1. Introduction
Nitrogen-containing heterocycles play a very important role in medicinal chemistry because they are the structural core of a broad spectrum of biologically active molecules. Some are in preclinical trials, and many others are already commercially available [
1]. Among the most privileged
N-heterocycles are the 5-substituted-1
H-tetrazoles (5S-1
H-Ts), due mainly to their ability to surrogate the carboxylic acids (RCOOH) into living systems since they have close values of pKa (5S-1
H-Ts: 4.5 to 4.9, RCOOH: 4.2 to 4.4), similar topologies, and practically the same electrostatic potential and electron density [
2]. For these reasons, 5S-1
H-Ts are considered bioisosteres of carboxylic acids [
3]. In addition, 5S-1
H-Ts are at least ten times more lipophilic compared to carboxylic acids, a property that makes them even more valuable for designing new drug candidates with enhanced pharmacokinetic profiles [
4]. Indeed, to date 23 drugs containing the 5S-1
H-T system have been approved by the FDA for safe use in humans, including antivirals, antiallergics, analgesics, anti-inflammatories, and especially antihypertensives such as the blockbuster drug losartan (
1,
Figure 1) [
5]. Another
N-heterocycle of high interest in medicinal chemistry, due also to its presence in many bioactive compounds and some marketed drugs such as the anticancer alkylating agent altretamine (
2,
Figure 1) [
6], is the 1,3,5-triazine, known as
s-triazine [
7]. This compound is a six-membered aromatic heterocycle containing three nitrogen atoms that are able to form strong H-bonds, and three C-
sp2 positions that can be decorated a la carte [
8]. Finally, the pyrrolo[3,4-
b]pyridin-5-one is a fused-type
bis-heterocycle that is also of high interest in medicinal chemistry. It includes the antidiabetic agent BMS-767778 (
3,
Figure 1) [
9] which is an N-
sp2-containing surrogate of the naturally occurring isoindolin-1-one (
3′,
Figure 1) [
10]. The pyrrolo[3,4-
b]pyridin-5-one system is very special in our research group. It has been the most essential molecule because it can be smartly assembled in a one-pot manner using multicomponent reactions (MCRs).
MCRs are privileged synthetic tools that involve sequential additions of at least three reagents into the same reactor (one pot). Given the nature of MCRs, they can be accessed quickly, in comparison to linear synthesis, to a series of highly functionalized and structurally complex products, leading to high atomic economy, eventually in good chemical yields and in short reaction times, needing only one work-up and in most cases just one purification process [
11]. MCRs (together with click reactions) are considered one of the most robust synthetic methodologies because a large variety of polyheterocyclic compounds of high interest in medicinal chemistry and optics have been synthesized in record time, satisfying the majority of the 12 principles of green chemistry [
12].
In 2001, J. Zhu and co-workers reported a new and elegant variant of the truncated Ugi reaction, recently recognized by us as the Ugi-Zhu three-component reaction (UZ-3CR) [
13], where primary amines react sequentially with aldehydes, and isocyanoacetamides (derived from amino acids) to lead to trisubstituted 5-aminooxazoles under mild reaction conditions [
14]. The most plausible reaction mechanism consists of the condensation between aldehydes
4 with amines
5 to give rise to the corresponding Schiff bases
6, which, depending on the substituents (
R1 and
R2), may or may not require activation by an acid, thus favoring the α-nucleophilic attack of the isocyanides
7, leading to formation of nitrilium cations
8. Then, the intermediate
8′ rapidly tautomerizes via a non-prototropic chain-ring process to the corresponding oxazoles
9 (
Scheme 1).
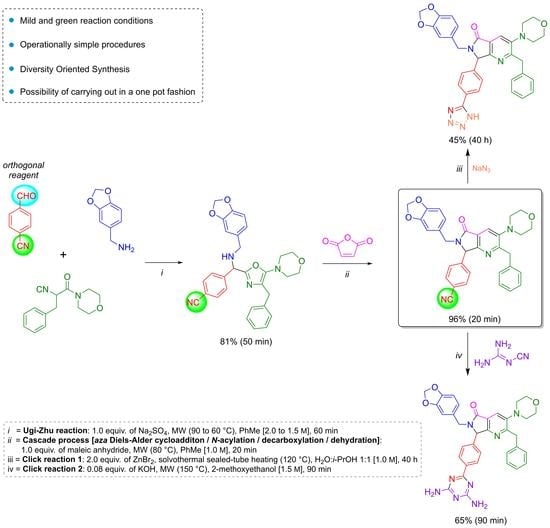
Thus, as a part of our ongoing program to synthesize novel and complex polyheterocyclic compounds, we have developed several methodologies based on UZ-3CR as key synthetic tool for preparing series of pyrrolo[3,4-
b]pyridin-5-ones either bound, fused or linked to various heterocyclic frameworks of interest in optics and medicinal chemistry. However, for the present work, the synthetic strategy behind a couple of new
tris-heterocycles, from the diversity-oriented synthesis (DOS) approach, started with an Ugi-Zhu reaction performed stepwise to first obtain a Schiff base, and then, the corresponding 5-aminooxazole. This pivotal heterocycle then reacts with maleic anhydride via a cascade process (
aza Diels–Alder cycloaddition/
N-acylation/decarboxylation/dehydration) to assemble the pyrrolo[3,4-
b]pyridin-5-one core [
15]. Finally, click reactions with different reagents and conditions provide the polyheterocyclic products.
