution for the signals since the diastereomerization barrier is frozen.
On raising the temperature above +25 °C, the AB system coalesced around +90 °C and became a singlet at +114 °C (Figure S7). This dynamic phenomenon is due to the more hindered B aryl group that exchanges the two conformational enantiomers still present when the syn/anti exchange is fast. Line shape simulation of the 1H NMR spectra allowed the determination of the enantiomerization energy barrier as 74.0 kJ/mol (17.7 kcal/mol).
2.4. Dynamic HPLC
Diastereomerization and enantiomerization processes for compounds 1a and 2a could, in principle, be detected using Dynamic CSP-HPLC at lower temperatures with respect to NMR. For compound 1a, we attempted dynamic-CSP-HPLC at low temperatures, down to −63 °C, but the amylose-based stationary phase did not provide valuable data. On the other hand, four broad peaks were observed for compound 2a at −63 °C, confirming the presence of four stereoisomers (Figure 4). However, the online chiro-optical detection at −63 °C was unfeasible, and the fast rotation at room temperature prevented the assignment of each enantiomer.
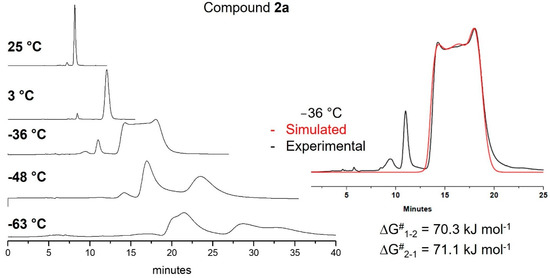
Figure 4.
Dynamic CSP-HPLC of 2a with simulated chromatogram at −36 °C.
As the temperature increased, the lowest barrier was exceeded, and the A-aryl group was free to rotate, separating two broad peaks at −48 °C. With further temperature increase, a chromatographic trace recorded at −36 °C showed a typical plateau between two peaks. When the B-aryl group was free to rotate, a single peak was detectable over +3 °C. The energy of the enantiomerization barrier (TS3A-FIX-B-EXT of Figure S6) was estimated to be 70.7 kJ/mol (16.90 kcal/mol) through simulation of the chromatogram trace [33,34,35,36]. Also, in this case, the calculated enthalpy energy barrier was in perfect agreement with the experimental data.
2.5. Separation and Characterization of Atropisomeric Compounds 1b and 2b
In the case of compounds 1b and 2b, the conformational features are similar to their corresponding 1a and 2a, respectively. However, the 2-Methylnaphthyl substituent significantly increases the rotational energy barrier [37,38,39]. The calculated energy barrier is higher than 30 kcal/mol, which is high enough to allow the separation of the syn/anti isomers and atropisomers resolution at room temperature.
For compound 1b, we were able to separate the syn/anti diasteroisomers using CSP-HPLC on (R,R)-Whelk-O1 5 μm column (150 × 4.6 mm L × ID) with n-hexane/dichloromethane 95/5 as eluent. The anti and syn diastereoisomers were assigned based on the opposite CD signals for the two anti-enantiomers, while the peak of the syn stereoisomers did not show any CD signal (Figure 5).
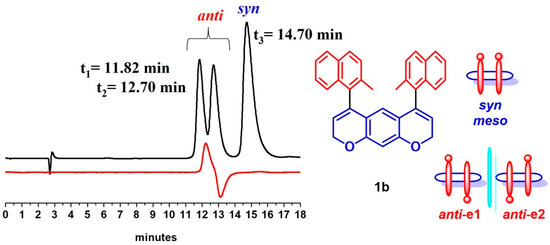
Figure 5.
CSP-HPLC separation on (R,R)-Whelk-O1 5 micron (150 × 4.6 mm L × ID) and using hexane/dichloromethane 95/5 as eluent. Flow 1.0 mL/min, detectors: UV 254 nm (black trace), CD 280 nm (red trace).
Compound 2b was resolved into the four available stereoisomers using CSP-HPLC on a Chiralpak IB-N5 5 μm column (250 × 10 mm L × ID) with n-hexane/chloroform 85/15 as eluent (Figure S26 in SI). After separation, 1H-NMR spectra of the four stereoisomers were acquired, and the syn/anti-assignment was unambiguously achieved using the double pulsed field gradient spin-echo NOE (DPFGSE-NOE) sequence [40,41,42,43]. In this case, signals corresponding to the ortho-methyl groups are the most useful for the NOE analysis. In the anti-configuration, the methyl groups are too far away to yield any NOE enhancement. For the second and the third eluted stereoisomers, besides having identical 1H NMR spectra, the signals, at 2.16 ppm and 2.38 ppm, respectively, did not show any NOE effect, attesting to their enantiomeric relationship of the anti-isomer. However, the first and fourth peaks obtained through CSP-HPLC separation showed an NOE effect elucidating that the methyl groups are on the same side as for the syn atropisomer. Specifically, when the methyl at 2.44 ppm for the syn atropisomer was irradiated, a slight NOE effect on the second methyl signal at 2.30 ppm was detected (Figure 6).
