
Gaseous- and Condensed-Phase Activities of Some Reactive P- and N-Containing Fire Retardants in Polystyrenes

 , , ,
, , , 
Abstract
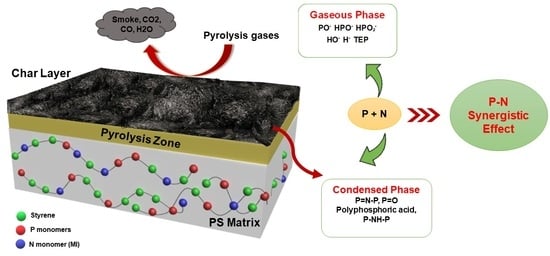
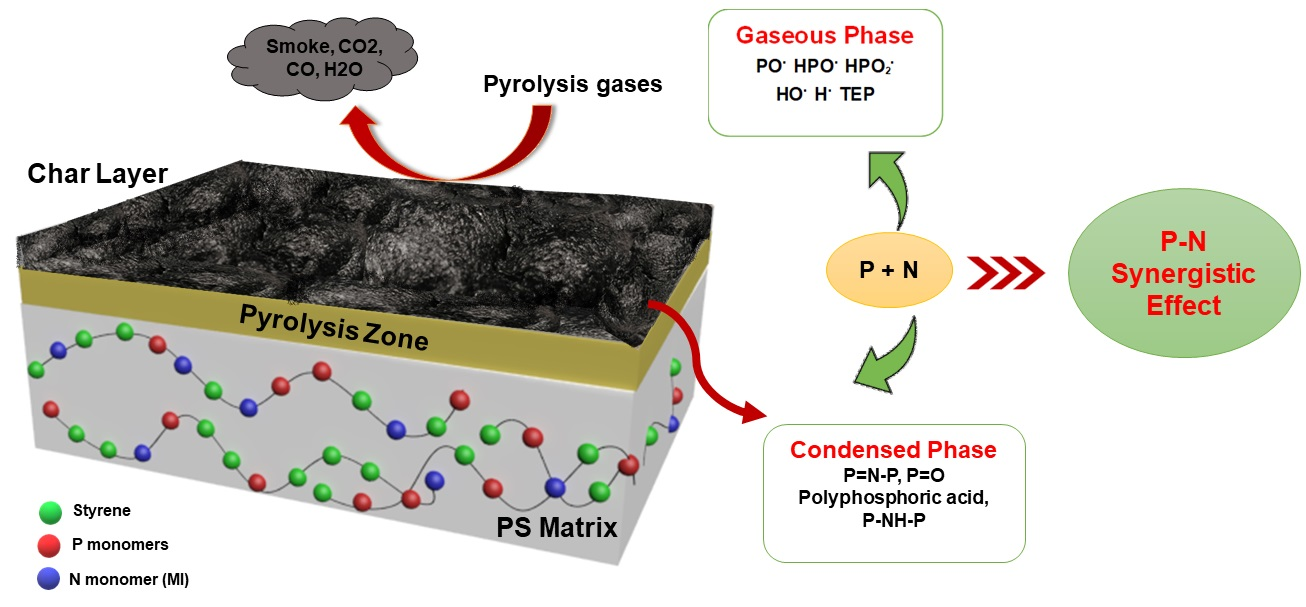
1. Introduction
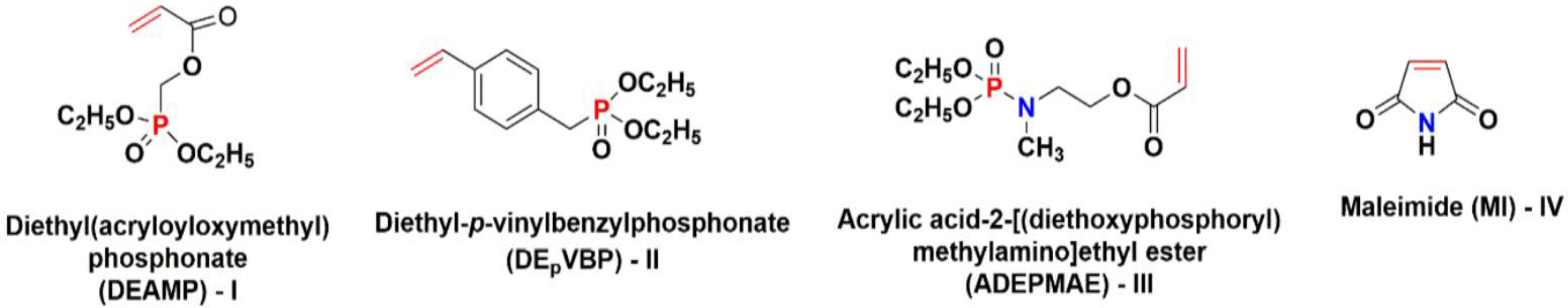
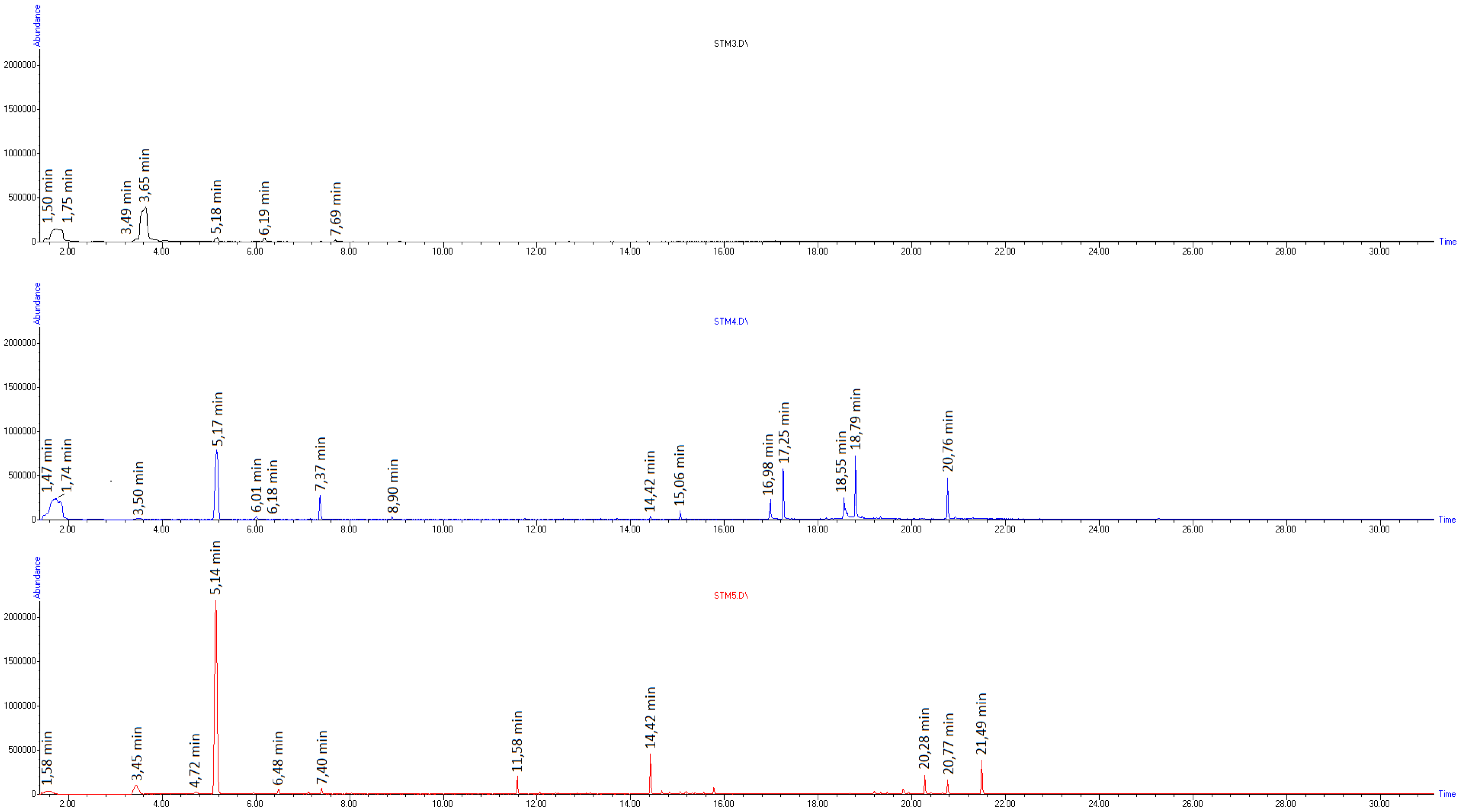
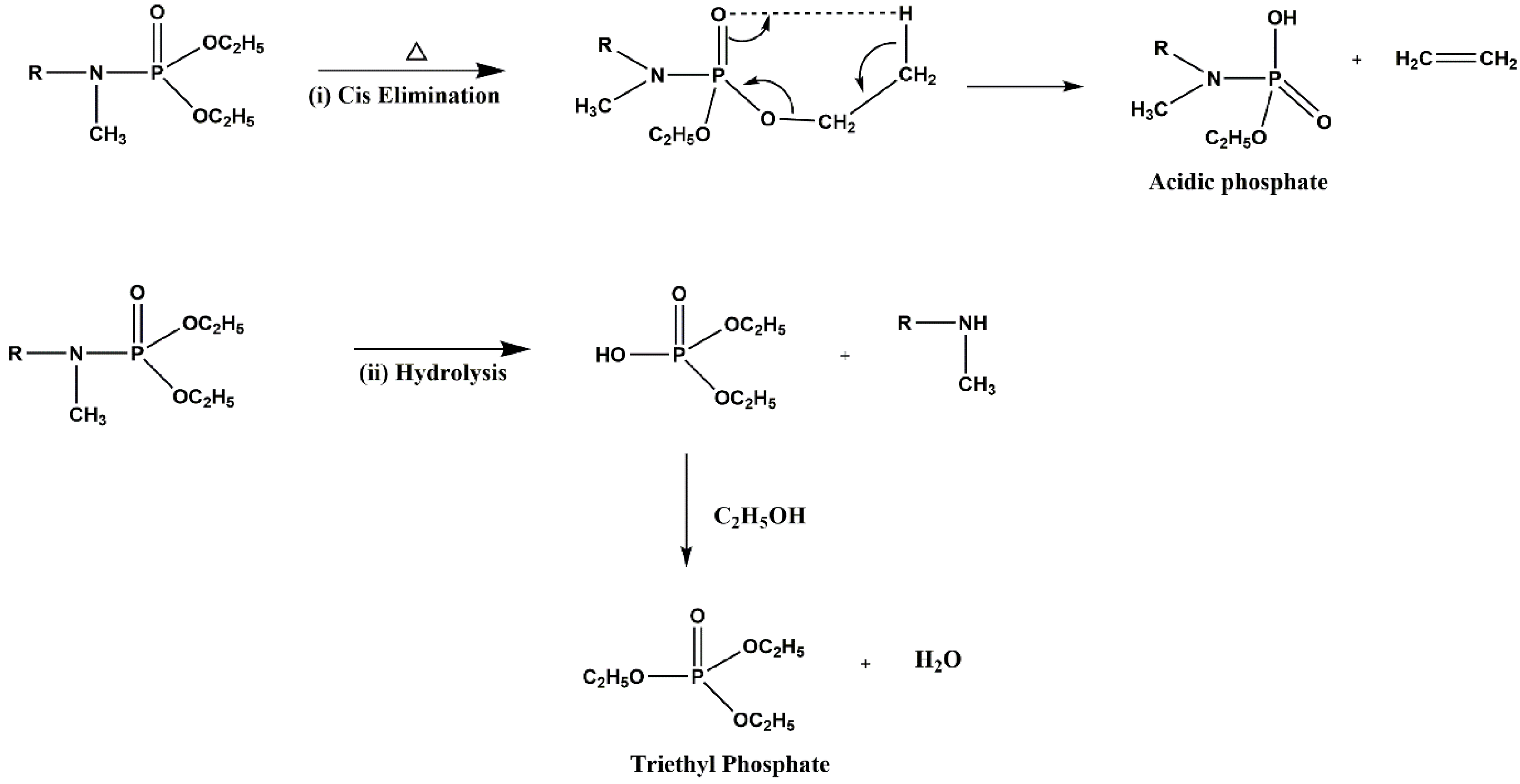
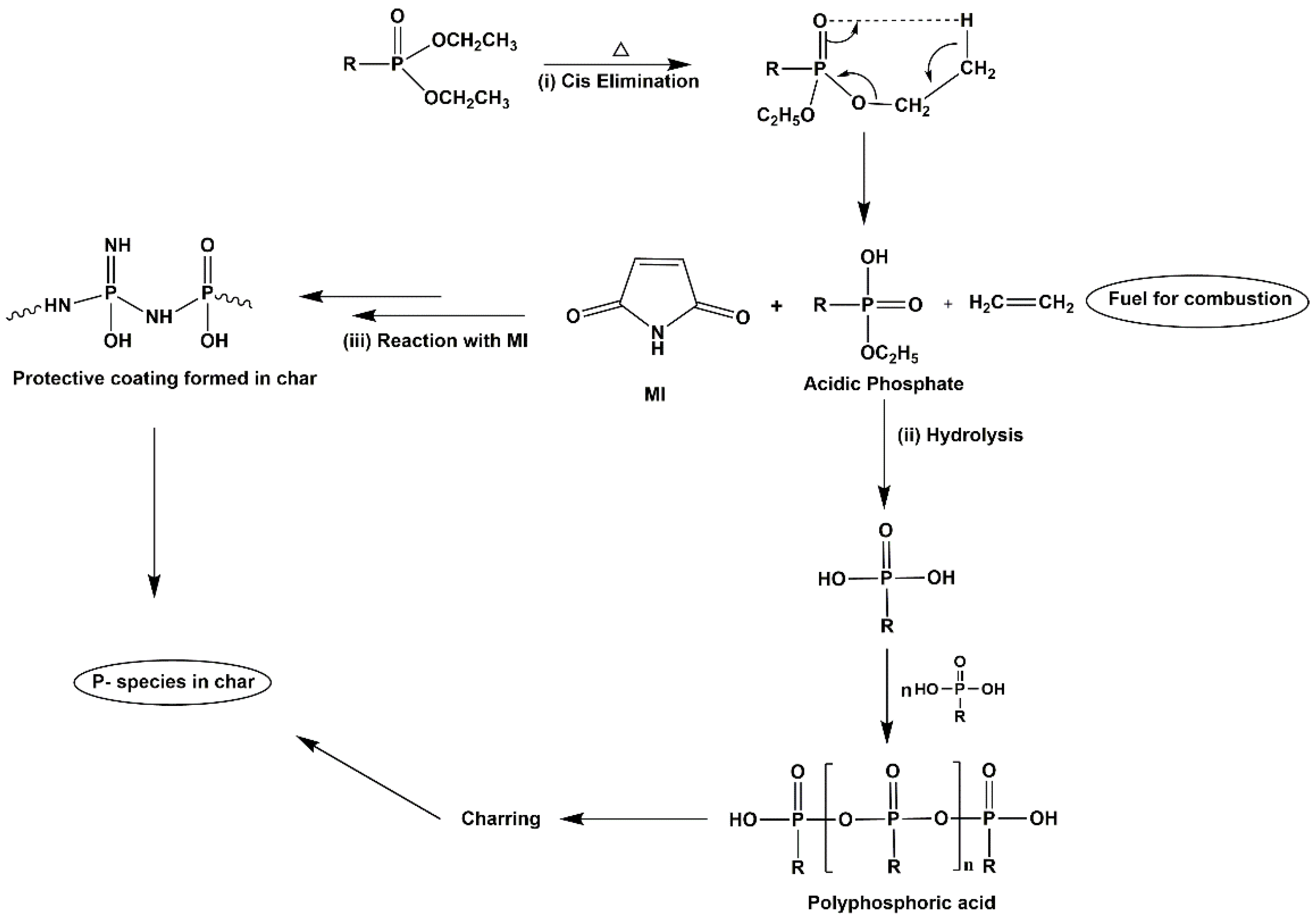
2. Results and Discussion
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Characteriztion Techniques
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Lynwood, C. Polystyrene: Synthesis, Characteristics, and Applications; Nova Science Publishers: New York, NY, USA, 2014. [Google Scholar]
- Weil, E.D.; Levchik, S.V. Flame retardants for polystyrenes in commercial use or development. J. Fire Sci. 2007, 25, 241–265. [Google Scholar] [CrossRef]
- Joseph, P.; Tretsiakova-McNally, S. Melt-flow behaviours of thermoplastic materials under fire conditions: Recent experimental studies and some theoretical approaches. Materials 2015, 8, 8793–8803. [Google Scholar] [CrossRef] [PubMed]
- Fire Safe Europe. Available online: https://firesafeeurope.eu/fseu-publications/ (accessed on 18 November 2022).
- Joseph, P.; Tretsiakova-McNally, S. Reactive modifications of some chain- and step-growth polymers with phosphorus-containing compounds: Effects on flame retardance—A review. Polym. Adv. Technol. 2011, 22, 395–406. [Google Scholar] [CrossRef]
- Baby, A.; Tretsiakova-McNally, S.; Arun, M.; Joseph, P.; Zhang, J. Reactive and additive modifications of styrenic polymers with phosphorus-containing compounds and their effects on fire retardance. Molecules 2020, 25, 3779. [Google Scholar] [CrossRef] [PubMed]
- Jakab, E.; Uddin, M.A.; Bhaskar, T.; Sakata, Y. Thermal decomposition of flame-retarded high-impact polystyrene. J. Anal. Appl. Pyrolysis 2003, 68–69, 83–99. [Google Scholar] [CrossRef]