2. Results and Discussion
The reaction sequence started with the synthesis of the Schiff base
12 via a condensation of the 4-formylbenzonitrile (
10) with piperonylamine (
11). It is worthy to note that the reagent
10 contains two functional groups, aldehyde, and nitrile. The first one is used in the UZ-3CR, and the second in further click reactions. Indeed,
10 is considered an orthogonal reagent. Thus, some experiments were performed with the aim to find the optimal reaction conditions. The first entry was carried out at room temperature, under continuous stirring, without additives or catalysts, and using methanol as solvent. The reaction was monitored with thin layer chromatography (TLC) until the amount of aldehyde in the reaction mixture was consumed or considerably decreased. However, after 24 hours, there was no appreciable change. Therefore, some additional experiments were performed with varying temperatures, additives, and solvents. The optimal conditions turned out to be using anhydrous toluene as solvent at 2.0 M concentration, one equivalent of anhydrous sodium sulfate as drying agent, and under microwave heating conditions at 90 °C for 30 min (see
Table S1 in the Supplementary Material for further details). It is essential to highlight that the addition of anhydrous sodium sulfate to the reaction mixture in the formation of the Schiff base allowed obtaining the condensation product
12 in 90% yield. This dehydrating salt is especially useful when reactions assisted by microwaves are carried out in closed-vessel systems, where Dean–Stark traps cannot be implemented. The next step was a nucleophilic α-addition of the isocyanide
13 [
16] to the imine
12 to provide the key 5-aminoxazole
14. The first tests behind optimal reaction parameters were carried out under relatively mild conditions since it was found previously that isocyanide
13 can undergo an acid-catalyzed process of chain-ring tautomerization leading to the 5-aminooxazole
15 as by-product. However, the desired 5-aminooxazole
14 was not detected by TLC. Thus, by increasing the temperature, traces of
14 were observed but the main isolated compound was
15, again. In this context, Lewis acids were used to activate the imine
12, so catalytic amounts of a panel of them in different proportions were added, and the reaction mixture was heated at 60 °C for 5 min before addition of the isocyanide
13. Then, the new resulting mixture was heated for 25 minutes using microwaves as heat source. The use of ytterbium triflate allowed obtaining the 5-aminooxazole
14 with the best yield (90%). The substantial improvement in the performance using ytterbium (III) compared to the other Lewis acids is because it is the most polarizable due to its size compared to scandium (III), indium (III), and aluminum (III). Indeed, it is a softer acid according to the HSAB Pearson principle, and therefore, it coordinates preferentially with the imine, activating it better. It is worth mentioning that experimentally, it was found that both indium chloride and scandium triflate reach coordination with isocyanide and even with the nitrile group, avoiding an efficient coordination with the imine (see