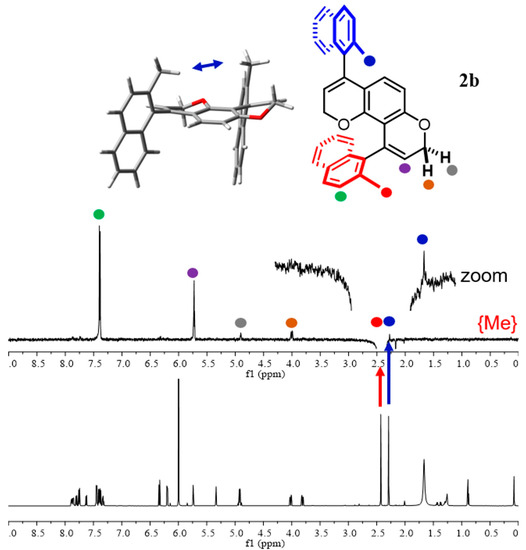
Figure 6.
Bottom: 1H NMR (600 MHz in C2D2Cl4) of the first eluted stereoisomer of 2b. Top: DPFGSE-NOE NMR by irradiating the singlet signal of methyl at 2.44 ppm. The 3D structure of M,M-syn 2b is also reported.
2.6. Kinetic Studies for Compounds 1b and 2b
Being very high in the energy barrier involved, rate constants of the syn/anti interconversion for compounds 1b and 2b were derived from kinetic studies using either HPLC or 1H NMR as the monitoring techniques. In detail, for the compound 1b, syn/anti-stereo-stability was investigated starting from the pure syn 1b in cis/trans-decaline solution and monitoring the appearance of anti 1b at different temperatures (+110 °C, +120 °C and +130 °C respectively, see Figures S18–S21 and details in SI). The diastereomerization processes were completed in almost 4 h, providing energy barriers of about 129.3 kJ/mol (30.9 kcal/mol), matching the prediction from DFT calculations. The ΔG# value results were slightly affected by the temperature attesting a low contribution of entropic term and confirmed by Eyring plot analysis, where the extrapolated ΔS was −12 u.e.
For compound 2b, the monitoring over the time of the 1H NMR methyl signal of the pure anti-isomer in a C2D2Cl4 solution kept at different temperatures allowed valid results (Figure S25 in SI). The methyl 1H NMR signal of anti-decreases while that one of the syn began to appear. After 48 h at 120 °C the equilibrium was reached with a syn/anti ratio of 50:50. Using a first-order kinetics equation for a process at equilibrium, we obtained the rate constants at different temperatures, hence the energy barrier (133.5 kJ/mol, 31.9 kcal/mol).
2.7. Assignment of Absolute Configuration for Compounds 1b and 2b
Compounds 1b and 2b do not contain heavy atoms in their structures, making it impossible to assign their absolute configuration (AC) using anomalous dispersion X-ray diffraction with Mo-Kα radiation. Instead, we used chiroptical properties, namely electronic circular dichroism spectroscopy (ECD), in synergy with time-dependent density functional theory (TD-DFT) [44]. For compound anti-1b, the ECD spectra recorded in acetonitrile of the first eluted CSP-HPLC (Figure 7) showed a small positive band at 280 nm, a large negative cotton effect at 235 nm and a large positive band at 215 nm due to the interactions between the dipoles of pyrano-chromene and the 2-methyl-naphthyl moieties (Figure S29). Being the three ground states for the anti-conformation very close in energy, the ECD spectrum for each ground state was calculated (Figure S22), and the weighted average was obtained considering the Boltzmann population (Figure 7). ECD simulations were obtained using four different functionals for redundancy and the 6-311++G(2d,p) basis set with acetonitrile as solvent (IEF-PCM approach) [31,32]. The calculated ECD for P,P showed a good agreement with the experimental spectrum of the second eluted CSP-HPLC; consequently, we assigned the M,M AC to the first eluted atropisomer anti-1b.
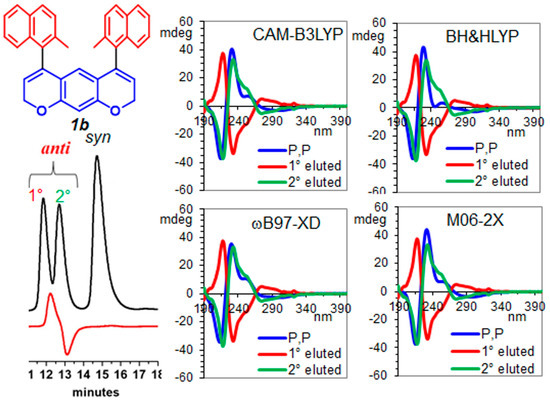
Figure 7.
Experimental ECD spectra for Compound anti-1b, recorded in ACN, are red lines (first eluted) and green lines (second eluted). Their comparison with the calculated spectra of P,P-anti-1b are shown (blue lines). To match the experimental spectra, the calculated spectra were red-shifted by 12 nm for CAM-B3LYP and M06-2X, 15 nm for BH&HLYP and 14 nm for ωB97x-D, and vertically scaled by 0.19 for CAM-B3LYP, 0.15 for BH&HLYP, 0.2 for ωB97x-D and 0.2 for M06-2X.
The relative configurations of compound 2b were assigned by the NOE effect (see Figure 5); subsequently, the AC was assigned in the same way as compound 1b. The details were reported in the supporting information. The syn PA,PB AC was assigned to the first eluted, the syn MA,MB AC to the fourth, while the anti PA,MB AC was assigned to the second eluted and the anti MA,PB AC to the third (Figures S27, S28, S30 and S31 for Mos involved in the UV transitions).