- Baby, A.; Tretsiakova-McNally, S.; Joseph, P.; Arun, M.; Zhang, J.; Pospiech, D. The influence of phosphorus- and nitrogen- containing groups on the thermal stability and combustion characteristics of styrenic polymers. J. Therm. Anal. Calorim. 2022, 2022, 1–13. [Google Scholar] [CrossRef]
- Ezechiá, M.; Covino, S.; Cajthaml, T. Ecotoxicity and biodegradability of new brominated flame retardants: A review. Ecotoxicol. Environ. Saf. 2014, 110, 153–167. [Google Scholar] [CrossRef]
- Morgan, A.B. The future of flame retardant polymers—Unmet needs and likely new approaches. Polym. Rev. 2019, 59, 25–54. [Google Scholar] [CrossRef]
- Joseph, P.; Arun, M.; Bigger, S.; Guerrieri, M.; Pospiech, D.; Harnisch, C. Mode of action of condensed- and gaseous-phase fire retardation in some phosphorus-modified polymethyl methacrylate- and polystyrene-based bulk polymers. Polymers 2021, 13, 3402. [Google Scholar] [CrossRef]
- Levchik, V.; Weil, D. A review of recent progress in phosphorus-based flame retardants. J. Fire Sci. 2006, 24, 345–364. [Google Scholar] [CrossRef]
- Joseph, P.; Tretsiakova-McNally, S. Combustion behaviours of chemically modified polyacrylonitrile polymers containing phosphorylamino groups. Polym. Degrad. Stab. 2012, 97, 2531–2535. [Google Scholar] [CrossRef]
- Tretsiakova-McNally, S.; Joseph, P. Pyrolysis combustion flow calorimetry studies on some reactively modified polymers. Polymers 2015, 7, 453–467. [Google Scholar] [CrossRef]
- Ebdon, J.R.; Price, D.; Hunt, B.J.; Joseph, P.; Gao, F.; Milnes, G.J.; Cunliffe, L.K. Flame retardance in some polystyrenes and poly(methyl methacrylate)s with covalently bound phosphorus-containing groups: Initial screening experiments and some laser pyrolysis mechanistic studies. Polym. Degrad. Stab. 2000, 69, 267–277. [Google Scholar] [CrossRef]
