

Coupling Effect of Non-Ignition Impact and Heat on the Decay of FOX-7

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methodology
2.1. Reactive Force Field
2.2. Hirshfeld Surface
2.3. Compute Details
3. Results and Discussion
3.1. Validation of ReaxFF
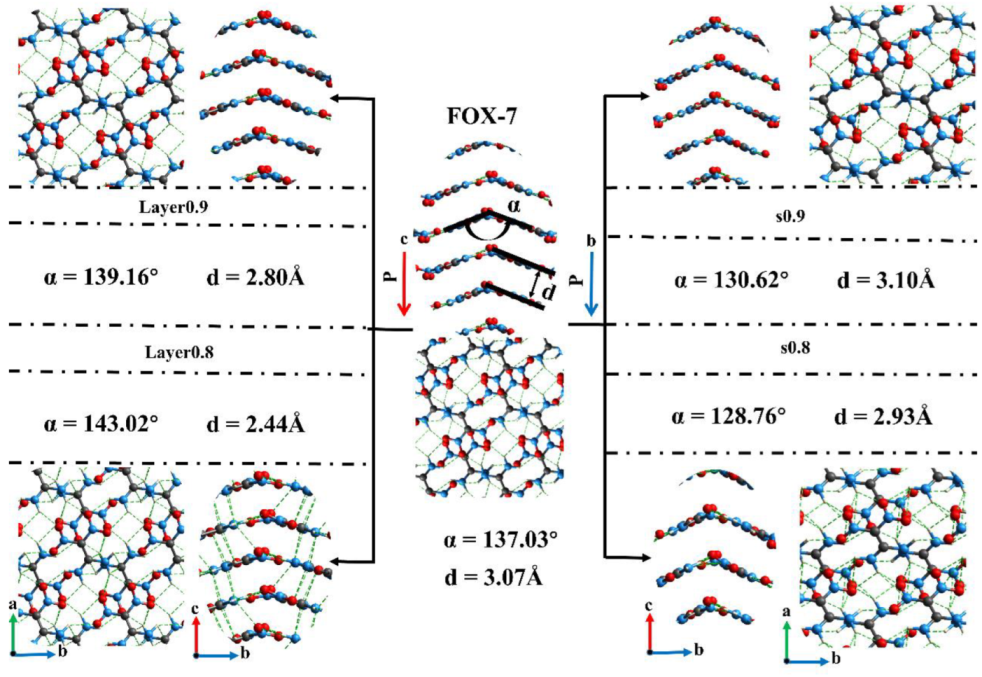
3.2. Intermolecular Interactions
3.3. Evolution of Chemical Species
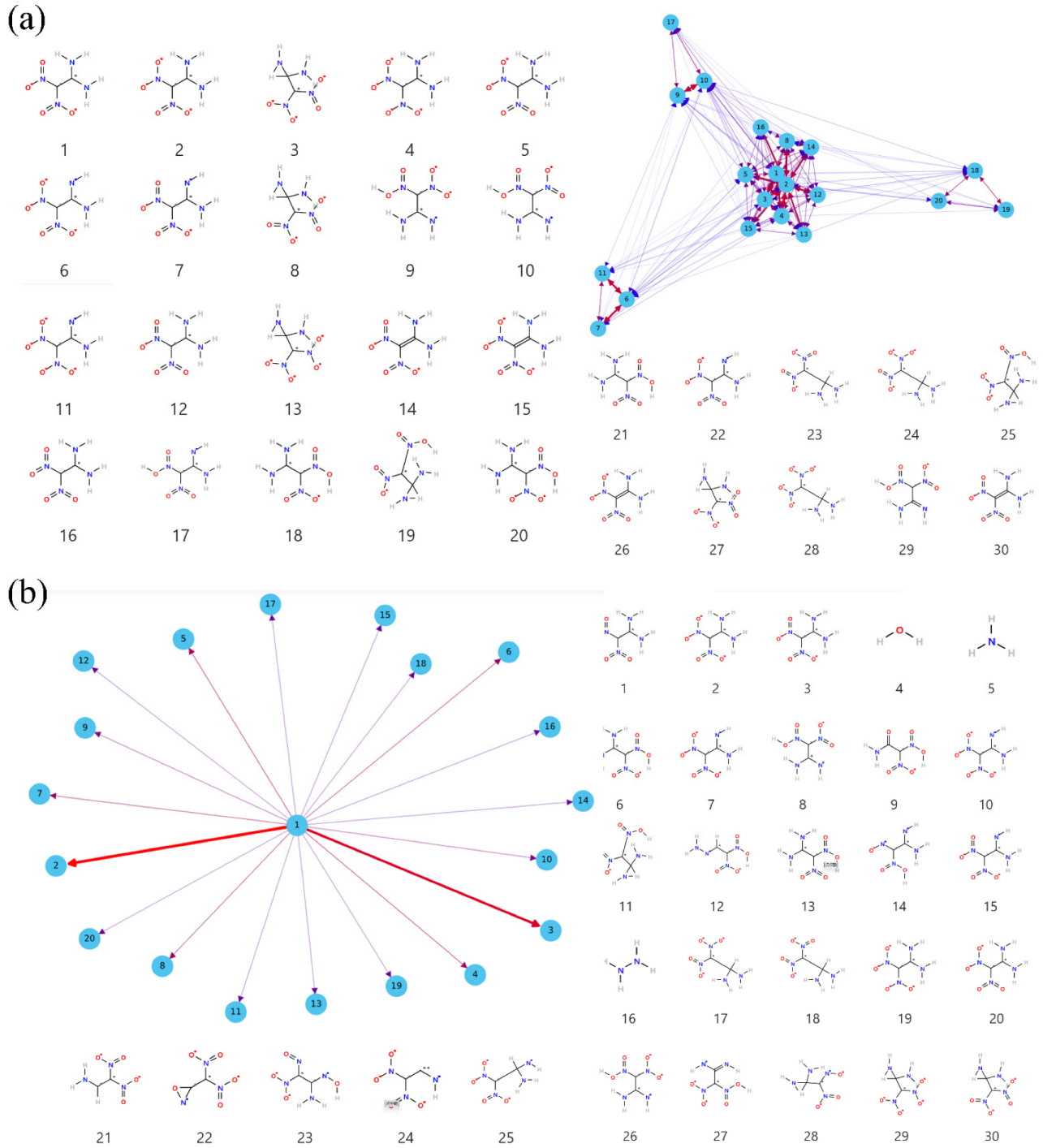
3.4. Details of Decomposition Reaction Network
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wu, J.; Huang, Y.; Yang, L.; Geng, D.; Wang, F.; Wang, H.; Chen, L. Reactive Molecular Dynamics Simulations of the Thermal Decomposition Mechanism of 1,3,3-Trinitroazetidine. ChemPhysChem 2018, 19, 2683–2695. [Google Scholar] [CrossRef]
- Lan, G.; Li, J.; Zhang, G.; Ruan, J.; Lu, Z.; Jin, S.; Cao, D.; Wang, J. Thermal Decomposition Mechanism Study of 3-Nitro-1,2,4-Triazol-5-One (NTO): Combined TG-FTIR-MS Techniques and ReaxFF Reactive Molecular Dynamics Simulations. Fuel 2021, 295, 120655. [Google Scholar] [CrossRef]
- Strachan, A.; Kober, E.M.; van Duin, A.C.T.; Oxgaard, J.; Goddard, W.A. Thermal Decomposition of RDX from Reactive Molecular Dynamics. J. Chem. Phys. 2005, 122, 054502. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Xue, X.; Chi, Y.; Li, H.; Long, X.; Zhang, C. Nature of the Enhanced Self-Heating Ability of Imperfect Energetic Crystals Relative to Perfect Ones. J. Phys. Chem. C 2017, 121, 12101–12109. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, F.; Xu, S.; Ju, X. Molecular Reaction Dynamics Simulation of Thermal Decomposition for Aluminiferous RDX Composites. Comput. Mater. Sci. 2020, 177, 109556. [Google Scholar] [CrossRef]
- Sikder, A.K.; Sikder, N. A Review of Advanced High Performance, Insensitive and Thermally Stable Energetic Materials Emerging for Military and Space Applications. J. Hazard. Mater. 2004, 112, 1–15. [Google Scholar] [CrossRef]
- Bemm, U.; Östmark, H. 1,1-Diamino-2,2-Dinitroethylene: A Novel Energetic Material with Infinite Layers in Two Dimensions. Acta Crystallogr. C 1998, 54, 1997–1999. [Google Scholar] [CrossRef]
- Badgujar, D.M.; Talawar, M.B.; Asthana, S.N.; Mahulikar, P.P. Advances in Science and Technology of Modern Energetic Materials: An Overview. J. Hazard. Mater. 2008, 151, 289–305. [Google Scholar] [CrossRef]
- Mullay, J. A Relationship between Impact Sensitivity and Molecular Electronegativity. Propellants Explos. Pyrotech. 1987, 12, 60–63. [Google Scholar] [CrossRef]