Table S2 in the Supplementary Material for further details). Then, the 5-aminoxazole
14 was reacted with maleic anhydride (
16) into a cascade process (
aza Diels-Alder cycloaddition/
N-acylation/decarboxylation/dehydration) to generate the corresponding pyrrolo[3,4-
b]pyridin-5-one
17. Different entries were carried out using toluene as solvent but varying temperature, finding that at 80 °C in 20 minutes the reaction was accomplished in 96% yield (see
Table S3 in the Supplementary Material for further details). It is worth mentioning that a couple of additional experiments were performed at temperatures just over 80 °C but decomposition and formation of many uncharacterized by-products were observed, in addition to a very difficult work-up. The next step was the transformation of nitrile group from the pyrrolo[3,4-
b]pyridin-5-one
17 into two nitrogenated heterocycles, tetrazole and triazine, both via click reactions. Thus, it has been widely documented that the most common reactions to synthesize 5-substituted-1
H-tetrazoles are the (3+2) cycloadditions [
17] between nitriles and some azide source as the 4π-component, catalyzed by Bronsted or Lewis acids [
18,
19,
20,
21]. In addition, it is known that aromatic nitriles as 2π-components in [3+2] cycloadditions react very low, unless they have strong EWGs [
22], or if the [3+2] cycloadditions are catalyzed by acids. One of the rare examples of non-catalyzed [3+2] cycloadditions involving 4π-components to nitriles (2π-components) was reported by Jasinski and co-workers in 2020 [
23]. In the compound
17, the nitrile group is placed in the
para-position relative to a methine group, so the [3+2] cycloaddition occurred due to a less steric hindrance, but it was not easy. Thus, a first attempt was carried out using the methodology described by Harasawa and co-workers in 2013 [
24], which is useful for the synthesis of 5-substituted-1
H-tetrazoles from electronically deactivated aromatic nitriles with some azide source. Unfortunately, the reaction did not proceed, probably due to a protonation of the N(
sp2)-atom from pyridine
17. Therefore, the strategy was changed, and the methodology published in 2001 by Demko and Sharpless was used, employing ZnBr
2 in equimolar amounts, and sodium azide (
18) [
25] in a mixture 1:1
v/
v of H
2O/
i-PrOH at reflux using conventional heating, or under microwave heating conditions. To our delight, the desired polyheterocycle
19 was obtained in both cases but in low yields, 9% and 6%, respectively, in addition to the fact that the work-up became laborious. Based on these results, it was considered that more drastic conditions were required to improve the yield, so the reaction was repeated, but using a pressure sealed tube. These conditions allowed performance of the cycloaddition but using a solvothermal sealed tube. Thus, the corresponding compound
19 was obtained in 45% yield, and, with respect to the work-up, it became trivial because once the reaction was finished, the tube reactor was left to cool, and a mixture of hexanes/EtOAc led to the precipitation of tetrazole. Finally, the polyheterocycle
