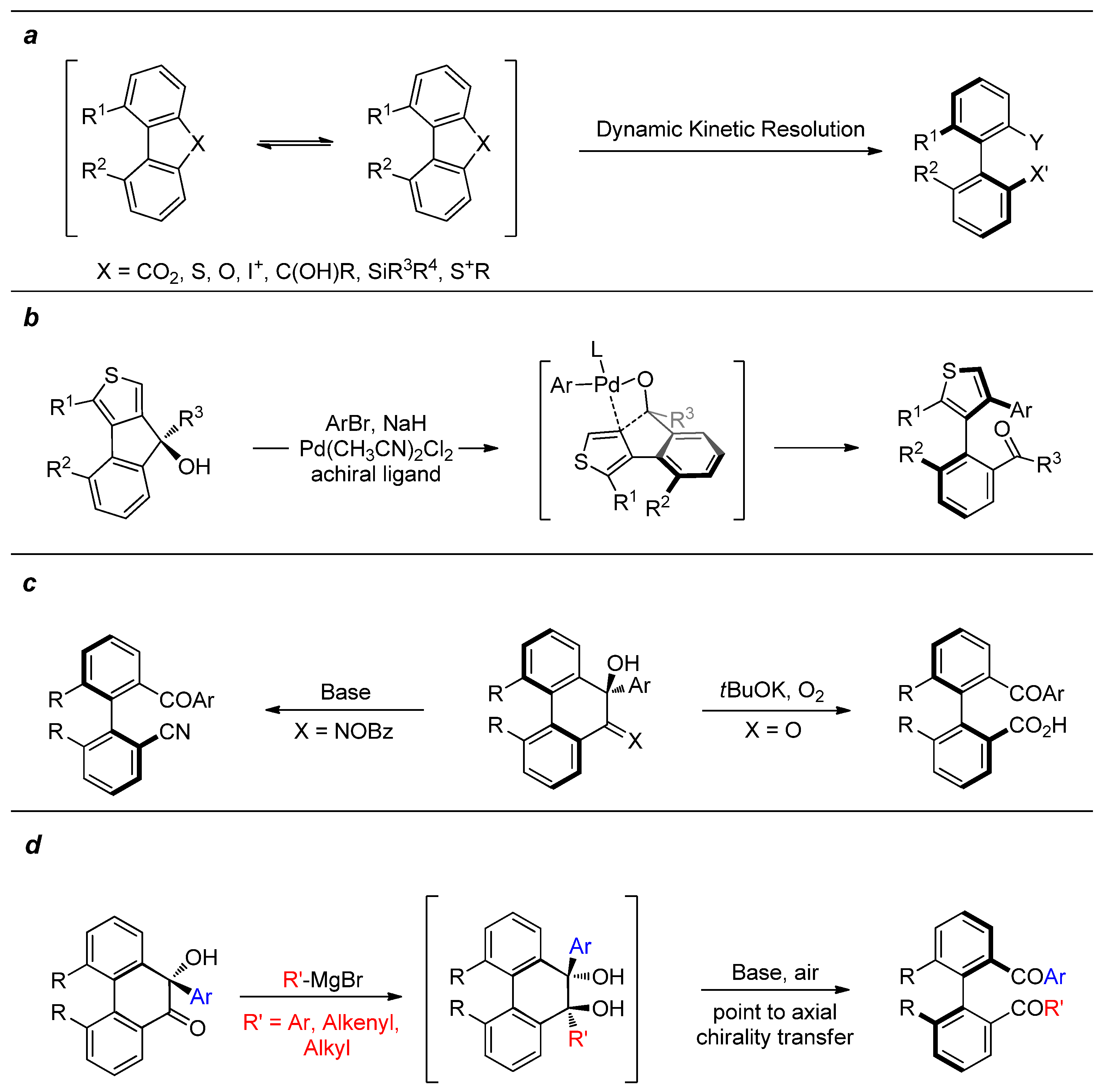
3.1.1. General Procedure for the Synthesis of Target Compounds 3a–3w
Under a nitrogen atmosphere, R’-MgBr (1.0 M, 0.60 mL, 0.60 mmol, 3.0 equiv) was added dropwisely to a solution of 1a–1w (0.20 mmol, 1.0 equiv) in anhydrous THF (3.0 mL) at 0 °C. After being stirred at 25 °C for 4 h, the reaction was quenched with water (15 mL) and extracted with EtOAc (10 mL × 3). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated to afford the crude diol, which was used in next step without further purification.