- Bliss, D.E.; Christian, S.L.; Wilson, W.S. Impact Sensitivity of Polynitroaromatics. J. Energetic Mater. 1991, 9, 319–348. [Google Scholar] [CrossRef]
- Kamlet, M.J.; Adolph, H.G. The Relationship of Impact Sensitivity with Structure of Organic High Explosives. II. Polynitroaromatic Explosives. Propellants Explos. Pyrotech. 1979, 4, 30–34. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, X.; Huang, H. π-Stacked Interactions in Explosive Crystals: Buffers against External Mechanical Stimuli. J. Am. Chem. Soc. 2008, 130, 8359–8365. [Google Scholar] [CrossRef] [PubMed]
- Lan, G.; Jin, S.; Chen, M.; Li, J.; Du, L.; Wang, J.; Chen, K.; Li, L. Preparation and Thermal Properties Study of HNIW/FOX-7 Based High Energy Polymer Bonded Explosive (PBX) with Low Vulnerability to Thermal Stimulations. J. Energ. Mater. 2020, 38, 83–97. [Google Scholar] [CrossRef]
- Pravica, M.; Galley, M.; Park, C.; Ruiz, H.; Wojno, J. A High Pressure, High Temperature Study of 1,1-Diamino-2,2-Dinitro Ethylene. High Press. Res. 2011, 31, 80–85. [Google Scholar] [CrossRef]
- Bishop, M.M.; Chellappa, R.S.; Pravica, M.; Coe, J.; Liu, Z.; Dattlebaum, D.; Vohra, Y.; Velisavljevic, N. 1,1-Diamino-2,2-Dinitroethylene under High Pressure-Temperature. J. Chem. Phys. 2012, 137, 174304. [Google Scholar] [CrossRef]
- Jeong, K.; Jeon, Y.; Kwon, S. Assessment of Various DFT, DFT-D, and MP2 Methods for Studying FOX-7 Detonation Properties. J. Mol. Model. 2017, 23, 250. [Google Scholar] [CrossRef]
- Chaoyang, Z.; Yuanjie, S.; Xiaodong, Z.; Haishan, D.; Xinfeng, W. Computational Investigation on HEDM of Azoic and Azoxy Derivatives of DAF, FOX-7, TATB, ANPZ and LLM-105. J. Mol. Struct. THEOCHEM 2005, 728, 129–134. [Google Scholar] [CrossRef]
- Bishop, M.M.; Chellappa, R.S.; Liu, Z.; Preston, D.N.; Sandstrom, M.M.; Dattelbaum, D.M.; Vohra, Y.K.; Velisavljevic, N. High Pressure-Temperature Polymorphism of 1,1-Diamino-2,2-Dinitroethylene. J. Phys. Conf. Ser. 2014, 500, 052005. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, H. High-Pressure Behavior of Crystalline FOX-7 by Density Functional Theory Calculations. Comput. Mater. Sci. 2008, 42, 698–703. [Google Scholar] [CrossRef]
- Dreger, Z.A.; Stash, A.I.; Yu, Z.; Chen, Y.; Tao, Y.; Gupta, Y.M. High-Pressure Structural Response of an Insensitive Energetic Crystal: 1,1-Diamino-2,2-Dinitroethene (FOX-7). J. Phys. Chem. C 2016, 120, 27600–27607. [Google Scholar] [CrossRef]