19 was washed with ethyl acetate and hot water and dried under high vacuum (see
Table S4 in the Supplementary Material for further details). Finally, the preparation of a second polyheterocycle also from the chemical scaffold
17 consisted of the annulation reaction between
17 and dicyandiamide (
20) via a [3+2] cycloaddition assisted by microwave radiation. Thus, the reaction was carried out using DMSO as solvent and KOH as catalyst, according to the methodology reported by Fang and co-workers [
26]. For the first attempt at 100 °C, the reaction did not proceed, so, the temperature was increased to 125 °C, and 150 °C but, in both cases only traces of the product
21 were detected. It is worth mentioning that from 150 °C, the DMSO decomposes into methane, sulfur dioxide, and other volatile substances [
27,
28]. The reaction was repeated, replacing the DMSO by 2-methoxyethan-1-ol at 150 °C again, but this time the desired product
21 was obtained in 65% yield,
Scheme 2 (see
Table S5 in the Supplementary Material for further details).
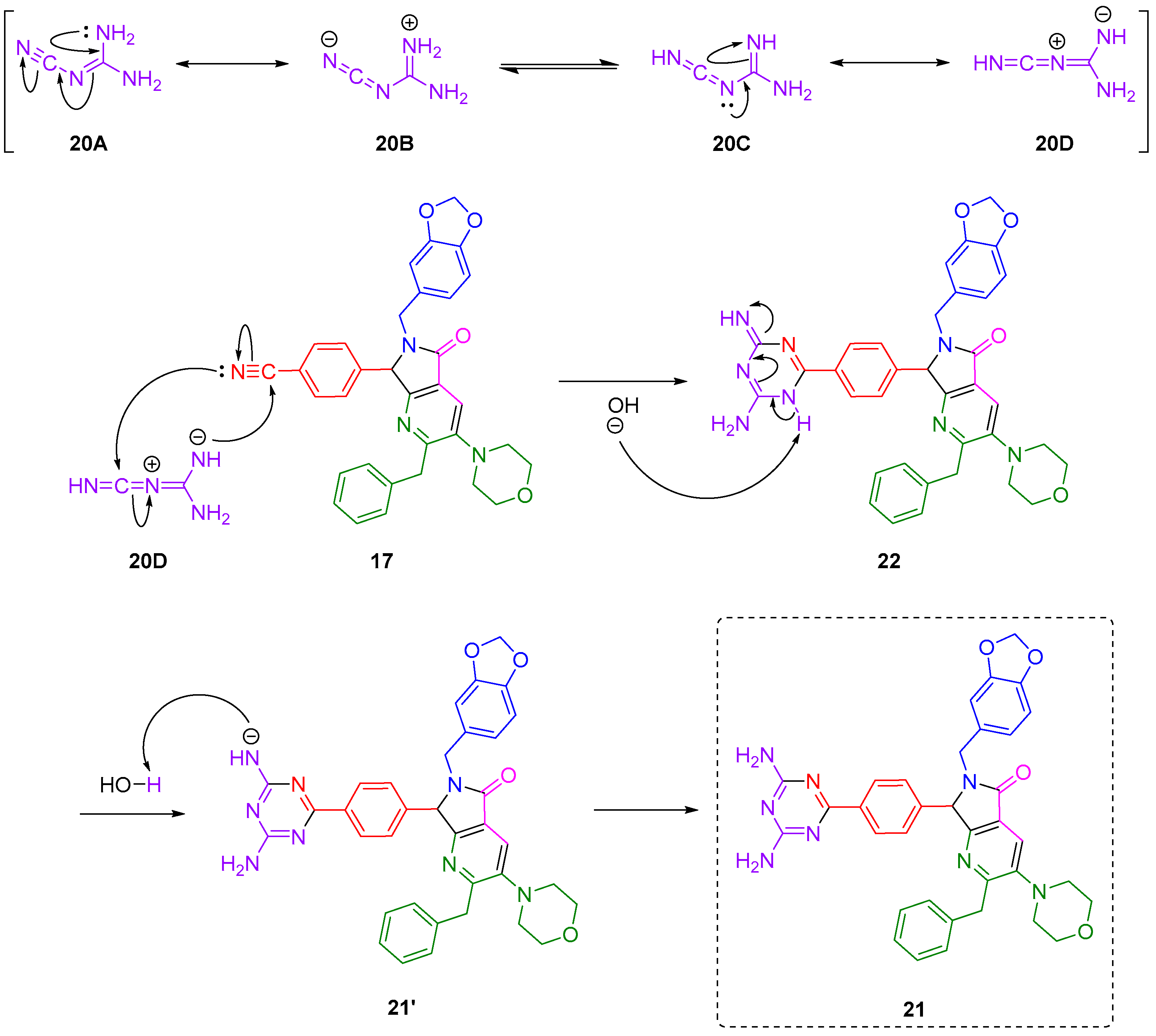
Although there are some reports about the synthesis of triazines from nitriles and dicyandiamide via [4+2] cycloadditions, to our best knowledge, there is no proposals of mechanisms. Thus, in
Scheme 3, a plausible reaction mechanism is proposed. Dicyandiamide (
20) exists in different canonic forms, with its zwitterionic form
20D the one that carries out the intermolecular [4+2] cycloaddition, leading to the formation of the six-membered polyheterocycle
22. This intermediate subsequently undergoes a base-assisted aromatization to get finally the 2,4-diamino-1,3,5-triazine-pyrrolo[3,4-
b]pyridin-5-one
21, after recovering a H-atom from water. It is noteworthy that despite
Scheme 3 suggesting a synchronous one step mechanism between
20D and
17, in fact the [4+2] cycloadditions can occur stepwise, even by non-polar [
29], or polar [
30] mechanisms. As a matter of fact, all kind of Diels–Alder cycloadditions, and, in general click reactions, remain controversial mainly due to their various possible reaction mechanisms. In this way, theoretical and experimental studies behind reaction mechanisms will be worthy of research.
Having the optimal conditions for the step-by-step synthesis of compounds
12,
14,
17 and
21, and with the idea of evaluating the robustness of this methodology from the one pot chemistry approach, the synthesis of the final compound
21 was carried out via a domino process coupling in sequential manner the Ugi-Zhu reaction with the
N-acylation, decarboxylation, dehydration, and the [4+2] cycloaddition, yielding 19%. Of course, this yield seems to be low, but it is not true at all considering the high number of formed bonds, the atom economy (89%), and that only a couple of water molecules and one carbon dioxide were released in the entire domino process (
Scheme 4). In addition, only one work up and purification process was needed. In the same way, only one solvent shift was performed, but by evaporation/addition, the reaction mixture was maintained in the same MW-sealed reaction tube.