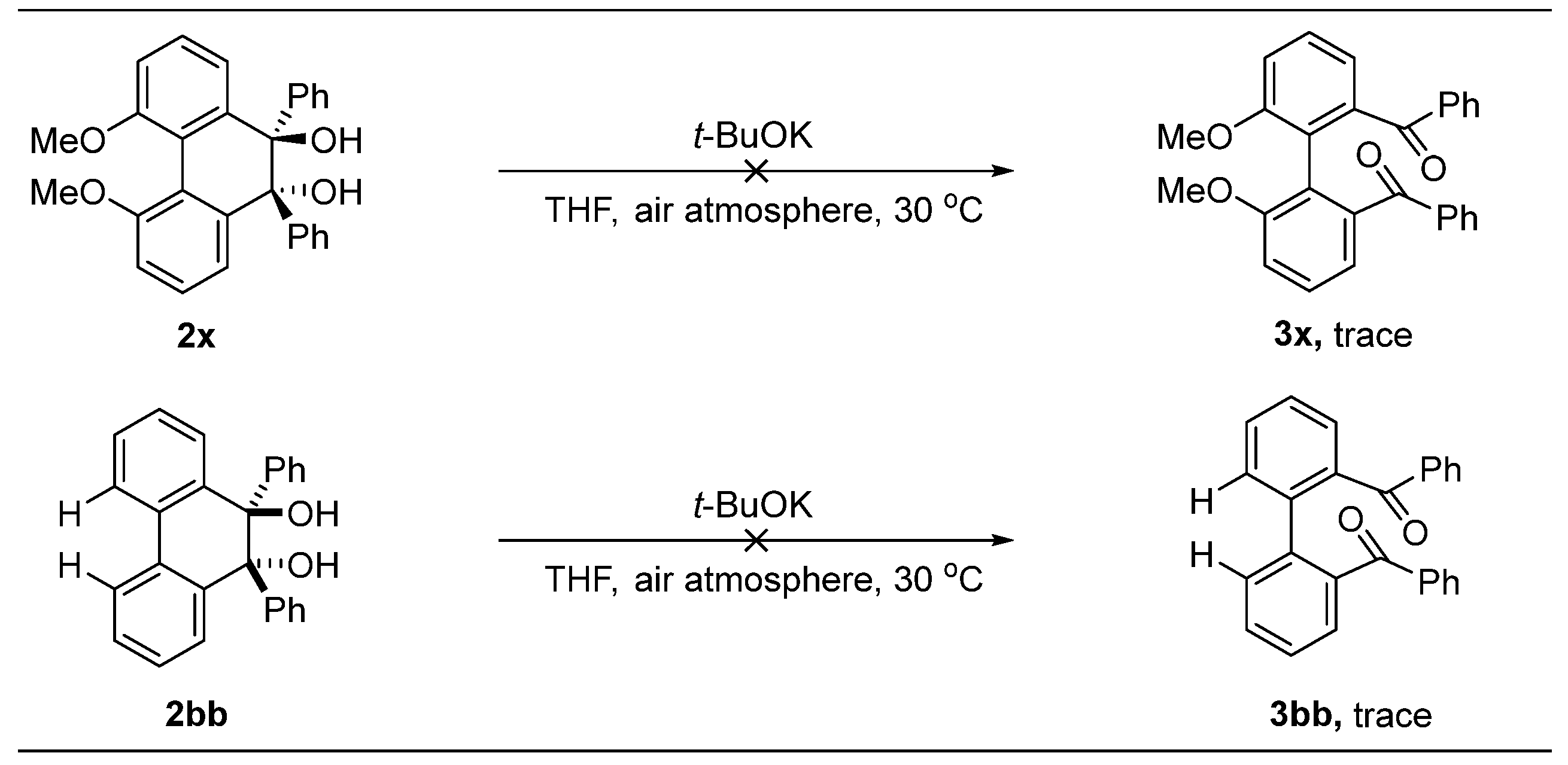
Under an air atmosphere to a mixture of the above crude diol in anhydrous THF (5.0 mL), t-BuOK (67.3 mg, 0.60 mmol, 3.0 equiv) was added at room temperature and stirred for 30 min. The solvent was removed, and the residue was purified by flash chromatography on silica gel (PE/EtOAc) to afford 3a–3w.
The reaction of 1a (62.8 mg, 0.20 mmol, 95% ee, 1.0 equiv) afforded product 3a (73.9 mg, 95%, 95% ee, ST = 100%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. − 3.10 (c 1.50, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 5:95, flow: 0.8 mL/min, λ = 254 nm, tR = 6.9 min (minor), 8.3 min (major). 1H NMR (500 MHz, CDCl3) δ 7.46–7.43 (m, 4H), 7.43–7.40 (m, 2H), 7.29–7.25 (m, 2H), 7.25–7.22 (m, 2H), 7.16–7.12 (m, 2H), 7.09–7.04 (m, 4H). 2.16 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 197.1, 139.0, 138.6, 137.2, 136.9, 132.3, 131.8, 130.2, 127.6, 126.8, 126.1, 20.2. HRMS (ESI) calcd for C28H23O2 [M + H]+ 391.1693, found 391.1696.