- Xu, K.; Song, J.; Zhao, F.; Heng, S.; Ding, L.; Wang, Y.; Hu, R. Non-Isothermal Decomposition Kinetics, Specific Heat Capacity and Adiabatic Time-to-Explosion of a Novel High Energy Material: 1-Amino-1-Methylamino-2,2-Dinitroethylene (AMFOX-7). J. Chin. Chem. Soc. 2009, 56, 524–531. [Google Scholar] [CrossRef]
- Bu, R.; Xie, W.; Zhang, C. Heat-Induced Polymorphic Transformation Facilitating the Low Impact-Sensitivity of 2,2-Dinitroethylene-1,1-Diamine (FOX-7). J. Phys. Chem. C 2019, 123, 16014–16022. [Google Scholar] [CrossRef]
- Jiang, L.; Fu, X.; Zhou, Z.; Zhang, C.; Li, J.; Qi, F.; Fan, X.; Zhang, G. Study of the Thermal Decomposition Mechanism of FOX-7 by Molecular Dynamics Simulation and Online Photoionization Mass Spectrometry. RSC Adv. 2020, 10, 21147–21157. [Google Scholar] [CrossRef] [PubMed]
- Bishop, M.M.; Velisavljevic, N.; Chellappa, R.; Vohra, Y.K. High Pressure–Temperature Phase Diagram of 1,1-Diamino-2,2-Dinitroethylene (FOX-7). J. Phys. Chem. A 2015, 119, 9739–9747. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Dreger, Z.A.; Gupta, Y.M. High-Pressure Stability of 1,1-Diamino-2,2-Dinitroethene (FOX-7): H/D Isotope Effect. Chem. Phys. Lett. 2015, 624, 59–63. [Google Scholar] [CrossRef]
- Kuklja, M.M.; Rashkeev, S.N. Shear-Strain-Induced Chemical Reactivity of Layered Molecular Crystals. Appl. Phys. Lett. 2007, 90, 151913. [Google Scholar] [CrossRef]
- Zeng, C.; Yang, Z.; Wen, Y.; He, W.; Zhang, J.; Wang, J.; Huang, C.; Gong, F. Performance Optimization of Core-Shell HMX@(Al@GAP) Aluminized Explosives. Chem. Eng. J. 2021, 407, 126360. [Google Scholar] [CrossRef]
- Han, Q.; Zhu, W. Effect of Particle Size on the Thermal Decomposition of Nano ε-CL-20 by ReaxFF-Lg Molecular Dynamics Simulations. Chem. Phys. Lett. 2020, 761, 138067. [Google Scholar] [CrossRef]
- Zhao, Y.; Mei, Z.; Zhao, F.; Xu, S.; Ju, X. Thermal Decomposition Mechanism of 1,3,5,7-Tetranitro-1,3,5,7-Tetrazocane Accelerated by Nano-Aluminum Hydride (AlH3 ): ReaxFF-Lg Molecular Dynamics Simulation. ACS Omega 2020, 5, 23193–23200. [Google Scholar] [CrossRef]
- Fantauzzi, D.; Bandlow, J.; Sabo, L.; Mueller, J.E.; van Duin, A.C.T.; Jacob, T. Development of a ReaxFF Potential for Pt–O Systems Describing the Energetics and Dynamics of Pt-Oxide Formation. Phys. Chem. Chem. Phys. 2014, 16, 23118–23133. [Google Scholar] [CrossRef]