3. Materials and Methods
3.1. Materials
All reagents and solvents, HPLC and ACS grade, were purchased from Sigma Aldrich (Milan, Italy). HPLC gradient grade solvents were filtered on 0.45 μm Omnipore filters (Merck Millipore, Darmstadt, Germany) before use. Analytical-grade solvents and commercially available reagents were used as received. Deuterated solvents (CDCl3, C2D2Cl4, DMSO-d6) for NMR spectra were obtained from EurisoTop. The following stationary phases were employed for the chromatography: silica gel 60 Å F254 (Merck) for TLC and silica gel 60 Å (230−400 mesh, Sigma-Aldrich) for atmospheric pressure chromatography. The glassware used in these reactions was placed in an oven at 70 °C for at least 3 h immediately before use.
3.2. Semipreparative HPLC
A Waters 600 HPLC pump, a Rheodyne7012 injector (loop of 1 mL) and a Waters 2487 UV detector (Waters, Milford, MA, USA) with a wavelength set at 254 nm were used to purify the products at semi-preparative level. The stationary phases employed are reported in the product characterizations.
3.3. HPLC System and Simulation of Dynamic Profile
HPLC analyses were performed on the Jasco HPLC system equipped with a PU-980 HPLC pump, a Rheodyne 7725i injector (loop of 20 µL), a 975 series UV detector and a 995-CD series detector. Chromatographic data were recorded and processed with Borwin software (Version 1.50, Jasco Europe, Italy). Variable low-temperature HPLC chromatograms were obtained on a Jasco HPLC by submerging the column in a dewar containing dry ice and acetone. The uncertainty in temperature measurements can be estimated as ±0.5 °C. Dynamic HPLC plots were simulated with the Auto-D-HPLC-Y2 K software based on a stochastic model. This software performs a line-shape simulation by the simplex algorithm to optimize chromatographic and kinetic parameters, thus obtaining the best agreement between the experimental and simulated dynamic profiles. The error in determining the activation-free energies can be estimated as ±0.2 kcal/mol.
3.4. HPLC Analysis
Sample 1a: unresolved at −63 °C.
Sample 2a was analyzed on Chiralpak-IA (250 × 4.6 mm, L × I.D.) with eluent n-hexane-dichloromethane 98/2 + 0.5% ethanol at 1.0 mL/min and 254 nm UV detection (sharp peak at 8.1 min and 25 °C). Chromatographic traces recorded at 25 °C, 3 °C, −36 °C, −48 °C and −63 °C.
Sample 1b was investigated on (R,R)-Whelk-O1 5µm column (150 × 4.6 mm, L × I.D.) with n-hexane-dichloromethane 95/5 at the flow rate of 1.0 mL/min, detectors UV at 254 nm and CD at 280 nm (retention time of 1b-(−)-anti 13.6 min, 1b-(+)-anti 14.8 min and 1b-syn 17.8 min). Semipreparative conditions: (S,S)-Whelk-O2 10µm (250 × 10 mm) with n-hexane-dichloromethane 95/5 at a 4.0 mL/min flow rate and 254 nm UV detection.
Sample 2b was analyzed on Chiralpak IB-N5 5 µm column (250 × 10 mm L × ID) with n-hexane/chloroform 85/15 as eluent.
3.5. Off-Line Kinetic Procedure
Compound 1b
An aliquot of a pure atropisomer was dissolved in 1 mL of cis/trans decalin (boiling point = +189 °C – +191 °C) using a test tube with a screw cap. Then the sample was heated in a thermostatic oil bath (±2 °C) at the desired temperature. Small samples were taken at different times directly in a syringe containing the mobile phase. The enantioselective HPLC then allowed the determination of the enantiomeric ratio.
Compound 2b
An aliquot of syn atropisomer was dissolved in 0.7 mL of C2D2Cl4 (boiling point = +146 °C) using an NMR tube. Then the sample was heated in a thermostatic oil bath (±2 °C) at the desired temperature. After cooling fast at ambient temperature, the 1H NMR was acquired at different times. The anti-atropisomer formation was monitored, and the integral of methyls was taken to calculate the ratio syn/anti. A first-order kinetic equation was then used to derive the rate constant for diastereomerization and, hence, the activation barrier using the Eyring equation.
3.6. NMR Experiments
NMR spectra were recorded using a spectrometer operating in a field of 14.4 T (600 MHz) for 1H, (151 MHz) for 13C. Chemical shifts are given in parts per million relative to the internal standard tetramethylsilane (1H and 13C) or relative to the residual peak of the solvents. The 151 MHz 13C spectra were acquired under proton decoupling conditions with a 36,000 Hz spectral width, 5.5 μs (60° tip angle) pulse width, 1 s acquisition time, and a 5 s delay time. The 13C signals were assigned by distortionless enhancement by polarization transfer spectra (DEPT 1.5).
Dynamic NMR. Temperature calibrations were performed using a digital thermometer and a Cu/Ni thermocouple in an NMR tube filled with 1,1,2,2-tetrachloroethane. The experimental conditions were kept as equal as possible with all subsequent work. The uncertainty in temperature measurements can be estimated as ±1 °C. Line shape simulations were performed using a PC version of the QCPE DNMR6 program. Electronic superimposition of the original spectrum and the simulated one enabled the determination of the most reliable rate constant. The reported values are relative to the exchange from the less populated to the more populated conformation. The rate constants afforded the free energy of activation ΔG# at each temperature by applying the Eyring equation. Within the experimental uncertainty due to the exact temperature determination, the activation energies were found to be invariant, thus implying a small activation entropy ΔS#.