The reaction of 1b (70.0 mg, 0.20 mmol, 93% ee, 1.0 equiv) afforded product 3b (74.2 mg, 88%, 92% ee, ST = 99%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 47.4 (c 1.60, CH2Cl2). HPLC conditions: Chiralcel IC-3, isopropanol/hexane = 3:97, flow: 0.8 mL/min, λ = 254 nm, tR = 15.7 min (major), 18.4 min (minor). 1H NMR (500 MHz, CDCl3) δ 7.45–7.39 (m, 2H), 7.33–7.28 (m, 4H), 7.27–7.22 (m, 2H), 7.12 (dd, J = 7.5, 1.5 Hz, 2H), 6.83 (d, J = 8.0 Hz, 4H), 2.24 (s, 6H), 2.17 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 196.7, 143.0, 139.1, 138.3, 137.1, 134.5, 131.5, 130.5, 128.2, 126.3, 126.1, 21.4, 20.2. HRMS (ESI) calcd for C30H26O2Na [M+Na]+ 441.1825, found 441.4823.
The reaction of 1c (66.4 mg, 0.20 mmol, 92% ee, 1.0 equiv) afforded product 3c (70.8 mg, 83%, 88% ee, ST = 96%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. − 104 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 2:98, flow: 0.8 mL/min, λ = 254 nm, tR = 10.6 min (minor), 19.3 min (major). 1H NMR (500 MHz, CDCl3) δ 7.48–7.46 (m, 2H), 7.46–7.42 (m, 4H), 7.31–7.22 (m, 2H), 7.12 (dd, J = 8.0, 1.0 Hz, 2H), 6.81–6.72 (m, 4H), 2.15 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 195.4, 166.4 (d, J = 255.4 Hz), 139.2, 138.4, 136.6, 133.5 (d, J = 3.0 Hz), 132.9 (d, J = 9.3 Hz), 132.0, 126.4, 126.3, 114.8 (d, J = 21.8 Hz), 20.2. 19F NMR (471 MHz, CDCl3) δ −105.8. HRMS (ESI) calcd for C28H21F2O2 [M + H]+ 427.1504, found 427.1510.
The reaction of 1d (69.6 mg, 0.20 mmol, 93% ee, 1.0 equiv) afforded product 3d (75.3 mg, 82%, 89% ee, ST = 96%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 55.0 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 3:97, flow: 0.8 mL/min, λ = 254 nm, tR = 8.8 min (minor), 12.0 min (major). 1H NMR (500 MHz, CDCl3) δ 7.44 (d, J = 7.5 Hz, 2H), 7.38–7.36 (m, 2H), 7.36–7.33 (m, 2H), 7.30–7.24 (m, 2H), 7.14–7.09 (m, 2H), 7.10–7.02 (m, 4H), 2.15 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 195.7, 139.32, 139.26, 138.3, 136.4, 135.3, 132.0, 131.6, 128.0, 126.4, 126.3, 20.1. HRMS (ESI) calcd for C28H21Cl2O2 [M + H]+ 459.0913, found 459.0915.
The reaction of 1e (78.4 mg, 0.20 mmol, 95% ee, 1.0 equiv) afforded product 3e (87.7 mg, 80%, 92% ee, ST = 97%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. − 2.87 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 2:98, flow: 0.8 mL/min, λ = 254 nm, tR = 8.8 min (minor), 11.1 min (major). 1H NMR (500 MHz, CDCl3) δ 7.49–7.47 (m, 2H), 7.47–7.46 (m, 2H), 7.45–7.43 (m, 2H), 7.32–7.29 (m, 2H), 7.29–7.27 (m, 2H), 7.17 (dd, J = 7.5, 1.5 Hz, 2H), 7.12–7.07 (m, 4H), 2.19 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 197.1, 139.0, 138.6, 137.3, 136.9, 132.3, 131.8, 130.3, 127.6, 126.8, 126.2, 20.2. HRMS (ESI) calcd for C28H20Br2O2Na [M+Na]+ 568.9722, found 568.9731.
The reaction of 1f (68.8 mg, 0.20 mmol, 89% ee, 1.0 equiv) afforded product 3f (77.4 mg, 86%, 88% ee, ST = 99%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 184 (c 1.40, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 3:97, flow: 0.8 mL/min, λ = 254 nm, tR = 17.0 min (minor), 24.2 min (major). 1H NMR (500 MHz, CDCl3) δ 7.41 (d, J = 7.5 Hz, 2H), 7.33 (d, J = 8.5 Hz, 4H), 7.26–7.21 (m 2H), 7.13–7.03 (m, 2H), 6.54–6.38 (m, 4H), 3.70 (s, 6H), 2.18 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 195.7, 162.8, 139.3, 138.1, 137.2, 132.6, 131.3, 129.9, 126.1, 125.9, 112.7, 55.0, 20.2. HRMS (ESI) calcd for C30H26O4Na [M+Na]+ 473.1723, found 473.1722.