- Price, D.; Cunliffe, L.K.; Bullett, K.J.; Hull, T.R.; Milnes, G.J.; Ebdon, J.R.; Hunt, B.J.; Joseph, P. Thermal behaviour of covalently bonded phosphate and phosphonate flame retardant polystyrene systems. Polym. Degrad. Stab. 2007, 92, 1101–1114. [Google Scholar] [CrossRef]
- Tretsiakova-McNally, S.; Joseph, P. Thermal and calorimetric evaluations of polyacrylonitrile containing covalently-bound phosphonate groups. Polymers 2018, 10, 131. [Google Scholar] [CrossRef] [PubMed]
- Grand, A.F. Fire Retardancy of Polymeric Materials; Taylor & Francis Milton Park: Abingdon-on-Thames, UK, 2000. [Google Scholar]
- Ramgobin, A.; Fontaine, G.; Bourbigot, S. A case study of polyether ether ketone (I): Investigating the thermal and fire behaviour of a high-performance material. Polymers 2020, 12, 1789. [Google Scholar] [CrossRef] [PubMed]
- Salmeia, K.A.; Fage, J.; Liang, S.; Gaan, S. An overview of mode of action and analytical methods for evaluation of gas phase activities of flame retardants. Polymers 2015, 7, 504–526. [Google Scholar] [CrossRef]
- Qiu, T.; Ge, F.; Li, C.; Lu, S. Study of the thermal degradation of flame-retardant polyester GFRP using TGA and TG-FTIR-GC/MS. J. Therm. Anal. Calorim. 2022, 147, 5743–5760. [Google Scholar] [CrossRef]
- Jiao, L.; Sun, J. A thermal degradation study of insulation materials extruded polystyrene. Procedia Eng. 2014, 71, 622–628. [Google Scholar] [CrossRef]
- Ahmad, Z.; Al-Sagheer, F.; Al-Awadi, N.A. Pyro-GC/MS and thermal degradation studies in polystyrene–poly(vinyl chloride) blends. J. Anal. Appl. Pyrolysis 2010, 87, 99–107. [Google Scholar] [CrossRef]
- Garrison, A.W.; Boozer, C.E. The acid-catalyzed hydrolysis of a series of phosphoramidates. J. Am. Chem. Soc. 1968, 90, 3489–3494. [Google Scholar] [CrossRef]
- Ablouh, E.H.; Brouillette, F.; Taourirte, M.; Sehaqui, H.; El Achaby, M.; Belfkira, A. A highly efficient chemical approach to producing green phosphorylated cellulosic macromolecules. RSC Adv. 2021, 11, 24206–24216. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Arun, M.; Moinuddin, K.; Joseph, P. Mechanistic aspects of condensed- and gaseous-phase activities of some phosphorus-containing fire retardants. Polymers 2020, 12, 1801. [Google Scholar] [CrossRef]