3.7. DFT Calculations
DFT calculations: Ground state optimizations and transition states were obtained by DFT calculations performed by the Gaussian16 software suite [30] using standard parameters. Full optimization and frequency analysis for ground and transition states employed the B3LYP and the 6-31G(d) basis set. The IEFPCM approach was used to account for the solvent contribution. The analysis of the vibrational frequencies showed the absence of imaginary frequencies for the ground states and the presence of one imaginary frequency for each transition state. Visual inspection of the corresponding normal mode validated the identification of the transition states.
3.8. ECD Measurements
The ECD spectra of compounds 1b/2b were acquired in the 190–400 nm region using a JASCO J-810 spectropolarimeter in far-UV HPLC-grade acetonitrile solution. Concentration was about 1 × 10−4 M, tuned by dilution to have a maximum absorbance between 0.8 and 1 with a cell path of 0.2 cm. The spectra were obtained by the average of 6 scans at 50 nm∙min−1 scan rate.
3.9. Synthesis and Characterization
3.9.1. Synthesis of 1,3-bis((3-(2-methylnaphthalen-1-yl)prop-2-yn-1-yl)oxy)benzene 5
In a 50 mL round bottom flask equipped with a magnetic stirring bar, resorcinol (144.8 mg, 1.3 mmol, 1 equiv.) was dissolved in DMF (6 mL) at room temperature. Then K2CO3 (545 mg, 3.95 mmol, 3 equiv.) was added, and after 15 min 1-(3-bromoprop-1-yn-1-yl)-2-methylnaphthalene (750 mg, 2.89 mmol, 2.2 equiv.) was added to the mixture. The reaction was monitored by TLC until the disappearance of the starting material, then diluted with Et2O and washed with NaHSO4 (×2) and brine (×2). The organic extract was dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by chromatography on SiO2 (25–40 μm), eluting with a 98/2 (v/v) n-hexane/AcOEt mixture to obtain 589.0 mg (97% yield) of 1,3-bis((3-(2-methylnaphthalen-1-yl)prop-2-yn-1-yl)oxy)benzene 5.
5: pale yellow solid; 97% yield; 1H NMR (400 MHz) (CDCl3) δ 8.25 (d, J = 8.4 Hz, 2 H), 7.79 (d, J = 8.1 Hz, 2 H), 7.52 (t, J = 7.3 Hz, 2 H), 7.42 (t, J = 7.5 Hz, 2 H), 7.36 – 7.28 (m, 3 H), 6.97 (m, 1 H), 6.84 (dd, J1 = 8.2 Hz, J2 = 2.2 Hz, 2 H), 5.17 8s, 4 H), 2.60 8s, 6 H); 13C{1H} NMR (101 MHz) (CDCl3) δ 159.0 (q), 139.8 (q), 133.7 (q), 131.4 (q), 130.0 (CH), 128.5 (CH), 128.0 (CH), 127.9 (CH), 126.9 (CH), 125.8 (CH), 125.5 (CH), 118.3 (q), 108.3 (CH), 103.2 (CH), 57.0 (CH2), 21.3 (CH3).
3.9.2. Synthesis of 4,6-bis(2-bromophenyl)-2,9-dihydropyrano [4,3-g]chromene 1a and 4,10-bis(2-bromophenyl)-2H,8H-pyrano [2,3-f]chromene 2a
Compounds 1a and 2a were prepared as reported [24]. Linear and angulated 1a/2a were separated by semipreparative HPLC on silica. Column: Silica Adamas (250 × 10 mm ID), eluent Hex/DCM 50/50, flow: 1 mL/min, detector: UV 254 nm.
1a (mixture of stereoisomers): white solid; m.p. = 159–161 °C; IR (neat): 2925, 2837, 1676, 1576, 1488, 1427 cm−1; 1H-NMR (400.13 MHz, CDCl3, +25 °C): δ = 7.45 (m, 2 H), 7.19–7.16 (m, 3 H), 7.06 (dt, J1 = 7.6 Hz, J2 = 1.8 Hz, 3 H), 6.42 (s, 1 H), 5.80–5.74 (m, 1 H), 5.55 (s, 2 H), 4.95–4.88 (m, 4 H); 13C{1H} NMR (101 MHz, DMSO-d6, +80 °C): δ = 155.4, 138.7, 136.0, 132.6, 131.2, 129.1, 127.2, 123.7, 123.2, 118.5, 116.4, 104.0, 65.7; HRMS: m/z [M + H]+ calcd for C24H17Br2O2: 496.9569; found: 496.9565.
2a (mixture of stereoisomers): yellow oil; IR (neat): 2930, 2835, 1676, 1575, 1490, 1427 cm−1; 1H NMR (400 MHz) (CDCl3): δ = 7.55–7.47 (m, 2 H), 7.27–6.98 (m, 6 H), 6.36 (s, 2 H), 5.65 (bs, 1 H), 5.39 (bs, 1 H), 4.76–4.61 (m, 2 H), 4.42–4.33 (m, 1 H), 4.22–4.14 (m, 1 H); 13C{1H} NMR (101 MHz) (CDCl3): δ = 155.7, 150.6, 142.6, 139.4, 136.5, 135.3, 133.0, 132.0, 131.4, 130.0, 129.3, 128.3, 127.5, 127.0, 126.3, 123.8, 122.6, 122.2, 118.9, 117.8, 112.4, 109.3, 65.0, 64.9; HRMS: m/z [M + H]+ calcd for C24H17Br2O2: 496.9569; found: 496.9565.