The reaction of 1g (79.6 mg, 0.20 mmol, 97% ee, 1.0 equiv) afforded product 3g (80.5 mg, 72%, 95% ee, ST = 98%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 8.75 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 2:98, flow: 0.8 mL/min, λ = 254 nm, tR = 8.5 min (minor), 10.2 min (major). 1H NMR (500 MHz, CDCl3) δ 7.53–7.49 (m, 4H), 7.46 (d, J = 7.5 Hz, 2H), 7.31–7.26 (m, 2H), 7.14 (dd, J = 8.0, 1.5 Hz, 2H), 6.94 (d, J = 8.5 Hz, 4H), 2.16 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 195.4, 152.2, 139.2, 138.5, 136.4, 135.3, 132.3, 132.2, 126.6, 126.4, 120.1 (q, J = 259 Hz), 119.2, 20.1. 19F NMR (471 MHz, CDCl3) δ -57.80. HRMS (ESI) calcd for C30H21F6O4 [M + H]+ 559.1339, found 559.1348.
The reaction of 1h (65.6 mg, 0.20 mmol, 95% ee, 1.0 equiv) afforded product 3h (72.1 mg, 86%, 94% ee, ST = 99%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 40.5 (c 1.40, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 2:98, flow: 0.8 mL/min, λ = 254 nm, tR = 7.6 min (minor), 8.7 min (major). 1H NMR (500 MHz, CDCl3) δ 7.43 (d, J = 7.5 Hz, 2H), 7.31–7.27 (m, 2H), 7.27–7.23 (m, 2H), 7.14–7.09 (m, 4H), 7.07 (d, J = 7.5 Hz, 2H), 7.00–6.96 (m, 2H), 2.19 (s, 6H), 2.04 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 197.3, 139.2, 138.5, 137.4, 137.2, 137.0, 133.1, 131.7, 131.0, 127.6, 127.2, 126.6, 126.2, 21.0, 20.1. HRMS (ESI) calcd for C30H26O2Na [M+Na]+ 441.1825, found 441.1828.
The reaction of 1i (71.2 mg, 0.20 mmol, 94% ee, 1.0 equiv) afforded product 3i (87.8 mg, 92%, 92% ee, ST = 98%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. − 5.75 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 2:98, flow: 0.8 mL/min, λ = 254 nm, tR = 7.6 min (minor), 8.7 min (major). 1H NMR (500 MHz, CDCl3) δ 7.46–7.42 (m, 2H), 7.41–7.37 (m, 2H), 7.28–7.23 (m, 2H), 7.21–7.19 (m, 2H), 7.18–7.16 (m, 2H), 7.16–7.13 (m, 2H), 7.01–6.95 (m, 2H), 2.70 (p, J = 6.9 Hz, 2H), 2.19 (s, 6H), 1.09 (d, J = 4.0 Hz, 6H), 1.07 (d, J = 4.0 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 197.5, 148.3, 139.0, 138.8, 137.5, 137.1, 131.8, 130.5, 128.2, 128.1, 127.5, 127.0, 126.1, 33.6, 23.7, 23.5, 20.1. HRMS (ESI) calcd for C34H35O2 [M + H]+ 475.2632, found 475.2640.
The reaction of 1j (66.4 mg, 0.20 mmol, 94% ee, 1.0 equiv) afforded product 3j (66.5 mg, 78%, 94% ee, ST = 100%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 6.45 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 3:97, flow: 0.8 mL/min, λ = 254 nm, tR = 8.1 min (minor), 11.3 min (major). 1H NMR (500 MHz, CDCl3) δ 7.45 (d, J = 7.5 Hz, 2H), 7.31–7.26 (m, 2H), 7.25–7.21 (m, 2H), 7.21–7.17 (m, 2H), 7.17–7.14 (m, 2H), 7.12–7.05 (m, 2H), 7.04–6.99 (m, 2H), 2.15 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 195.5 (d, J = 2.1 Hz), 162.1 (d, J = 248 Hz), 139.3 (d, J = 6.3 Hz), 139.1, 138.4, 136.4, 132.2, 129.3 (d, J = 7.4 Hz), 126.6, 126.4, 126.21, 126.18, 119.5 (d, J = 21.5 Hz), 116.6 (d, J = 22.3 Hz), 20.1. 19F NMR (471 MHz, CDCl3) δ −112.5. HRMS (ESI) calcd for C28H21F2O2 [M + H]+ 427.1504, found 427.1510.
The reaction of 1k (69.6 mg, 0.20 mmol, 96% ee, 1.0 equiv) afforded product 3k (68.9 mg, 75%, 96% ee, ST = 100%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 40.8 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 3:97, flow: 0.8 mL/min, λ = 254 nm, tR = 9.4 min (minor), 13.8 min (major). 1H NMR (500 MHz, CDCl3) δ 7.45 (d, J = 7.5 Hz, 2H), 7.41–7.38 (m, 2H), 7.36–7.32 (m, 2H), 7.31–7.28 (m, 2H), 7.28–7.25 (m, 2H), 7.17–7.10 (m, 2H), 7.09–7.03 (m, 2H), 2.15 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 195.5, 139.2, 138.7, 138.4, 136.3, 134.2, 132.5, 132.3, 130.0, 129.1, 128.3, 126.54, 126.50, 77.2, 20.1. HRMS (ESI) calcd for C28H21Cl2O2 [M + H]+ 459.0913, found 459.0915.