- Ghanadpour, M.; Wicklein, B.; Carosio, F.; Wågberg, L. All-natural and highly flame-resistant freeze-cast foams based on phosphorylated cellulose nanofibrils. Nanoscale 2018, 10, 4085–4095. [Google Scholar] [CrossRef]
- Cade-Menun, B.J. Characterizing phosphorus in environmental and agricultural samples by 31P Nuclear Magnetic Resonance spectroscopy. Talanta 2005, 66, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Salmeia, K.A.; Gaan, S. An overview of some recent advances in DOPO-derivatives: Chemistry and flame retardant applications. Polym. Degrad. Stab. 2015, 113, 119–134. [Google Scholar] [CrossRef]
- Zheng, Z.; Liu, S.; Wang, B.; Yang, T.; Cui, X.; Wang, H. Preparation of a novel phosphorus- and nitrogen-containing flame retardant and its synergistic effect in the intumescent flame-retarding polypropylene system. Polym. Compos. 2015, 36, 1606–1619. [Google Scholar] [CrossRef]
- Yang, S.; Hu, Y.; Zhang, Q. Synthesis of a phosphorus–nitrogen-containing flame retardant and its application in epoxy resin. High Perform. Polym. 2019, 31, 186–196. [Google Scholar] [CrossRef]
- You, G.; Cheng, Z.; Peng, H.; He, H. The synthesis and characterization of a novel phosphorus–nitrogen containing flame retardant and its application in epoxy resins. J. Appl. Polym. Sci. 2014, 131, 1–8. [Google Scholar] [CrossRef]
- Smith, M.; Scudiero, L.; Espinal, J.; McEwen, J.S.; Garcia-Perez, M. Improving the deconvolution and interpretation of XPS spectra from chars by Ab Initio calculations. Carbon 2016, 110, 155–171. [Google Scholar] [CrossRef]
- Bulusheva, L.G.; Arkhipov, V.E.; Popov, K.M.; Sysoev, V.I.; Makarova, A.A.; Okotrub, A.V. Electronic structure of nitrogen-and phosphorus-doped graphenes grown by chemical vapor deposition method. Materials 2020, 13, 1173. [Google Scholar] [CrossRef] [PubMed]