3.9.3. Synthesis of 4,6-bis(2-methylnaphthalen-1-yl)-2H,8H-pyrano [3,2-g]chromene 1b and 4,10-bis(2-methylnaphthalen-1-yl)-2H,8H-pyrano [2,3-f]chromene 2b
In a 50 mL Carousel Tube Reactor, (Radely Discovery Technology) containing a magnetic stirring bar 1,3-bis((3-(2-methylnaphthalen-1-yl)prop-2-yn-1-yl)oxy)benzene (121 mg, 0.26 mmol, 1 equiv.) was dissolved in CH2Cl2 (2 mL) at room temperature. Then [tris(2,4-di-tert-butyl-phenyl)phosphite]gold(I) chloride (9.1 mg, 0.01 mmol, 0.04 equiv.) was added, followed by AgSbF6 (3.6 mg, 0.01 mmol, 0.04 equiv.). The mixture was allowed to stir for an hour, and then CH2Cl2 was evaporated under reduced pressure. The residue was purified by chromatography on SiO2 (25–40 μm), eluting with a 97/3 (v/v) n-hexane/AcOEt mixture to obtain a mixture of 4,6-bis(2-methylnaphthalen-1-yl)-2H,8H-pyrano [3,2-g]chromene 1b and 4,10-bis(2-methylnaphthalen-1-yl)-2H,8H-pyrano [2,3-f]chromene 2b in a ratio of 87/13. For product characterization, see the following sections.
Syn/Anti stereoisomers were separated by semipreparative HPLC on (S,S) Whelk-O2 10 micron (250 × 10 mm L × ID) by using hexane/dichloromethane 95/5 + 0.1% ethanol at a flow rate of 4.0 mL/min. Detector UV 254 nm. In the analytical version, the geometry of the column was 150 × 4.6 mm L × ID (chromatographic trace in Figure S1 (black trace). Anti and syn stereoisomers were assigned based on CD signals (see the red trace in the figure). In addition, changing chiral stationary phase (R,R)-Whlek-O1 5 micron (150 × 4.6 mm L × ID) and using hexane/dichloromethane 95/5 as eluent, the separation of both enantiomers of anti 1b was obtained (see Figure S2, at flow of1.0 mL/min, detectors: UV 254 nm, CD 280 nm). After separation, 1H-NMR spectra of two stereoisomers were acquired.
1b syn: 1H NMR (600 MHz) (CDCl3): δ 7.51 (d, J = 5.4 Hz, 2H), 7.44 (d, J = 5.5 Hz, 2H), 7.37 (d, J = 5.6 Hz, 2H), 7.16–7.13 (m, 2H), 7.06 (d, J = 5.6 Hz, 2H), 6.97–6.94 (m, 2H), 6.48 (s, 1H), 5.48 (t, J = 2.4 Hz, 2H), 5.16 (s, 1H), 5.00–4.94 (m, 4H), 2.13 (s, 6H); 13C{1H} NMR (150 MHz) (CDCl3): d (ppm) 155.4, 133.8, 133.0, 132.8, 131.9, 131.4, 127.7, 127.3, 127.1, 125.5, 125.1, 124.5, 123.0, 118.5, 117.1, 103.6, 65.9, 19.9.
1b anti: 1H NMR (600 MHz) (CDCl3): δ 7.68 (d, J = 5.3 z, 2H), 7.63 (d, J = 5.5 Hz, 2H), 7.46 (d, J = 5.6 Hz, 2H), 7.35–7.27 (m, 4H), 6.86 (d, J = 5.6 Hz, 2H), 6.48 (s, 1H), 5.48 (t, J = 2.4 Hz, 2H), 5.14 (s, 1H), 5.03–4.94 (m, 4H), 1.79 (s, 6H); 13C{1H} (150 MHz) (CDCl3): δ (ppm) 155.5, 133.7, 133.6, 132.6, 132.0, 131.7, 128.0, 127.6, 127.0, 125.6, 125.5, 124.5, 123.0, 118.5, 117.1, 103.6, 65.9, 19.6.
2b syn: 1H NMR (600 MHz) (C2D2Cl4): d (ppm) 7.81–7.76 (m, 2 H), 7.72 (d, J = 5.3 Hz, 1 H), 7.67 (d, J = 5.5 Hz, 2 H), 7.55 (d, J = 5.6 Hz, 1 H), 7.38–7.35 (m, 2 H), 7.33–7.29 (m, 3 H), 7.26–7.24 (m, 2 H), 6.26 (d, J = 5.6 Hz, 1 H), 6.12 (d, J = 5.6 Hz, 1 H), 5.66 (t, J = 2.7 Hz, 1 H), 5.26 (t, J = 2.5 Hz, 1 H), 4.87–4.81 (m, 2 H), 3.94 (dd, J1 = 9.8 Hz, J2 = 2.6 Hz, 1 H), 3.73 (dd, J1 = 9.8 Hz, J2 = 2.5 Hz, 1 H), 2.35 (s, 3 H), 2.21 (s, 3 H); 13C{1H} NMR (150 MHz) (C2D2Cl4): d (ppm) 155.6, 151.2, 137.4, 134.1, 134.0, 133.8, 132.7, 132.6, 132.5, 131.9, 131.8, 128.8, 128.6, 128.0, 127.9, 127.6, 126.6, 126.2, 126.0, 125.9, 125.6, 125.1, 124.6, 122.3, 119.7, 118.7, 113.4, 109.1, 65.3, 64.7, 20.9, 20.4.