- Roshan, K.A.; Talkhoncheh, M.K.; Mueller, J.E.; Goddard, W.A. ReaxFF Reactive Force Field Development for Cu/Si Systems and Application to Copper Cluster Formation During Cu Diffusion Inside Silicon. arXiv 2021, arXiv:2102.00943. [Google Scholar]
- van Duin, A.C.T.; Dasgupta, S.; Lorant, F.; Goddard, W.A. ReaxFF: A Reactive Force Field for Hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef]
- Liu, L.; Liu, Y.; Zybin, S.V.; Sun, H.; Goddard, W.A. ReaxFF- l g: Correction of the ReaxFF Reactive Force Field for London Dispersion, with Applications to the Equations of State for Energetic Materials. J. Phys. Chem. A 2011, 115, 11016–11022. [Google Scholar] [CrossRef]
- Larentzos, J.P.; Rice, B.M.; Byrd, E.F.C.; Weingarten, N.S.; Lill, J.V. Parameterizing Complex Reactive Force Fields Using Multiple Objective Evolutionary Strategies (MOES). Part 1: ReaxFF Models for Cyclotrimethylene Trinitramine (RDX) and 1,1-Diamino-2,2-Dinitroethene (FOX-7). J. Chem. Theory Comput. 2015, 11, 381–391. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-Atom Fragments for Describing Molecular Charge Densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A Program for Hirshfeld Surface Analysis, Visualization and Quantitative Analysis of Molecular Crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Zeng, T.; Yang, R.; Li, J.; Tang, W.; Li, D. Thermal Decomposition Mechanism of Nitroglycerin by ReaxFF Reactive Molecular Dynamics Simulations. Combust. Sci. Technol. 2021, 193, 470–484. [Google Scholar] [CrossRef]
- Cheng, X.; Wang, Q.; Li, J.; Wang, J.; Li, X. ReaxFF Molecular Dynamics Simulations of Oxidation of Toluene at High Temperatures. J. Phys. Chem. A 2012, 116, 9811–9818. [Google Scholar] [CrossRef]
- Zeng, J.; Cao, L.; Chin, C.; Ren, H.; Zhang, J.Z.H.; Zhu, T. ReacNetGenerator: An Automatic Reaction Network Generator for Reactive Molecular Dynamics Simulations. Phys. Chem. Chem. Phys. 2020, 22, 683–691. [Google Scholar] [CrossRef]
- She, C.; Jin, S.; Chen, S.; Li, L.; Shu, Q.; Chen, Y.; Wang, J.; Wu, N.; Chen, M.; Chen, K. Reactive Molecular Dynamics Simulation of Thermal Decomposition for Nano-FOX-7. Appl. Phys. A 2021, 127, 881. [Google Scholar] [CrossRef]
- Li, S.; Bu, R.; Gou, R.; Zhang, C. Hirshfeld Surface Method and Its Application in Energetic Crystals. Cryst. Growth Des. 2021, 21, 6619–6634. [Google Scholar] [CrossRef]