- Skorupska, M.; Kamedulski, P.; Lukaszewicz, J.P.; Ilnicka, A. The improvement of energy storage performance by sucrose-derived carbon foams via incorporating nitrogen atoms. Nanomaterials 2021, 11, 760. [Google Scholar] [CrossRef] [PubMed]
- Ayiania, M.; Smith, M.; Hensley, A.J.R.; Scudiero, L.; McEwen, J.S.; Garcia-Perez, M. Deconvoluting the XPS spectra for nitrogen-doped chars: An analysis from first principles. Carbon 2020, 162, 528–544. [Google Scholar] [CrossRef]
- Wang, C.; Hu, F.; Yang, H.; Zhang, Y.; Lu, H.; Wang, Q. 1.82 wt. % Pt/N, P co-doped carbon overwhelms 20 wt. % Pt/C as a high-efficiency electrocatalyst for hydrogen evolution reaction. Nano Res. 2017, 10, 238–246. [Google Scholar] [CrossRef]
- Wu, J.; Zheng, X.; Jin, C.; Tian, J.; Yang, R. Ternary doping of phosphorus, nitrogen, and sulfur into porous carbon for enhancing electrocatalytic oxygen reduction. Carbon 2015, 92, 327–338. [Google Scholar] [CrossRef]
- NIST XPS Database, Spectrum Search Result. Available online: https://srdata.nist.gov/xps (accessed on 26 May 2022).
- Gaan, S.; Sun, G.; Hutches, K.; Engelhard, M.H. Effect of nitrogen additives on flame retardant action of tributyl phosphate: Phosphorus-nitrogen synergism. Polym. Degrad. Stab. 2008, 93, 99–108. [Google Scholar] [CrossRef]
- Yung, H.; Shih, P.Y.; Liu, H.S.; Chin, T.S. Nitridation effect on properties of stannous-lead phosphate glasses. J. Am. Ceram. Soc. 1997, 80, 2213–2220. [Google Scholar] [CrossRef]
- Markwart, J.C.; Battig, A.; Zimmermann, L.; Wagner, M.; Fischer, J.; Schartel, B.; Wurm, F.R. Systematically controlled decomposition mechanism in phosphorus flame retardants by precise molecular architecture: P–O vs P–N. ACS Appl. Polym. Mater. 2019, 1, 1118–1128. [Google Scholar] [CrossRef]