2b anti: 1H NMR (600 MHz) (C2D2Cl4): δ 7.78–7.76 (m, 2 H), 7.73 (d, J = 5.0 Hz, 1 H), 7.67 (d, J = 5.6 Hz, 2 H), 7.62 (d, J = 5.5 Hz, 1 H), 7.36–7.28 (m, 6 H), 7.26–7.24 (m, 2 H), 6.26 (d, J = 5.6 Hz, 1 H), 6.12 (d, J = 5.6 Hz, 1 H), 5.66 (t, J = 2.7 Hz, 1 H), 5.25 (, t, J = 2.5 Hz, 1 H), 5.25 (s, 1 H), 4.87–4.81 (m, 2 H), 3.96 (dd, J1 = 9.8 Hz, J2 = 2.6 Hz, 1 H), 3.66 (dd, J1 = 9.8 Hz, J2 = 2.4 Hz, 1 H), 2.39 (s, 3 H), 2.16 (s, 3 H); 13C{1H} NMR (150 MHz) (C2D2Cl4): δ (ppm) 155.6, 151.2, 137.4, 134.2, 134.0, 133.8, 132.7, 132.63, 132.60, 132.5, 131.9, 131.8, 128.8, 128.7, 128.0, 127.9, 127.6, 126.6, 126.2, 126.0, 125.9, 125.8, 125.6, 125.1, 124.6, 122.3, 119.7, 118.7, 113.4, 109.1, 65.3, 64.7, 21.0, 20.4.
3.10. ESI-HRMS
High-resolution Spectra were recorded on an Exactive Orbitrap Spectrometer (Thermo Scientific) with ESI source. Samples were dissolved in acetonitrile (c: 10−4M).
Supplementary Materials
The supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28134915/s1. Page S2. Synthesis of compound 5. Figure S1. 1H NMR and 13C NMR spectra of compound 5 at +25 °C. Page S4 and Figure S2. HPLC separation of compounds 1a and 2a. Page S5. Characterization of compound 1a. Figure S3. 1H NMR and 13C NMR spectra of compound 1a at +25 °C. Figure S4. 1H NMR and 13C NMR spectra of compound 1a at +80 °C. Page S8. Characterization of compound 2a. Figure S5. 1H NMR and 13C NMR spectra of compound 2a at +25 °C. Figure S6. Predicted DFT (6-311++G(d,p), PCM=chloroform) conformations of syn/anti for compounds 2a are shown. Figure S7. Simulated and experimental dynamic 1H NMR (600 MHz) spectra in C2D2Cl4 of compound 2a for the enantiomerization process. Figure S8. Simulated and experimental dynamic 1H NMR (600 MHz) spectra in C2D2Cl4 of compound 2a for the diastereomerization process. Figure S9. 1H NMR spectra of 2a at -10 °C (263 K) in CDCl3 and CD3CN. Page S14. Synthesis of compounds 1b and 2b. Figure S10. 1H NMR and 13C NMR spectra of compound 1b mixture of isomers at +25 °C. Figure S11. HPLC separation of syn/anti 1b. Figure S12. 1H NMR and 13C NMR spectra of compound syn-1b at +25 °C. Figure S13. 1H NMR and 13C NMR spectra of compound anti-1b at +25 °C. Figures S14 and S15. DFT calculations. Figures S16-S21. Kinetic studies of compound 1b. Figure S22. ECD studies of compound anti-1b. Page S27. Characterization of compound 2b. Figure S23. 1H NMR and 13C NMR spectra of compound anti-2b at +25 °C. Figure S24. 1H NMR and 13C NMR spectra of compound syn-2b at +25 °C. Figure S25. Kibetic studies of compound 2b. Figure S26. CSP-HPLC separation of compound 2b. Figures S27–S31. ECD studies of compound 2b. Pages S37–S89 DFT calculations.
Author Contributions
Conceptualization, A.C. and M.M.; methodology, A.F., N.M. and A.S.; software, N.M. and A.S.; formal analysis, A.F.; investigation, A.S. and N.M.; data curation, M.M.; writing—original draft preparation, N.M. and A.S.; writing—review and editing, A.C., M.M. and A.M.; supervision, A.M. and G.F. All authors have read and agreed to the published version of the manuscript.
Funding
The University of Bologna is gratefully acknowledged (RFO Funds 2021 and 2022). A.M., M.M., and N.M. thank Alchemy SrL, Bologna, for the generous gift of chemicals. The authors of the Department of Chemistry and Drug Technologies are gratefully acknowledged to Sapienza (RM1221816BF705A8 found).