The reaction of 1l (68.8 mg, 0.20 mmol, 95% ee, 1.0 equiv) afforded product 3l (86.5 mg, 96%, 91% ee, ST = 96%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 27.6 (c 1.60, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 10:90, flow: 0.8 mL/min, λ = 254 nm, tR = 7.7 min (minor), 8.5 min (major). 1H NMR (500 MHz, CDCl3) δ 7.44 (d, J = 7.5 Hz, 2H), 7.28–7.24 (m, 2H), 7.16 (dd, J = 8.0, 1.5 Hz, 2H), 7.00–6.97 (m, 4H), 6.97–6.93 (m, 2H), 6.85–6.80 (m, 2H), 3.63 (s, 6H), 2.18 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 196.8, 158.9, 139.1, 138.48, 138.47, 136.9, 131.8, 128.6, 126.6, 126.2, 123.2, 119.4, 113.7, 55.0, 20.1. HRMS (ESI) calcd for C30H27O4 [M + H]+ 454.1904, found 451.1903.
The reaction of 1m (72.0 mg, 0.20 mmol, 83% ee, 1.0 equiv) afforded product 3m (71.4 mg, 74%, 83% ee, ST = 100%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 20.0 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 3:97, flow: 0.8 mL/min, λ = 254 nm, tR = 10.5 min (minor), 12.0 min (major). 1H NMR (500 MHz, CDCl3) δ 7.44 (d, J = 7.5 Hz, 2H), 7.30–7.27 (m, 2H), 7.27–7.24 (m, 2H), 7.18–7.15 (m, 2H), 7.15–7.14 (m, 2H), 7.14–7.10 (m, 2H), 7.01–6.91 (m, 2H), 2.32 (s, 6H), 2.17 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 196.7, 139.1, 138.8, 138.5, 137.8, 136.7, 132.0, 130.2, 127.9, 126.9, 126.8, 126.7, 126.3, 20.1, 15.2. HRMS (ESI) calcd for C30H27S2O4 [M + H]+ 483.1447, found 483.1453.
The reaction of 1n (78.0 mg, 0.20 mmol, 94% ee, 1.0 equiv) afforded product 3n (99.7 mg, 92%, 93% ee, ST = 99%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 62.8 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 3:97, flow: 0.8 mL/min, λ = 254 nm, tR = 9.9 min (minor), 11.9 min (major). 1H NMR (500 MHz, CDCl3) δ 7.66–7.61 (m, 2H), 7.48–7.40 (m, 6H), 7.32–7.28 (m, 2H), 7.28–7.27 (m, 2H), 7.27–7.26 (d, J = 3.0 Hz, 2H), 7.26–7.25 (m, 2H), 7.25–7.24 (m, 2H), 7.24–7.21 (m, 2H), 7.21–7.16 (m, 2H), 7.10–7.04 (m, 2H), 2.21 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 197.1, 140.5, 139.6, 139.2, 138.7, 137.7, 136.8, 132.0, 130.9, 128.91, 128.90, 128.6, 128.2, 127.4, 126.9, 126.8, 126.3, 20.2. HRMS (ESI) calcd for C40H31O2 [M + H]+ 543.2319, found 543.2318.
The reaction of 1o (68.1 mg, 0.20 mmol, 92% ee, 1.0 equiv) afforded product 3o (73.5 mg, 83%, 90% ee, ST = 98%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 24.6 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 2:98, flow: 0.8 mL/min, λ = 254 nm, tR = 8.0 min (minor), 9.3 min (major). 1H NMR (500 MHz, CDCl3) δ 7.44 (d, J = 7.5 Hz, 2H), 7.42–7.39 (m, 2H), 7.31–7.29 (m, 2H), 7.28–7.27 (m, 2H), 7.27–7.22 (m, 2H), 7.16–7.10 (m, 2H), 7.03–6.97 (m, 2H), 6.52–6.35 (m, 2H), 5.55 (d, J = 17.5 Hz, 2H), 5.15 (d, J = 11.0 Hz, 2H), 2.18 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 197.0, 139.1, 138.5, 137.4, 137.2, 136.9, 135.8, 131.9, 130.0, 129.6, 127.94, 127.87, 126.6, 126.3, 114.7, 20.1. HRMS (ESI) calcd for C32H27O2 [M + H]+ 443.2006, found 443.2011.
The reaction of 1p (68.4 mg, 0.20 mmol, 94% ee, 1.0 equiv) afforded product 3p (67.9 mg, 76%, 93% ee, ST = 99%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 80.2 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel IC-3, isopropanol/hexane = 1:99, flow: 0.8 mL/min, λ = 254 nm, tR = 13.0 min (major), 16.4 min (minor). 1H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 7.5 Hz, 2H), 7.26–7.21 (m, 2H), 7.10–7.04 (m, 2H), 6.95 (s, 4H), 6.87 (s, 2H), 2.19 (s, 6H), 2.01 (s, 12H). 13C NMR (126 MHz, CDCl3) δ 197.5, 139.3, 138.4, 137.3, 137.2, 133.9, 131.5, 128.1, 126.4, 126.2, 20.8, 20.1. HRMS (ESI) calcd for C32H31O2 [M + H]+ 447.2319, found 447.2325.