- Wang, P.; Cai, Z. Highly efficient flame-retardant epoxy resin with a novel DOPO-based triazole compound: Thermal stability, flame retardancy and mechanism. Polym. Degrad. Stab. 2017, 137, 138–150. [Google Scholar] [CrossRef]
- Zhu, Z.-M.; Wang, L.-X.; Lin, X.-B.; Dong, L.-P. Synthesis of a novel phosphorus-nitrogen flame retardant and its application in epoxy resin. Polym. Degrad. Stab. 2019, 169, 108981. [Google Scholar] [CrossRef]
- Valencoso, M.; Battig, A.; Markwart, J.C.; Schartel, B.; Wurm, F.R. Molecular firefighting—How modern phosphorus chemistry can help solve the challenge of flame retardancy. Angew. Chem. Int. Ed. 2018, 57, 10450–10467. [Google Scholar] [CrossRef] [PubMed]
- Liepins, R.; Surles, J.R.; Morosoff, N.; Stannett, V.; Duffy, J.J.; Day, F.H. Localized radiation grafting of flame retardants to polyethylene terephthalate. II. Vinyl phosphonates. J. Appl. Polym. Sci. 1978, 22, 2403–2414. [Google Scholar] [CrossRef]
- Ebdon, J.R.; Hunt, B.J.; Joseph, P.; Wilkie, T.K. Flame retardance of polyacrylonitriles covalently modified with phosphorus- and nitrogen-containing groups. In Fire Retardancy of Polymers: New Strategies and Mechanisms; Hull, T.R., Kandola, B.K., Eds.; Royal Society of Chemistry: Cambridge, UK, 2009; pp. 331–340. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
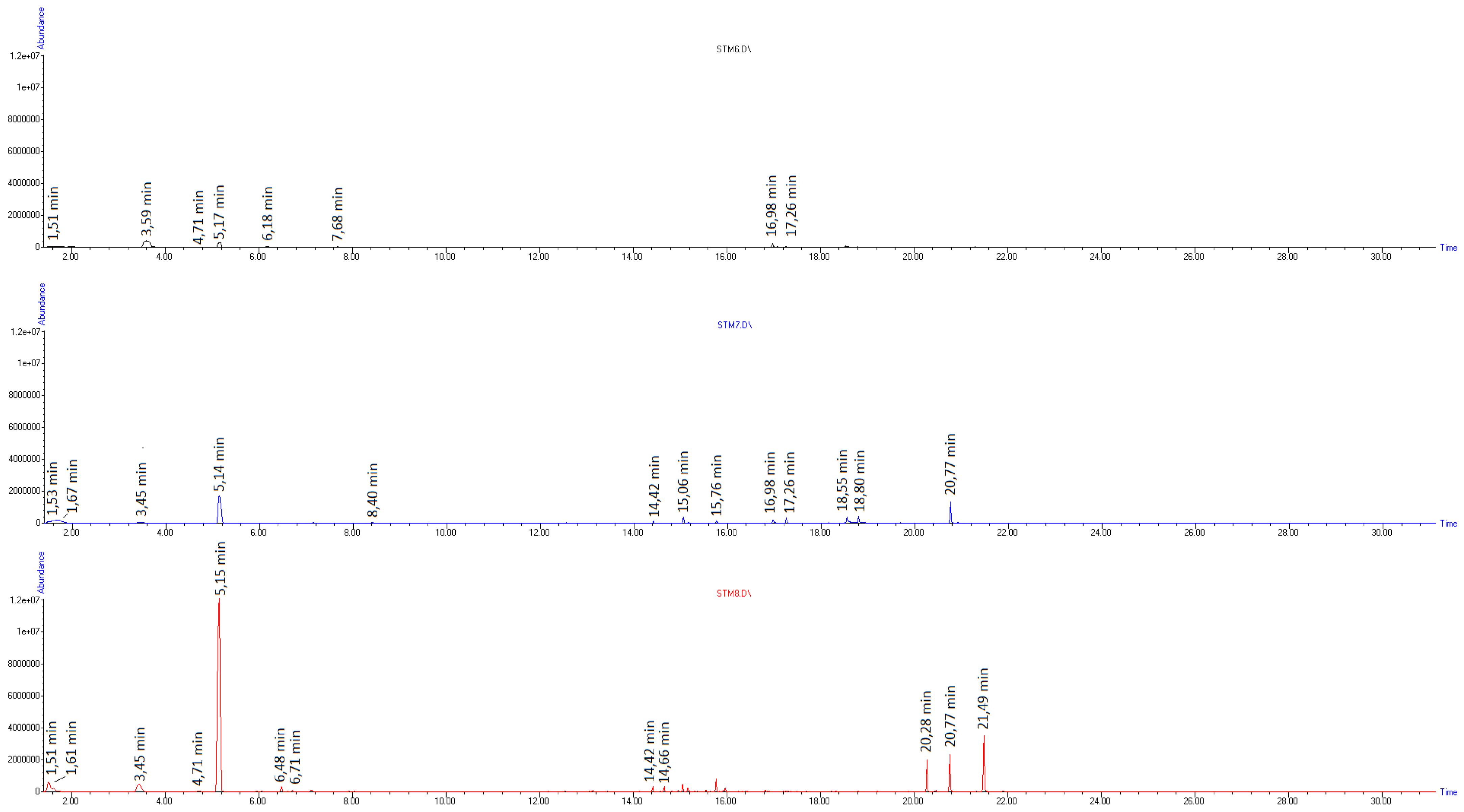
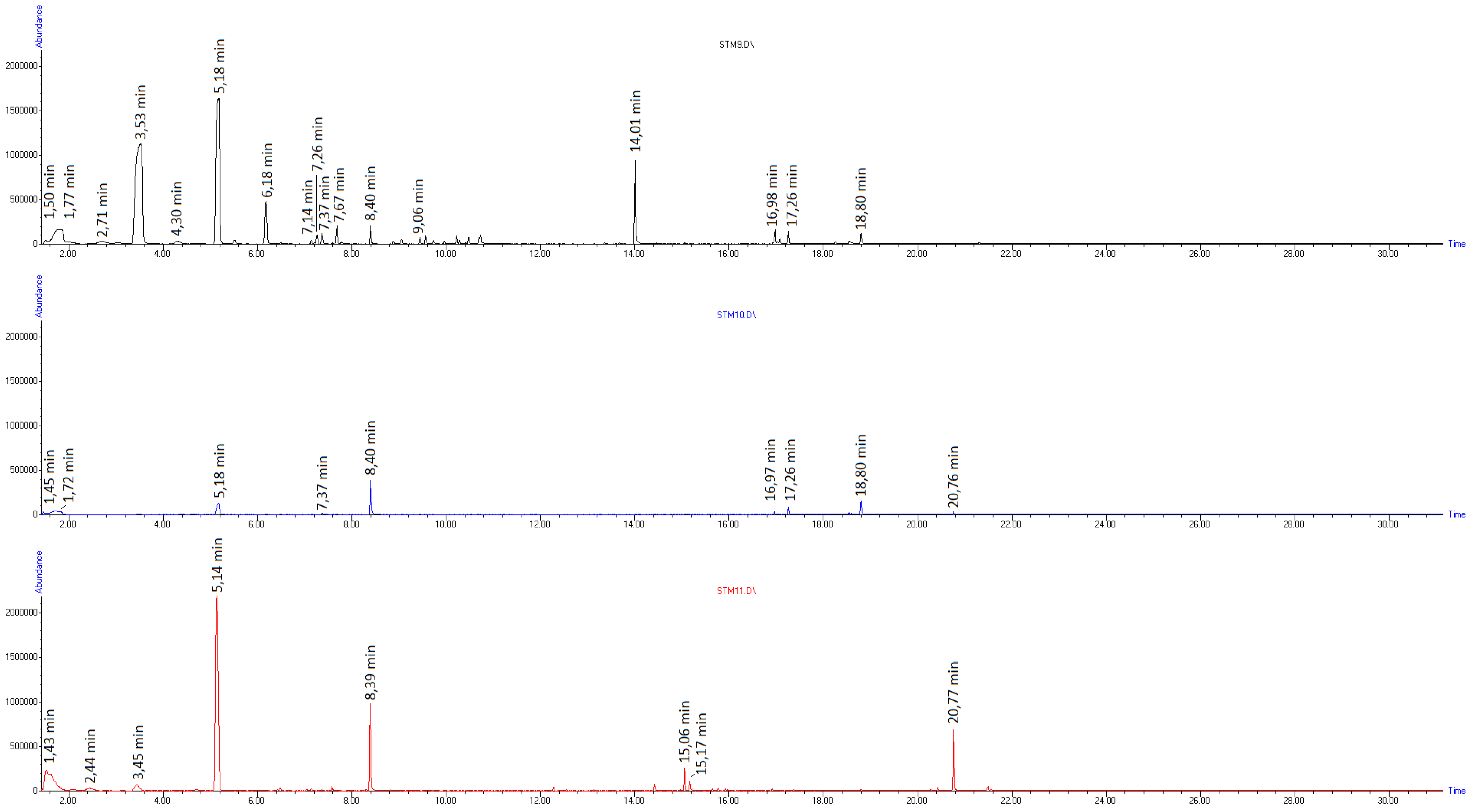
| Compound Name | m/z | Chemical Structure | RT (min) at 260 °C | RT (min) at 415 °C |
|---|---|---|---|---|
| Toluene | 92 |  | 3.44 | 3.44 |
| Ethylbenzene | 106 |  | - | 4.71 |
| Styrene-monomer | 104 |  | 5.17 | 5.15 |
| Benzaldehyde | 106 |  | 6.17 | 6.16 |
| α-Methylstyrene | 106 |  | - | 6.48 |
| Limonene | 136 |  | - | 7.15 |
| Phenylacetaldehyde | 120 |  | 7.37 | 7.35 |
| Acetophenone | 120 |  | 7.68 | 7.68 |
| Dihydroxyacetophenone | 152 |  | 7.76 | - |
| Bibenzyl | 182 |  | - | 13.13 |
| Diphenylpropane | 196 |  | - | 14.42 |
| Stilbene | 180 |  | - | 14.96 |