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Cheng, J.K.; Xiang, S.H.; Li, S.; Ye, L.; Tan, B. Recent Advances in Catalytic Asymmetric Construction of Atropisomers. Chem. Rev. 2021, 121, 4805–4902. [Google Scholar] [CrossRef] [PubMed]
- Mancinelli, M.; Bencivenni, G.; Pecorari, D.; Mazzanti, A. Stereochemistry and Recent Applications of Axially Chiral Organic Molecules. Eur. J. Org. Chem. 2020, 2020, 4070–4086. [Google Scholar] [CrossRef]
- Zilate, B.; Castrogiovanni, A.; Sparr, C. Catalyst-Controlled Stereoselective Synthesis of Atropisomers. ACS Catal. 2018, 8, 2981–2988. [Google Scholar] [CrossRef]
- Glunz, P.W. Recent encounters with atropisomerism in drug discovery. Bioorg. Med. Chem. Lett. 2018, 28, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Pecorari, D.; Mazzanti, A.; Mancinelli, M. Atropostatin: Design and Total Synthesis of an Atropisomeric Lactone–Atorvastatin Prodrug. Molecules 2023, 28, 3176. [Google Scholar] [CrossRef]
- Basilaia, M.; Chen, M.H.; Secka, J.; Gustafson, J.L. Atropisomerism in the Pharmaceutically Relevant Realm. Acc. Chem. Res. 2022, 55, 2904–2919. [Google Scholar] [CrossRef]
- Castrogiovanni, A.; Lotter, D.; Bissegger, F.R.; Sparr, C. JoyaPhos: An Atropisomeric Teraryl Monophosphine Ligand. Chem. Eur. J. 2020, 26, 9864–9868. [Google Scholar] [CrossRef]
- Bernardi, L.; Bolzoni, G.; Fochi, M.; Mancinelli, M.; Mazzanti, A. An Atropisomerically Enforced Phosphoric Acid for Organocatalytic Asymmetric Reactions. Eur. J. Org. Chem. 2016, 3208–3216. [Google Scholar] [CrossRef]
- Prati, L.; Mancinelli, M.; Ciogli, A.; Mazzanti, A. Tetrasubstituted cyclopentadienones as suitable enantiopure ligands with axial chirality. Org. Biomol. Chem. 2017, 15, 8720–8728. [Google Scholar] [CrossRef]
- Bai, X.-F.; Cui, Y.-M.; Cao, J.; Xu, L.-W. Atropisomers with Axial and Point Chirality: Synthesis and Applications. Acc. Chem. Res. 2022, 55, 2545–2561. [Google Scholar] [CrossRef]
- Dynamic Kinetic Resolution and Dynamic Kinetic Asymmetric Transformation of Atropisomers. In Science of Synthesis: Dynamic Kinetic Resolution (DKR) and Dynamic Kinetic Asymmetric Transformations (DYKAT); Bäckvall, J.-E., Ed.; Thieme Publishing Group: Stuttgart, Germany, 2023; Volume 1, pp. 441–483. [Google Scholar]
- Atropisomerism and Axial Chirality in Heteroaromatic Compounds in Advances. In Heterocyclic Chemistry; Katritzky, A., Ed.; Elsevier B.V.: Amsterdam, The Netherlands, 2012; Volume 105, pp. 1–188. [Google Scholar]
- Gawroński, J.; Kacprzak, K. Architecture and function of atropisomeric molecular triads. Chirality 2002, 14, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Kumarasamy, E.; Raghunathan, R.; Sibi, M.P.; Sivaguru, J. Nonbiaryl and Heterobiaryl Atropisomers: Molecular Templates with Promise for Atropselective Chemical Transformations. Chem. Rev. 2015, 115, 11239–11300. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Salamanca, P.; Fernández, R.; Hornillos, V.; Lassaletta, J.M. Asymmetric Synthesis of Axially Chiral C-N Atropisomers. Chem. Eur. J. 2022, 28, e202104442. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, N.; Righi, P.; Mazzanti, A.; Mancinelli, M.; Ciogli, A.; Bencivenni, G. Remote Control of Axial Chirality: Aminocatalytic Desymmetrization of N-Arylmaleimides via Vinylogous Michael Addition. J. Am. Chem. Soc. 2014, 136, 10250–10253. [Google Scholar] [CrossRef]
- Ciogli, A.; Kumar, S.V.; Mancinelli, M.; Mazzanti, A.; Perumal, S.; Severi, C.; Villani, C. Atropisomerism in 3-arylthiazolidine-2-thiones. A combined dynamic NMR and dynamic HPLC study. Org. Biomol. Chem. 2016, 14, 11137–11147. [Google Scholar] [CrossRef] [PubMed]
- Pecorari, D.; Giuliani, E.; Mazzanti, A.; Stagni, S.; Fiorini, V.; Vigarani, G.; Zinna, F.; Pescitelli, G.; Mancinelli, M. Synthesis and Stereody-namic and Emission Properties of Dissymmetric Bis-Aryl Carbazole Boranes and Identification of a CPL-Active B–C Atropisomeric Com-pound. J. Org. Chem. 2023, 88, 871–881. [Google Scholar] [CrossRef]
- Mazzanti, A.; Boffa, M.; Marotta, E.; Mancinelli, M. Axial Chirality at the Boron–Carbon Bond: Synthesis, Stereodynamic Analysis, and Atropisomeric Resolution of 6-Aryl-5,6-dihydrodibenzo[c,e][1,2]azaborinines. J. Org. Chem. 2019, 84, 12253–12258. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Rodriguez, J.; Bonne, D. Enantioselective Synthesis of Atropisomers with Multiple Stereogenic Axes. Angew. Chem. Int. Ed. 2020, 59, 12623–12634. [Google Scholar] [CrossRef]
- Ambrogi, M.; Ciogli, A.; Mancinelli, M.; Ranieri, S.; Mazzanti, A. Atropisomers of Arylmaleimides: Stereodynamics and Absolute Configuration. J. Org. Chem. 2013, 78, 3709–3719. [Google Scholar] [CrossRef]