3. Materials and Methods
3.1. General Information, Instrumentation, Software and Chemicals
1H and 13C nuclear magnetic resonance (NMR) spectra were acquired on a Bruker AMX Advance III spectrometer (500 MHz, Fällande, Uster, Switzerland). The solvents used for NMR experiments were deuterated chloroform (CDCl3) and deuterated dimethyl sulfoxide (DMSO-d6). Chemical shifts are reported in parts per million (d/ppm). Coupling constants are reported in Hertz (J/Hz). The internal reference for NMR spectra was tetramethyl silane (TMS) at 0.00 ppm. Multiplicities of the signals are reported using the standard abbreviations: singlet (s), doublet (d), triplet (t), quartet (q), and multiplet (m). NMR spectra were analyzed using the MestReNova software Ver. 12.0.0-20080 (A Coruña, Spain). The infrared (IR) spectrum was acquired on a Perkin-Elmer 2000 spectrometer (Norwalk, CT, USA) using the attenuated total reflectance (ATR) method. The maximum absorbance peaks are reported in reciprocal centimeters (νmax/cm−1), uncorrected. The IR spectrum was analyzed using the Origin software (Ver. 2018b, 9.55, OriginLab Corporations, Northampton, MA, USA). The high-resolution mass spectroscopy (HRMS) spectrum was acquired by electrospray ionization (ESI) on a Micro-TOF II spectrometer Bruker Daltonics GmbH (Bremen, Germany). The sample was injected directly (Apollo source) and analyzed by the time-of-flight method (TOF). The HRMS spectrum was analyzed using the Compass software (Ver. 1.5, Bruker Daltonik GmbH, Bremen, Germany). Microwave-assisted reactions were performed in closed-vessel mode on a CEM Discover SP MW-reactor (Matthews, North Carolina, CA, USA). Reaction progress was monitored by thin-layer chromatography (TLC), and the spots were visualized under ultraviolet (UV) light (254 or 365 nm). Glass preparative plates (20 × 20 cm) coated with silica-gel 60 doped with UV indicator (F254) were used to purify the products. All starting reagents and solvents were used as received (without further purification, distillation, or dehydration). Chemical structures were drawn using the ChemDraw software (Ver. 15.0.0.106 Professional, Perkin Elmer Informatics, Cambridge, MA, USA). The purity of all synthesized products (>96%) was assessed by NMR.
3.2. Synthesis of 4-(((Benzo[d] [1,3]dioxol-5-ylmethyl)imino)methyl)imino)methyl)benzonitrile 12
The 4-formylbenzonitrile (1.0 mmol, 1.0 equiv.), 1,3-benzodioxole-5-methylamine (1.0 mmol, 1.0 equiv.) and sodium sulfate anhydrous (1.0 mmol, 1.0 equiv.) were placed in a sealed CEM Discover microware reaction tube (10 mL) and diluted in anhydrous toluene (1.5 mL). The mixture was stirred and heated using microwave irradiation (90 °C, 150 W) for 30 min. Subsequently, the reaction mixture was filtered, and the resulting solid was washed with 15 mL of dichloromethane and 15 mL of acetone; then, it was dried under high vacuum, obtaining (237.86 mg, 90% yield) of a light yellow solid, Rf = 0.04 (EtOAc); 1H NMR (500 MHz, CDCl3): δ 8.37 (s, 1H, H-12), 7.86 (d, 2H, H-17, H-15, J = 8.0 Hz), 7.69 (d, 2H, H-18, H-14, J = 8.0 Hz), 6.84–6.76 (m, 3H, H-6, H-7, H-9), 5.94 (s, 2H, H-2), 4.76 (s, 2H, H-10) ppm; 13C NMR (125 MHz, CDCl3): δ 159.6 (C-12), 147.8 (C-4), 146.8 (C-5), 139.9 (C-13), 132.4 (C-15, C-17), 132.3 (C-8), 128.6 (C-14, C-18), 121.4 (C-7), 118.4 (C-19), 114.0 (C-16), 108.6 (C-6), 108.3 (C-9), 101.0 (C-2), 64.8 (C-10) ppm; HRMS: (ESI+) m/z calcd. for [M − H]+ C16H13N2O2+ 265.0972, found 265.0968 (error = 1.3 ppm); FT-IR (): 2835 (aryl H), 2242 (CN), 1480, 1442 (C=N), 1362, 1244 (C=C), 1179 (C–O), 1034, 916, 839 (C–C).