The reaction of 1q (72.8 mg, 0.20 mmol, 94% ee, 1.0 equiv) afforded product 3q (91.2 mg, 93%, 94% ee, ST = 100%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 282 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 2:98, flow: 0.8 mL/min, λ = 254 nm, tR = 12.9 min (minor), 15.8 min (major). 1H NMR (500 MHz, CDCl3) δ 7.58 (dd, J = 8.5, 1.5 Hz, 2H), 7.54–7.51 (m, 2H), 7.51–7.47 (m, 2H), 7.32–7.30 (m, 2H), 7.30–7.28 (m, 2H), 7.28–7.27 (m, 2H), 7.22 (d, J = 8.0 Hz, 2H), 7.20–7.18 (m, 2H), 7.18–7.14 (m, 2H), 7.12–7.07 (m, 2H), 2.27 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 197.1, 139.5, 138.4, 137.0, 134.7, 133.9, 133.4, 131.7, 131.3, 129.2, 127.8, 127.7, 126.9, 126.4, 126.3, 125.7, 124.4, 20.2. HRMS (ESI) calcd for C36H27O2 [M + H]+ 491.2006, found 491.2006.
The reaction of 1r (71.6 mg, 0.20 mmol, 90% ee, 1.0 equiv) afforded product 3r (86.1 mg, 90%, 90% ee, ST = 100%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 63.2 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 3:97, flow: 0.8 mL/min, λ = 254 nm, tR = 33.7 min (minor), 38.5 min (major). 1H NMR (500 MHz, CDCl3) δ 7.46–7.39 (m, 2H), 7.30–7.27 (m, 2H), 7.15–7.10 (m, 2H), 7.00–6.96 (m, 2H), 6.96–6.92 (m, 2H), 6.45 (d, J = 8.0 Hz, 2H), 5.96 (d, J = 1.5 Hz, 2H), 5.93 (d, J = 1.0 Hz, 2H), 2.17 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 195.1, 151.2, 147.2, 139.3, 138.1, 136.9, 131.8, 131.4, 127.7, 126.2, 125.9, 109.5, 107.0, 101.6, 20.2. HRMS (ESI) calcd for C30H23O6 [M + H]+ 479.1489, found 479.1497.
The reaction of 1s (80.9 mg, 0.20 mmol, 92% ee, 1.0 equiv) afforded product 3s (91.3 mg, 80%, 90% ee, ST = 98%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 238 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 2:98, flow: 0.8 mL/min, λ = 254 nm, tR = 16.7 min (minor), 23.4 min (major). 1H NMR (500 MHz, CDCl3) δ 7.64 (s, 2H), 7.56 (d, J = 8.5 Hz, 2H), 7.51 (d, J = 7.5 Hz, 2H), 7.43 (d, J = 7.5 Hz, 2H), 7.34–7.28 (m, 2H), 7.22–7.18 (m, 4H), 7.16 (d, J = 7.5 Hz, 2H), 7.09–7.02 (m, 2H), 6.85 (d, J = 7.5 Hz, 2H), 2.28 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 196.3, 158.0, 156.1, 139.7, 138.2, 137.0, 131.9, 131.6, 129.0, 127.3, 126.5, 125.9, 124.0, 123.3, 122.8, 122.7, 120.6, 111.2, 110.7, 77.2, 20.3. HRMS (ESI) calcd for C40H27O4 [M + H]+ 571.1904, found 571.1908.
The reaction of 1a (62.8 mg, 0.20 mmol, 95% ee, 1.0 equiv) with 3-MeOC6H4MgBr (0.60 mmol, 3.0 equiv) afforded product 3t (77.8 mg, 93%, 94% ee, ST = 99%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 15.6 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 3:97, flow: 0.8 mL/min, λ = 254 nm, tR = 9.0 min (minor), 10.8 min (major). 1H NMR (500 MHz, CDCl3) δ 7.45–7.43 (m, 2H), 7.42–7.39 (m, 2H), 7.31–7.27 (m, 1H), 7.27–7.23 (m, 2H), 7.17–7.11 (m, 2H), 7.11–7.05 (m, 2H), 7.03–6.97 (m, 2H), 6.96–6.91 (m 1H), 6.84–6.78 (m, 1H), 3.61 (s, 3H), 2.17 (s, 3H), 2.16 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 197.0, 196.8, 158.9, 139.0, 138.9, 138.6, 138.46, 138.45, 137.3, 136.9, 136.8, 132.2, 131.9, 131.7, 130.1, 128.6, 127.6, 126.9, 126.5, 126.2, 126.1, 123.3, 119.6, 113.6, 55.0, 20.14, 20.12. HRMS (ESI) calcd for C29H25O3 [M + H]+ 421.1798, found 421.1807.