| Styrene-dimer | 208 |  | - | 15.06 |
| Styrene-trimer | 314 |  | - | 20.77 |
| Poly(S-co-DEAMP) | Poly(S-co-ADEPMAE) | Poly(S-co-MI) | |||
|---|---|---|---|---|---|
| Ion Structure | m/z | Ion Structure | m/z | Ion Structure | m/z |
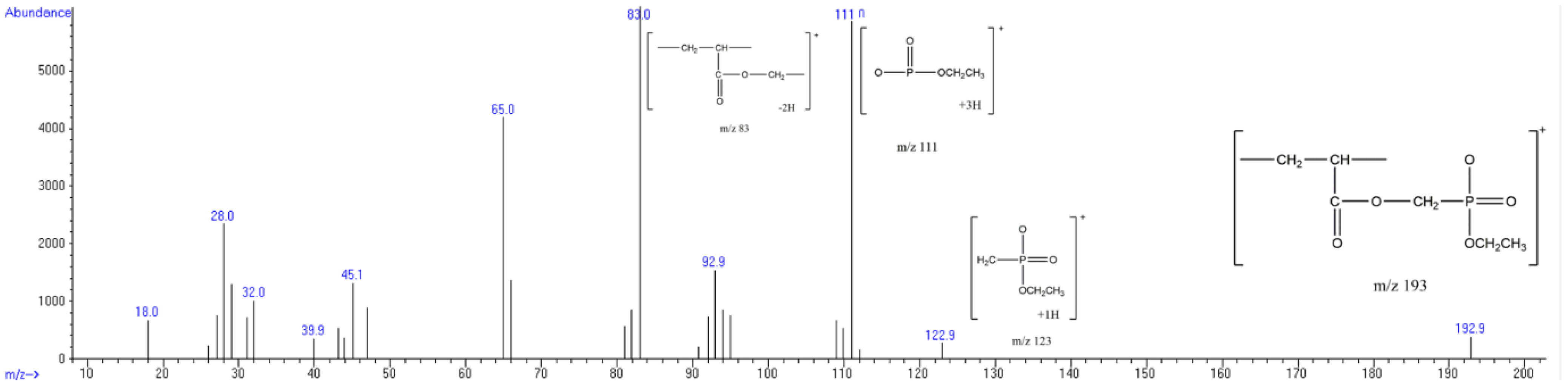
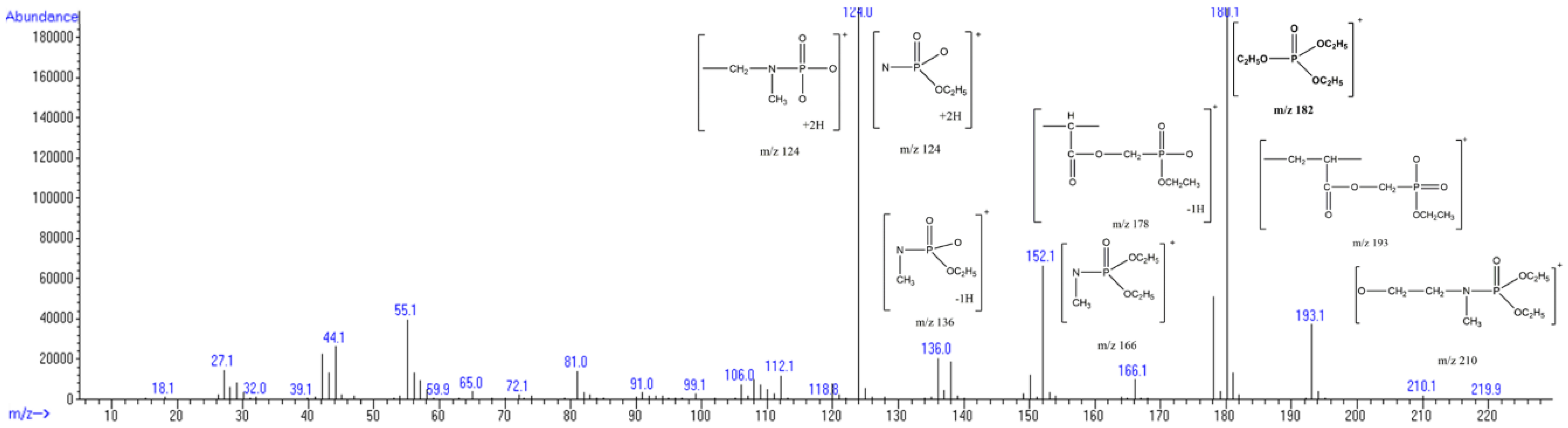
 | 83 |  | 124 |  | 115 |
 | 111 |  | 124 |  | 158 |
 | 123 |  | 136 |  | 172 |
 | 193 |  | 166 |  | 189 |
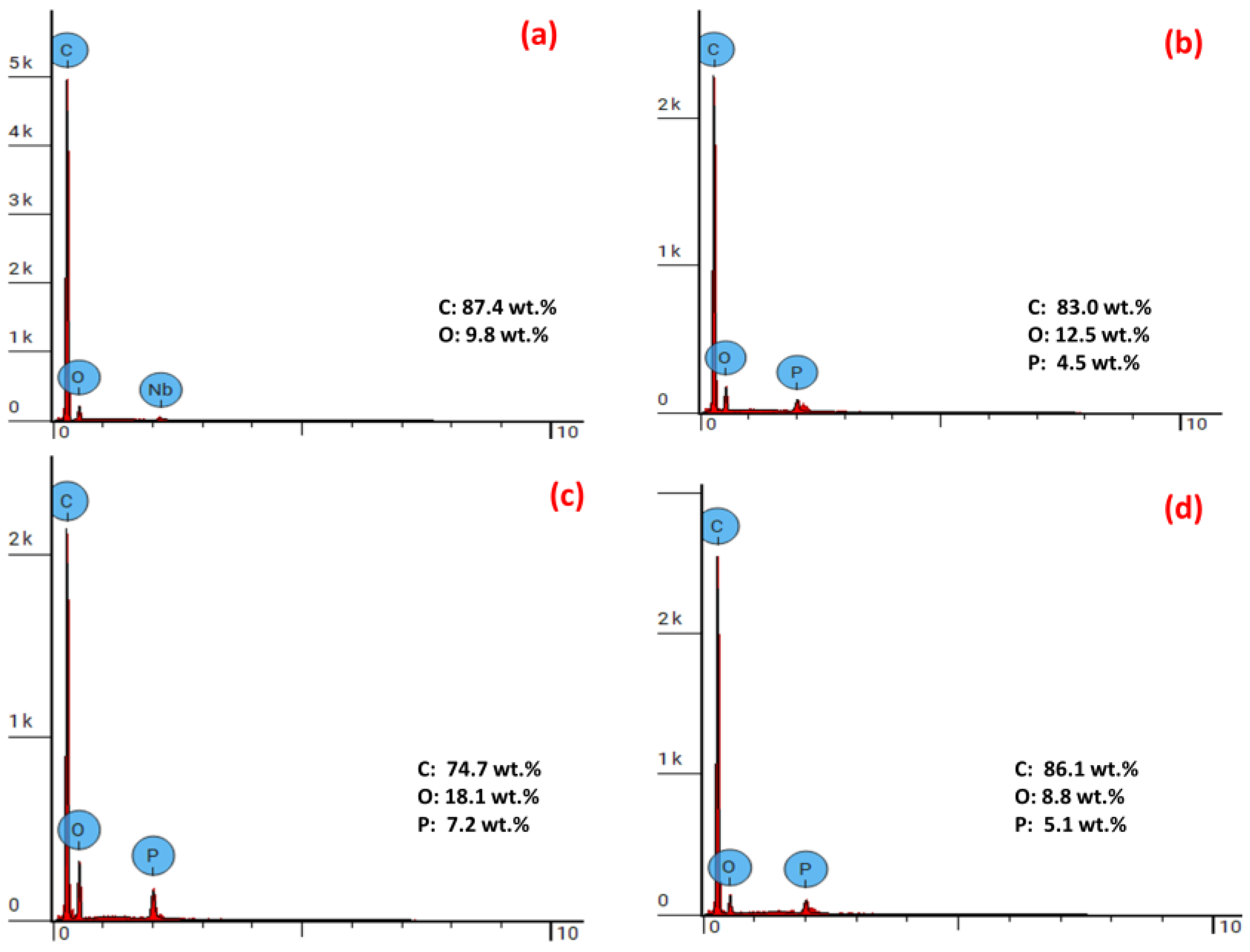
| Polymeric Sample | P Content in Polymer (wt. %) | Char Yield (wt. %) | P Content in Char (wt. %) * | Theoretical Pmax Loading in Char (wt. %) # | Actual P Retention in Char (wt. %) $ | P Content in Char from EDS (wt. %) |
|---|---|---|---|---|---|---|
| Poly(S-co-DEAMP) | 3.07 | 21.37 | 9.73 | 14.4 | 67.6 | - |
| Poly(S-co-DEpVBP) | 1.87 | 21.01 | 3.50 | 8.90 | 39.3 | - |
| Poly(S-co-ADEPMAE) | 2.55 | 14.29 | 6.10 | 17.8 | 34.5 | 4.5 |
| Poly(S-ter-DEAMP-ter-MI) | 2.95 | 33.00 | 3.47 | 8.94 | 38.8 | 7.2 |
| Poly(S-ter-DEpVBP-ter-MI) | 1.59 | 34.83 | 2.20 | 4.57 | 48.1 | 5.1 |
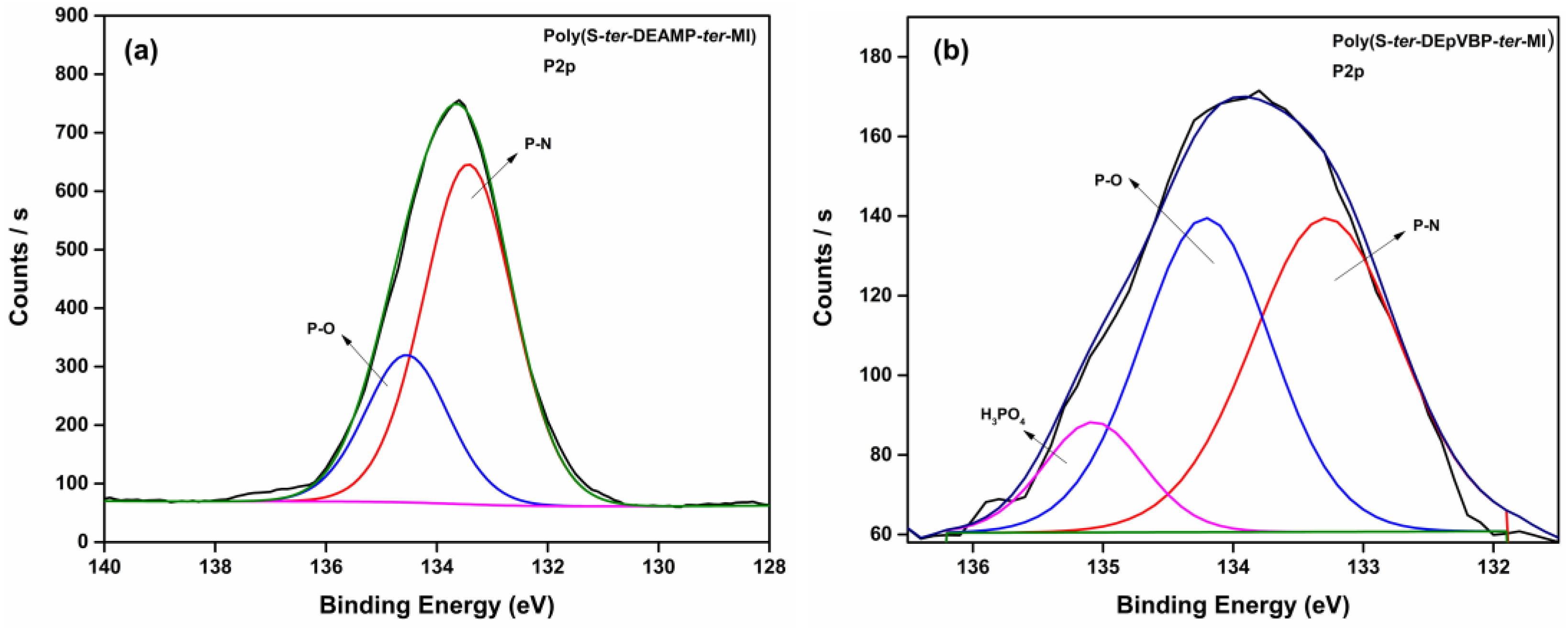
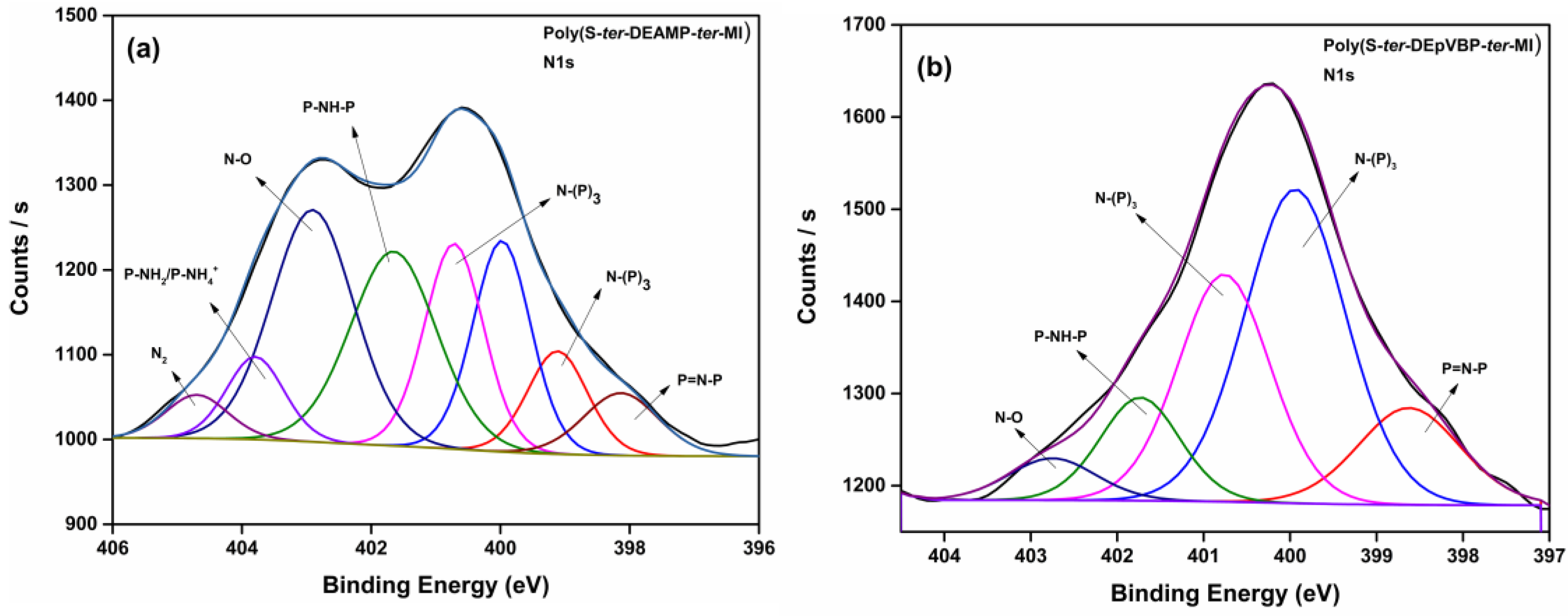
| Bonds | Assignment | Binding Energy (eV) | References |
|---|---|---|---|
| C1s | |||
| C-C/C=C | aliphatic/aromatic or sp2 bonded carbons | 284.7–284.8 | [33,34,35,36] |
| C-O | ether or hydroxyl bonded carbon | 285.9–286.0 | |
| C=O | carbonyl groups | 287.1 | |
| COO | carboxyl or ester groups | 288.1 | |
| O1s | |||
| O=C | carbonyl group | 531.1 | [33,35,36] |
| O-C | bonded to aliphatic | 532.2–532.6 | |
| O-C | bonded to aromatic | 533.2–533.7 | |
| O=N | bonded to nitrogen | 534.1–534.7 | |
| H-O-H | absorbed water | 535.2–535.5 | |
| Bonds | Binding Energy (eV) | References | |
|---|---|---|---|
| Poly(S-ter-DEAMP-ter-MI) | Poly(S-ter-DEpVBP-ter-MI) | ||
| P=N-P | 398.1 | 398.6 | [39,40,41] |
| N-(P)3 | 399.9–400.7 | 399.9–400.7 | |
| P-NH-P | 401.6 | 401.7 | |