- Kuklja, M.M.; Rashkeev, S.N. Molecular Mechanisms of Shear Strain Sensitivity of the Energetic Crystals DADNE and TATB. J. Energ. Mater. 2010, 28, 66–77. [Google Scholar] [CrossRef]
- Zhou, T.; Zybin, S.V.; Liu, Y.; Huang, F.; Goddard, W.A., III. Anisotropic Shock Sensitivity for B-Octahydro-1,3,5,7-Tetranitro-1,3,5,7- Tetrazocine Energetic Material under Compressive-Shear Loading from ReaxFF-Lg Reactive Dynamics Simulations. J. Appl. Phys. 2012, 12, 124904. [Google Scholar] [CrossRef]
- Chen, L.; Wang, H.; Wang, F.; Geng, D.; Wu, J.; Lu, J. Thermal Decomposition Mechanism of 2,2′,4,4′,6,6′-Hexanitrostilbene by ReaxFF Reactive Molecular Dynamics Simulations. J. Phys. Chem. C 2018, 122, 19309–19318. [Google Scholar] [CrossRef]
- Wang, F.; Chen, L.; Geng, D.; Wu, J.; Lu, J.; Wang, C. Thermal Decomposition Mechanism of CL-20 at Different Temperatures by ReaxFF Reactive Molecular Dynamics Simulations. J. Phys. Chem. A 2018, 122, 3971–3979. [Google Scholar] [CrossRef]
- Yang, L.; Wu, J.; Geng, D.; Wang, F.; Huang, Y.; Chen, L. Reactive Molecular Dynamics Simulation of the Thermal Decomposition Mechanisms of 4,10-Dinitro-2,6,8,12-Tetraoxa-4,10-Diazatetracyclo[5.5.0.05,9.03,11]Dodecane (TEX). Combust. Flame 2019, 202, 303–317. [Google Scholar] [CrossRef]
- Jiang, H.; Jiao, Q.; Zhang, C. Early Events When Heating 1,1-Diamino-2,2-Dinitroethylene: Self-Consistent Charge Density-Functional Tight-Binding Molecular Dynamics Simulations. J. Phys. Chem. C 2018, 122, 15125–15132. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
She, C.; Chen, K.; Chen, M.; Lu, Z.; Wu, N.; Li, L.; Wang, J.; Jin, S. Coupling Effect of Non-Ignition Impact and Heat on the Decay of FOX-7. Molecules 2022, 27, 8255. https://doi.org/10.3390/molecules27238255
She C, Chen K, Chen M, Lu Z, Wu N, Li L, Wang J, Jin S. Coupling Effect of Non-Ignition Impact and Heat on the Decay of FOX-7. Molecules. 2022; 27(23):8255. https://doi.org/10.3390/molecules27238255
Chicago/Turabian StyleShe, Chongchong, Kun Chen, Minglei Chen, Zhiyan Lu, Nana Wu, Lijie Li, Junfeng Wang, and Shaohua Jin. 2022. "Coupling Effect of Non-Ignition Impact and Heat on the Decay of FOX-7" Molecules 27, no. 23: 8255. https://doi.org/10.3390/molecules27238255
APA StyleShe, C., Chen, K., Chen, M., Lu, Z., Wu, N., Li, L., Wang, J., & Jin, S. (2022). Coupling Effect of Non-Ignition Impact and Heat on the Decay of FOX-7. Molecules, 27(23), 8255. https://doi.org/10.3390/molecules27238255