The reaction of 1a (62.8 mg, 0.20 mmol, 95% ee, 1.0 equiv) with 2-MeOC6H4MgBr (0.60 mmol, 3.0 equiv) afforded product 3u (65.4 mg, 78%, 94% ee, ST = 99%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 2.10 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 3:97, flow: 0.8 mL/min, λ = 254 nm, tR = 9.6 min (minor), 16.0 min (major). 1H NMR (500 MHz, CDCl3) δ 7.66–7.56 (m, 2H), 7.43–7.38 (m, 2H), 7.38–7.33 (m, 1H), 7.29–7.25 (m, 1H), 7.25–7.23 (m, 1H), 7.23–7.19 (m, 2H), 7.19–7.17 (m, 2H), 7.17–7.11 (m, 2H), 6.76–6.69 (m, 1H), 6.66–6.60 (m, 1H), 3.27 (s, 3H), 2.16 (s, 3H), 2.12 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 197.4, 196.6, 157.8, 139.4, 138.7, 138.5, 138.2, 137.8, 137.2, 132.4, 132.3, 132.1, 131.9, 131.2, 130.4, 128.6, 127.9, 127.6, 127.0, 126.4, 125.8, 119.7, 110.7, 54.6, 20.1, 20.0. HRMS (ESI) calcd for C29H24O3Na [M + Na]+ 443.1618, found 443.1620.
The reaction of 1v (68.4 mg, 0.20 mmol, 95% ee, 1.0 equiv) afforded product 3v (80.0 mg, 95%, 93% ee, ST = 98%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 13.5 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 3:97, flow: 0.8 mL/min, λ = 254 nm, tR = 6.2 min (major), 7.6 min (minor). 1H NMR (500 MHz, CDCl3) δ 7.47–7.43 (m, 4H), 7.31–7.26 (m, 2H), 7.25–7.23 (m, 2H), 7.10–7.03 (m, 4H), 6.96–6.91 (m, 2H), 2.32 (s, 6H), 2.15 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 197.3, 138.9, 137.3, 137.1, 135.7, 135.5, 132.7, 132.2, 130.2, 127.5, 127.2, 21.0, 20.1. HRMS (ESI) calcd for C30H27O2 [M + H]+ 419.2006, found 419.2012.
The reaction of 1w (68.4 mg, 0.20 mmol, 93% ee, 1.0 equiv) afforded product 3w (76.8 mg, 83%, 93% ee, ST = 100%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 46.5 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 5:95, flow: 0.8 mL/min, λ = 254 nm, tR = 13.5 min (major), 17.3 min (minor). 1H NMR (500 MHz, CDCl3) δ 7.99–7.84 (m, 4H), 7.57–7.49 (m, 4H), 7.48–7.42 (m, 4H), 7.38–7.33 (m, 4H), 7.26–7.19 (m, 2H), 7.09–7.01 (m, 4H). 13C NMR (126 MHz, CDCl3) δ 197.2, 137.2, 136.6, 136.0, 134.0, 133.7, 132.3, 130.0, 128.2, 128.0, 127.6, 127.4, 127.2, 126.9, 125.6. HRMS (ESI) calcd for C34H23O2 [M + H]+ 463.1693, found 463.1699.
3.1.2. General Procedure for the Synthesis of Target Compounds 3x–3aa
Under a nitrogen atmosphere, R’-MgBr (1.0 M, 0.60 mL, 0.60 mmol, 3.0 equiv) was added dropwisely to a solution of 1a or 1x (0.20 mmol, 1.0 equiv) in anhydrous THF (3.0 mL) at 0 °C. After being stirred at 25 °C for 4 h, the reaction was quenched with water (15 mL) and extracted with EtOAc (10 mL × 3). The combined organic phase was dried over anhydrous Na2SO4, filtered, and concentrated to afford the crude diol, which was used in next step without further purification.
Under a nitrogen atmosphere, sodium hypochlorite pentahydrate (99.3 mg, 0.60 mmol, 3.0 equiv) was added to a solution of the above crude diol and tetra(n-butyl)ammonium hydrogen sulfate (13.6 mg, 0.04 mmol, 20 mol%) in DCM (2.0 mL) and water (0.5 mL) at rt. After stirring for 1 h, the mixture was quenched with water (10 mL) and extracted with CH2Cl2 (15 mL × 2). The combined organic layer was dried over anhydrous Na2SO4, filtered, and then concentrated in vacuo. The residue was purified by flash chromatography on silica gel (PE/EtOAc) to deliver the product 3x–3aa.