3.3. Synthesis of 4-(((Benzo[d][1,3]dioxol-5-ylmethyl)amino)(4-benzyl-5-morpholinooxazol-2-yl)methyl)benzonitile 14
The 4-formylbenzonitrile (1.0 mmol, 1.0 equiv.), 1,3-benzodioxole-5-methylamine (1.0 mmol, 1.0 equiv.) and sodium sulfate anhydrous (1.0 mmol, 1.0 equiv.) were placed in a sealed CEM Discover microware reaction tube (10 mL) and diluted in anhydrous toluene (1.5 mL). The mixture was stirred and heated using microwave irradiation (90 °C, 150 W) for 30 min, and ytterbium (III) triflate (0.08 equiv.) was added. The mixture was stirred and heated using microwave irradiation (60 °C, 60 W) for 5 min, and then 2-isocyano-1-morpholino-3-phenylpropan-1-one (1.2 mmol, 1.2 equiv.) was added. The new mixture was stirred and again heated using microwave irradiation (70 °C, 150 W) for 25 min, time it took to complete the reaction. Then, the solvent was evaporated under reduced pressure and the reaction crude was diluted with ethyl acetate. A Na2CO3(aq.) (0.4 M) solution was added, and liquid–liquid extractions were carried out three times. The organic phases were collected and washed with brine three times (3 × 10 mL). The organic phase was dried with anhydrous MgSO4 and concentrated to dryness under vacuum. The resulting crude was purified by column chromatography using a mixture of hexanes: ethyl acetate 3:1 (v/v) as eluent to obtain a slightly yellowish oil (457.72 mg, 90% yield), Rf = 0.36 (Hex-EtOAc = 3:2 v/v); 1H NMR (500 MHz, CDCl3): δ 7.62 (d, 2H, H-33, H-35, J = 8.6 Hz), 7.54 (d, 2H, H-32, H-36, J = 8.6 Hz), 7.29–7.25 (m, 2H, H-27, H-29), 7.23–7.17 (m, 3H, H-28, H-26, H-30), 6.80 (dd, 1H, H-16, J = 1.7, 0.5 Hz), 6.71 (dd, 1H, H-22, J = 7.9, 0.5 Hz), 6.67 (dd, 1H, H-23, J = 7.9, 1.7 Hz), 5.91 (s, 2H, H-19), 4.88 (s, 1H, H-12), 3.81 (s, 1H, H-24), 3.70–3.67 (m, 4H, H-2, H-6), 3.65–3.61 (m, 2H, H-14), 2.92–2.89 (m, 4H, H-3, H-5) ppm; 13C NMR (125 MHz, CDCl3): δ 157.5 (C-9), 152.3 (C-7), 147.7 (C-17), 146.7 (C-21), 144.6 (C-31), 139.2 (C-25), 132.9 (C-15), 132.3 (C-33, C-35), 128.3 (C-26, C-30, C-27, C-29, C-32, C-36), 126.1 (C-28), 124.7 (C-37), 121.4 (C-22), 188.5 (C-11), 111.7 (C-31), 108.7 (C-16), 108.0 (C-23), 100.8 (C-19), 66.6 (C-2, C-6), 59.4 (C-12), 51.2 (C-14), 50.8 (C-3, C-5), 31.7 (C-24) ppm; HRMS: (ESI+) m/z calcd. for [M − H]+ C30H29N4O4+ 509.2183, found 509.2188 (error = 1.0 ppm); IR (): 3324 (aryl H), 2958, 2892, 2854 (C–H), 1630, 1607 (N–H), 2227 (CN), 1441 (C=N), 1374, 1298 (C=C), 1120 (C–O–C), 1032, 992, 920, 799, 732 (C–C).
3.4. Synthesis One Pot of 4-(6-(Benzo[d][1,3]dioxol-5-ylmethyl)-2-benzyl-3-morpholino-5-oxo-6,7-dihydro-5H-pyrrolo[3,4-b]pyridin-7-yl)benzonitrile 17
General Procedure (GP): 4-formylbenzonitrile (1.0 mmol, 1.0 equiv.), 1,3-benzodioxole-5-methylamine (1.0 mmol, 1.0 equiv.) and sodium sulfate anhydrous (1.0 mmol, 1.0 equiv.) were placed in a sealed CEM Discover microwave reaction tube (10 mL) and diluted in anhydrous toluene (1.5 mL). The mixture was stirred and heated using microwave irradiation (90 °C, 150 W) for 30 min, and ytterbium (III) triflate (0.08 equiv.) was added. The mixture was stirred and heated using microwave irradiation (60 °C, 60 W) for 5 min, and then 2-isocyano-1-morpholino-3-phenylpropan-1-one (1.2 mmol, 1.2 equiv.) was added. The new mixture was stirred and again heated using microwave irradiation (70 °C, 150 W) for 25 min, and then maleic anhydride (1.3 mmol, 1.0 equiv.) was added. Finally, the reaction mixture was stirred and heated using microwave irradiation (80 °C, 150 W) for 20 min. Then, the solvent was removed to dryness under vacuum. The crude was extracted using ethyl acetate (3 × 10.0 mL) and Na2CO3(aq.) (3 × 10.0 mL), and then washed with brine (3 × 10 mL). The organic layer was dried using anhydrous MgSO4, filtered, and