| N-O (oxidized N) | 402.9 | 402.7 | |
| P-NH2/P-NH4+ | 403.7 | - | |
| N2 | 404.7 | - | |
| Polymer Sample | P Content wt. % | N Content wt. % | [S]:[M1]:[M2] * in Polymer, mol % | Tpyrolysis, °C |
|---|---|---|---|---|
| PS | - | - | 100:0.00:0.00 | 260, 415 |
| Poly(S-co-DEAMP) | 3.07 | - | 88.3:11.7:0.00 | 210, 325, 380 |
| Poly(S-co-DEpVBP) | 1.87 | - | 93.1:6.90:0.00 | 240, 355, 445 |
| Poly(S-co-ADEPMAE) | 2.55 | 1.15 | 90.1:9.90:0.00 | 250, 300, 400 |
| Poly(S-co-MI) | - | 3.72 | 72.8:0.00:27.2 | 210, 420 |
| Poly(S-ter-DEAMP-ter-MI) | 2.95 | 3.07 | 63.7:11.0:25.3 | 240, 320, 375 |
| Poly(S-ter-DEpVBP-ter-MI) | 1.59 | 3.63 | 65.6:5.70:28.7 | 270, 375 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tretsiakova-McNally, S.; Baby, A.; Joseph, P.; Pospiech, D.; Schierz, E.; Lederer, A.; Arun, M.; Fontaine, G. Gaseous- and Condensed-Phase Activities of Some Reactive P- and N-Containing Fire Retardants in Polystyrenes. Molecules 2023, 28, 278. https://doi.org/10.3390/molecules28010278
Tretsiakova-McNally S, Baby A, Joseph P, Pospiech D, Schierz E, Lederer A, Arun M, Fontaine G. Gaseous- and Condensed-Phase Activities of Some Reactive P- and N-Containing Fire Retardants in Polystyrenes. Molecules. 2023; 28(1):278. https://doi.org/10.3390/molecules28010278
Chicago/Turabian StyleTretsiakova-McNally, Svetlana, Aloshy Baby, Paul Joseph, Doris Pospiech, Eileen Schierz, Albena Lederer, Malavika Arun, and Gaëlle Fontaine. 2023. "Gaseous- and Condensed-Phase Activities of Some Reactive P- and N-Containing Fire Retardants in Polystyrenes" Molecules 28, no. 1: 278. https://doi.org/10.3390/molecules28010278
APA StyleTretsiakova-McNally, S., Baby, A., Joseph, P., Pospiech, D., Schierz, E., Lederer, A., Arun, M., & Fontaine, G. (2023). Gaseous- and Condensed-Phase Activities of Some Reactive P- and N-Containing Fire Retardants in Polystyrenes. Molecules, 28(1), 278. https://doi.org/10.3390/molecules28010278