The reaction of 1x (69.2 mg, 0.20 mmol, 92% ee, 1.0 equiv) afforded product 3x (69.3 mg, 82%, 89% ee, ST = 97%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 88.3 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel AD-H, isopropanol/hexane = 10:90, flow: 1.0 mL/min, λ = 210 nm, tR = 16.4 min (minor), 30.8 min (major). 1H NMR (500 MHz, CDCl3) δ 7.81–7.75 (m, 4H), 7.43–7.37 (m, 2H), 7.30–7.26 (m, 4H), 7.26–7.22 (m, 2H), 7.05 (dd, J = 8.0, 1.0 Hz, 2H), 6.88 (dd, J = 8.5, 1.0 Hz, 2H), 3.48 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 196.4, 156.1, 139.7, 137.4, 132.2, 130.2, 128.0, 127.6, 124.3, 121.8, 112.8, 55.1. HRMS (ESI) calcd for C28H22O4Na [M+Na]+ 445.1410, found 445.1414.
The reaction of 1a (62.8 mg, 0.20 mmol, 95% ee, 1.0 equiv) with EtMgBr (0.60 mmol, 3.0 equiv) afforded product 3y (58.2 mg, 85%, 95% ee, ST = 100%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 1.52 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 2:98, flow: 0.8 mL/min, λ = 254 nm, tR = 8.6 min (minor), 9.4 min (major). 1H NMR (500 MHz, CDCl3) δ 7.68–7.63 (m, 2H), 7.51–7.47 (m, 2H), 7.44 (d, J = 7.5 Hz, 1H), 7.39–7.34 (m, 2H), 7.34–7.29 (m, 2H), 7.27–7.25 (m, 1H), 7.25–7.22 (m, 1H), 2.80 (dq, J = 18.0, 7.0 Hz, 1H), 2.38 (dq, J = 18.0, 7.0 Hz, 1H), 2.03 (s, 3H), 2.01 (s, 3H), 0.91 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 203.9, 197.2, 139.5, 138.1, 138.01, 137.98, 137.6, 137.4, 136.8, 132.7, 132.6, 131.8, 130.2, 128.0, 127.2, 126.6, 126.1, 125.7, 33.9, 20.2, 20.0, 8.0. HRMS (ESI) calcd for C24H22O2Na [M+Na]+ 365.1512, found 365.1519.
The reaction of 1a (62.8 mg, 0.20 mmol, 95% ee, 1.0 equiv) with 1-methylvinylmagnesium bromide (0.60 mmol, 3.0 equiv) afforded product 3z (53.9 mg, 76%, 96% ee, ST = 100%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. + 7.06 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel OD-3, isopropanol/hexane = 2:98, flow: 0.8 mL/min, λ = 254 nm, tR = 7.3 min (minor), 8.2 min (major). 1H NMR (500 MHz, CDCl3) δ 7.65–7.61 (m, 2H), 7.51–7.45 (m, 1H), 7.42 (dd, J = 7.5, 1.5 Hz, 1H), 7.38–7.34 (m, 2H), 7.34–7.32 (m, 1H), 7.30–7.26 (m 1H), 7.24–7.19 (m, 2H), 7.14 (dd, J = 7.5, 1.5 Hz, 1H), 5.38–5.34 (m, 1H), 5.25–5.21 (m, 1H), 2.11 (s, 3H), 2.09 (s, 3H), 1.69 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 199.1, 196.8, 143.9, 139.0, 138.8, 138.7, 137.9, 137.6, 137.1, 136.7, 132.5, 132.1, 131.4, 130.5, 130.1, 127.8, 127.1, 126.3, 126.13, 126.07, 20.12, 20.07, 17.3. HRMS (ESI) calcd for C25H22O2Na [M+Na]+ 377.1512, found 377.1524.
The reaction of 1a (62.8 mg, 0.20 mmol, 95% ee, 1.0 equiv) with cyclopropylmagnesium bromide (0.60 mmol, 3.0 equiv) afforded product 3aa (53.2 mg, 75%, 94% ee, ST = 99%) (eluent for column chromatography on silica gel PE/EtOAc = 10:1) as a white solid. − 11.7 (c 1.00, CH2Cl2). HPLC conditions: Chiralcel IC-3, isopropanol/hexane = 2:98, flow: 0.8 mL/min, λ = 254 nm, tR = 30.1 min (minor), 35.8 min (major). 1H NMR (500 MHz, CDCl3) δ 7.71–7.66 (m, 2H), 7.59 (dd, J = 7.5, 1.5 Hz, 1H), 7.52–7.47 (m, 1H), 7.43 (dd, J = 7.5, 1.5 Hz, 1H), 7.39–7.34 (m, 2H), 7.34–7.29 (m, 2H), 7.29–7.24 (m, 2H), 2.22–2.16 (m, 1H), 2.03 (s, 3H), 2.02 (s, 3H), 0.97–0.91 (m, 1H), 0.91–0.85 (m, 1H), 0.84–0.78 (m, 1H), 0.56–0.47 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 204.2, 197.1, 139.7, 139.3, 138.2, 137.6, 137.4, 137.1, 137.0, 132.6, 132.5, 132.0, 130.4, 128.0, 127.2, 126.9, 126.2, 125.8, 20.2, 20.1, 20.0, 12.4, 11.0. HRMS (ESI) calcd for C25H23O2 [M + H]+, 355.1693, found 355.1687.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