concentrated to dryness under vacuum. The new crude was purified by silica-gel column chromatography using mixtures of hexanes (Hex) and ethyl acetate (EtOAc) in 4:1 to 3:2 (v/v) proportions as mobile phase to isolate the corresponding pyrrolo[3,4-b]pyridin-5-one-7-yl benzonitrile (490.15 mg, 90% yield) as yellow oil, Rf = 0.30 (Hex-EtOAc = 3:2 v/v); 1H NMR (500 MHz, CDCl3): δ 7.90 (s, 1H, H-15), 7.67–7.64 (m, 2H, H-25, H-27), 7.28–7.25 (m, 2H, H-24, H-28), 7.18–7.10 (m, 5H, H-18, H-22), 6.70 (dd, 1H, H-37, J = 7.9, 0.4 Hz), 6.67 (d, 1H, H-31, J = 1.7 Hz), 6.57 (dd, 1H, H-38, J = 7.9, 1.7 Hz), 5.92 (d, 1H, H-34, J = 1.4 Hz), 5.91 (d, 1H, H-34’, J = 1.4 Hz), 5.33 (d, 1H, H-29, J = 14.5 Hz), 5.29 (s, 1H, H-11), 4.26 (d, 1H, H-16, J = 14.0 Hz), 4.15 (d, 1H, H-16’, J = 14.0 Hz), 3.81 (t, 4H, H-2, H-6, J = 4.6 Hz), 3.73 (d, 1H, H-29’, J = 4.6 Hz), 2.87–2.80 (m, 4H, H-3, H-5) ppm; 13C NMR (125 MHz, CDCl3): δ 167.0 (C-13), 162.3 (C-8), 159.2 (C-10), 148.2 (C-7), 148.1 (C-32), 147.3 (C-36), 141.1 (C-23), 139.0 (C-17), 132.7 (C-25, C-27), 130.0 (C-30), 128.7 (C-18, C-22), 128.6 (C-24, C-28), 128.2 (C-19, C-21), 126.2 (C-20), 123.9 (C-15), 123.5 (C-14), 121.8 (C-38), 118.3 (C-40), 112.5 (C-26), 108.7 (C-31), 108.3 (C-37), 101.2 (C-34), 67.0 (C-2, C-6), 63.7 (C-11), 53.0 (C-3, C-5), 44.0 (C-29), 39.9 (C-16) ppm; HRMS: (ESI+) m/z calcd. for [M − H]+ C33H29N4O4+ 545.2183, found 545.2192 (error = 1.6 ppm); IR (): 3419 (aryl H), 3005, 2968 (C–H), 1710 (C=O, ketone), 1490 (C=N), 1441 (C=C), 1416 (C–O–C), 1361, 1220, 1117, 1034, 905 (C–C).
3.5. Synthesis of 7-(4-(1H-Tetrazol-5-yl)phenyl)-6-(benzo[d][1,3]dioxol-5-ylmethyl)-2-benzyl-3-morpholino-6,7-dihydro-5H-pyrrolo[3,4-b]pyridin-5-one 19
According to the GP, compound 17 was synthesized, and once the reaction was finished, the solvent was evaporated to dryness, and the crude was transferred to a Pyrex pressure tube (5 mL), sodium azide (1.5 mmol, 1.5 equiv.) and zinc bromide (2.0 mmol, 2.0 equiv.) were added. The mixture was dissolved in 1.5 mL of H2O and 1.5 mL of isopropyl alcohol, and then the mixture of reaction was heated in an oil bath at 120 °C for 40 hours. Subsequently, the reaction mixture was allowed to cool to room temperature, and a mixture 1:1 v/v of hexanes with ethyl acetate was added at 0 °C, stirred for 10 min, and then filtered and washed with hot water and hot ethyl acetate. The resulting solid was dried under high vacuum overnight, obtaining 264.44 mg of a light brown solid (45% yield); mp > 250 °C; 1H NMR (500 MHz, DMSO-d6): δ 7.96 (d, 2H, H-25, H-27, J = 7.9 Hz), 7.92 (s, 1H, H-15), 7.16 (d, 2H, H-24, H-28, J = 7.9 Hz), 7.14–7.05 (m, 5H, H-18, H-22), 6.79 (d, 1H, H-37, J = 7.9 Hz), 6.72 (d, 1H, H-31, J = 1.7 Hz), 6.62–6.60 (m, 1H, H-38), 5.95 (d, 1H, H-34, J = 1.0 Hz), 5.93 (d, 1H, H-34’, J = 1.0 Hz), 5.44 (s, 1H, H-11), 5.00 (d, 1H, H-29, J = 15.1 Hz), 4.28 (d, 1H, H-16, J = 14.2 Hz), 4.06 (d, 1H, H-16’, J = 14.2 Hz), 3.76 (d, 1H, H-29’, J = 15.1 Hz), 3.72–3.66 (m, 4H, H-2, H-6), 2.89–2.76 (m, 4H, H-3, H-5) ppm; 13C NMR (125 MHz, DMSO-d6): δ 166.5 (C-13), 161.7 (C-8), 160.2 (C-10), 148.1 (C-7), 147.9 (C-32), 146.9 (C-36), 139.7 (C-17), 136.5 (C-26), 131.1 (C-30), 130.1 (C-40), 129.0 (C-18, C-22), 128.7 (C-24, C-28), 128.5 (C-19, C-21), 127.5 (C-25, C-27), 126.3 (C-20), 124.0 (C-15), 123.8 (C-14), 121.7 (C-38), 108.7 (C-31), 108.6 (C-37), 101.3 (C-34), 66.8 (C-2, C-6), 64.1 (C-11), 52.8 (C-3, C-5), 43.7 (C-29), 40.4 (C-16) ppm; HRMS: (ESI+) m/z calcd. for [M − H]+ C33H30N7O4+ 588.2354, found 588.2348 (error = 1.0 ppm); IR (: 2973, 2867 (C–H), 2368, 2187, 2156, 2090 (C=N), 1993, 1922, 1741 (C=C), 1701 (C=O, ketone), 1599, 1493, 1445, 1397, 1365 (C=N), 1290, 1246, 1220 (C–O–C), 1158, 1114, 1034, 1013, 915, 862, 796, 787, 729, 703, 650, 623 (C–C).