- Mancinelli, M.; Perticarari, S.; Prati, L.; Mazzanti, A. Conformational Analysis and Absolute Configuration of Axially Chiral 1-Aryl and 1,3-Bisaryl-xanthines. J. Org. Chem. 2017, 82, 6874–6885. [Google Scholar] [CrossRef]
- Lunazzi, L.; Mancinelli, M.; Mazzanti, A. Stereodynamics and Conformational Chirality of the Atropisomers of Ditolyl Anthrones and Anthraquinone. J. Org. Chem. 2008, 73, 5354–5359. [Google Scholar] [CrossRef]
- Lunazzi, L.; Mancinelli, M.; Mazzanti, A. Structure, Stereodynamics and Absolute Configuration of the Atropisomers of Hindered Arylanthraquinones. J. Org. Chem. 2009, 74, 1345–1348. [Google Scholar] [CrossRef]
- Borsley, S.; Kreidt, E.; Leigh, A.; Roberts, B.M.W. Autonomous fuelled directional rotation about a covalent single bond. Nature 2022, 604, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Toyoda, R.; Costil, R.; Feringa, B.L. Chemically Driven Rotatory Molecular Machines. Angew. Chem. Int. Ed. 2022, 61, e202206631. [Google Scholar] [CrossRef] [PubMed]
- Arcadi, A.; Fabrizi, G.; Fochetti, A.; Franzini, R.; Ghirga, F.; Goggiamani, A.; Iazzetti, A.; Marrone, F.; Serraiocco, A. Synthesis of polycyclic chromene cores through Gold (I)-catalyzed intramolecular hydroarylation reaction (IMHA). Eur. J. Org. Chem. 2021, 11, 1676–1687. [Google Scholar] [CrossRef]
- Le, C.M.; Menzies, P.J.C.; Petrone, D.A.; Lautens, M. Synergistic Steric Effects in the Development of a Palladium-Catalyzed Alkyne Carbohalogenation: Stereodivergent Synthesis of Vinyl Halides. Angew. Chem. 2015, 54, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Luchini, G.; Alegre-Requena, J.V.; Funes-Ardoiz, I.; Paton, R.S. GoodVibes: Automated Thermochemistry for Heterogeneous Computational Chemistry Data. F1000Research 2020, 9, 291. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Mennucci, B.; Cappelli, C.; Cammi, R.; Tomasi, J. Modeling Solvent Effects on Chiroptical Properties. Chirality 2011, 23, 717–729. [Google Scholar] [CrossRef]
- Wenzel, T.; Chisholm, C.D. Assignment of Absolute Configuration Using Chiral Reagentsand NMR Spectroscopy. Chirality 2011, 23, 190–214. [Google Scholar] [CrossRef]
- Gasparrini, F.; Misiti, D.; Pierini, M.; Villani, C. Enantiomerization barriers by dynamic HPLC. Stationary phase effects. Tetrahedron Asymm. 1997, 8, 2069–2073. [Google Scholar] [CrossRef]
- D’Acquarica, I.; Gasparrini, F.; Pierini, M.; Villani, C.; Zappia, G. Dynamic HPLC on chiral stationary phases: A powerful tool for the investigation of stereomutation processes. J. Sep. Sci. 2006, 29, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Menta, S.; Pierini, M.; Cirilli, R.; Grisi, F.; Perfetto, A.; Ciogli, A. Stereolability of Chiral Ruthenium Catalysts with Frozen NHC Ligand Conformations Investigated by Dynamic-HPLC. Chirality 2015, 27, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Belot, V.; Farran, D.; Jean, M.; Albalat, M.; Vanthuyne, N.; Roussel, C. Steric Scale of Common Substituents from Rotational Barriers of N-(o-Substituted aryl)thiazoline-2-thione Atropisomers. J. Org. Chem. 2017, 82, 10188–10200. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Li, H.-H.; Zhang, X.; Xiang, S.-H.; Li, S.; Tan, B. Organocatalytic Enantioselective Synthesis of Atropisomeric Aryl-p-Quinones: Platform Molecules for Diversity-Oriented Synthesis of Biaryldiols. Angew. Chem. Int. Ed. 2020, 59, 11374–11378. [Google Scholar] [CrossRef]
- Sweet, J.S.; Rajkumar, S.; Dingwall, P.; Knipe, P.C. Atroposelective Synthesis, Structure and Properties of a Novel Class of Axially Chiral N-Aryl Quinolinium Salt. Eur. J. Org. Chem. 2021, 3980–3985. [Google Scholar] [CrossRef]
- Stonehouse, J.; Adell, P.; Keeler, J.; Shaka, A.J. Ultrahigh-Quality NOE Spectra. J. Am. Chem. Soc. 1994, 116, 6037–6038. [Google Scholar] [CrossRef]
- Stott, K.; Stonehouse, J.; Keeler, J.; Hwang, T.L.; Shaka, A.J. Excitation Sculpting in High-Resolution Nuclear Magnetic Resonance Spectroscopy: Application to Selective NOE Experiments. J. Am. Chem. Soc. 1995, 117, 4199–4200. [Google Scholar] [CrossRef]
- Stott, K.; Keeler, J.; Van, Q.N.; Shaka, A.J. One-Dimensional NOE Experiments Using Pulsed Field Gradients. J. Magn. Reson. 1997, 125, 302–324. [Google Scholar] [CrossRef]
- Van, Q.N.; Smith, E.M.; Shaka, A.J. Observation of Long-Range Small-Molecule NOEs Using a Neoteric Sensitivity Enhancement Scheme. J. Magn. Reson. 1999, 141, 191–194. [Google Scholar] [CrossRef]
- Pescitelli, G.; Bruhn, T. Good Computational Practice in the Assignment of Absolute Configurations by TDDFT Calculations of ECD Spectra. Chirality 2016, 28, 466–474. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).