3.6. Synthesis of 6-(Benzo[d][1,3]dioxol-5-ylmethyl)-2-benzyl-7-(4-(4,6-diamino-1,3,5-triazin-2-yl)phenyl)-3-morpholino-6,7-dihydro-5H-pyrrolo[3,4-b]pyridin-5-one 21
According to the GP, compound 17 was synthesized, and once the reaction was finished, the solvent was evaporated to dryness, and to the crude, 1.5 mL of 2-methoxyethan-1-ol, dicyandiamide (1.2 mmol, 1.2 equiv.), and potassium hydroxide (0.008 mmol, 0.0008 equiv.) were added. The mixture was stirred and heated using microwave irradiation (150 °C, 250 W) for 90 min. Subsequently, the reaction mixture was allowed to cool to room temperature. Then, hot water was stirred for 10 min, and then filtered and washed with hot water and hot ethyl acetate. The resulting solid was dried under a high vacuum overnight, obtaining 408.65 mg of a yellow solid (65% yield); mp > 250 °C; 1H NMR (500 MHz, CDCl3): δ 8.28 (d, 2H, H-25, H-27, J = 8.6 Hz), 7.92 (s, 1H, H-11), 7.20 (d, 2H, H-24, H-28, J = 8.6 Hz), 7.15–7.09 (m, 5H, H-18, H-22), 6.70 (d, 1H, H-37, J = 7.9 Hz), 6.69 ( d, 1H, H-31, J = 1.7 Hz), 6.59 (dd, 1H, H-38, J = 7.8, 1.7 Hz), 5.92 (d, 1H, H-34, J = 1.4 Hz), 5.91 (d, 1H, H-34’, J = 1.4 Hz), 5.47 (bs, 4H, H-46, H-47), 5.33 (d, 1H, H-29, J = 14.8 Hz), 5.30 (s, 1H, H-11), 4.80 (d, 1H, H-16, J = 13.9 Hz), 4.12 (d, 1H, H-16’, J = 13.9 Hz), 3.81–3.77 (m, 4H, H-2, H-6), 3.66 (d, 1H, H-29’, J = 14.8 Hz), 2.86–2.77 (m, 4H, H-3, H-5) ppm; 13C NMR (125 MHz, CDCl3): δ 171.8 (C-40), 167.6 (C-42, C-44), 167.0 (C-13), 162.2 (C-8), 160.2 (C-10), 148.0 (C-17), 147.9 (C-32), 147.2 (C-36), 139.2 (C-7), 138.9 (C-23), 137.0 (C-26), 130.4 (C-30), 129.0 (C-25, C-27), 128.7 (C-18, C-22), 128.2 (C-24, C-28), 128.0 (C-19, C-21), 126.1 (C-20), 124.0 (C-15), 123.8 (C-14), 121.9 (C-38), 108.9 (C-37), 108.3 (C-31), 101.1 (C-34), 67.1 (C-2, C-6), 64.1 (C-11), 53.0 (C-3, C-5), 43.7 (C-29), 40.0 (C-16) ppm; HRMS: (ESI+) m/z calcd. for [M − H]+ C35H33N8O4+ 629.2619, found 629.2647 (error = 4.4 ppm); IR (): 3419 (aryl H), 3009, 2964 (C–H), 1715 (C=O, ketone), 1609, 1542 (N–H, secondary amine), 1485, 1440 (C=N), 1419, 1357 (C=C), 1220 (C–O–C), 1096, 1046, 902, 818, 778, 695.





 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




