3.1. Enthalpies of Reaction Characterizing the Antioxidant Activity
As noted in the Introduction, an antioxidant can transfer an H-atom to a free radical in one- or two-step processes. Again, to make the paper self-contained, let us be reminded that the three antioxidative mechanisms (HAT, SET-PT, and SPLET) and the corresponding reaction enthalpies (BDE, IP and PDE, and PA and ETE, respectively) can be expressed as follows:
Direct hydrogen atom transfer (HAT) [
39,
40,
41]
Stepwise electron transfer–proton transfer (SET-PT) [
42,
43]
Sequential proton loss–electron transfer (SPLET) [
44,
45]
In specific cases of interest for antioxidation, X stands for an O, N, or S atom. The above definitions should make clear that all aforementioned reaction enthalpies (in particular, IP) are adiabatic rather than vertical properties [
46,
47]; they should be evaluated at the global electronic energy minima of the various reaction products/reactants.
3.3. Implications of Theorem 1
The results presented in the literature on antioxidants violating Theorem 1 (and hence being incorrect) fall into two categories.
Studies reporting values different from belong to the first category. Because the main aim of this work is to draw attention to the necessary sanity tests rather than amply documenting incorrect results on antioxidative activity reported in the literature in the past, we will only give here a single but notorious example, to which, unpleasantly, we return to repeatedly below.
We named [
23], which is a notorious case because, unfortunately,
all values presented for the dietary vitamins investigated (A, B1, B3, B6, and C) fail to obey Theorem 1. This is obvious by inspecting the last column of
Table 1, where the pertaining results are collected. It is pure nonsense to settle between SET-PT and SPLET by “comparing” (as in [
23]) the combined enthalpies of the pertaining reactions
and
. Provided that they are correctly estimated, they should be strictly equal to each other, whatever the computational method utilized.
The second category comprises studies adjudicating between SET-PT and SPLET based on the reaction enthalpy of the first step. For example, SPLET is claimed to be the thermodynamically preferred pathway because . This is incorrect because it contradicts Corollary 1. If this was the case, thermodynamically speaking, the second, rate-determining step (electron transfer, ETE) would act as bottleneck of SPLET (, cf. Equation (6b)).
Equation (6) makes it clear that in all cases, it is the specific free radical and/or the reaction kinetics that settles whether SET-PL or SPLET prevails. Merely considering antioxidant’s enthalpies of reaction can never discriminate between these two pathways.
3.4. Implications of Theorem 2
There are at least two practical issues related to Theorem 2.
First, the theorem shows that it is incorrect to assign HAT as the preferred antioxidant pathway based on the fact that
is smaller than
(or
). Theorem 2 states that correctly estimated enthalpies should
always satisfy this inequality. This refutes the claim made in [
23] on this basis, that HAT is the thermodynamically preferred pathway for dietary vitamins to scavenge free radicals in aqueous solution.
Above, we said “correctly estimated” because, unfortunately, this is not always the case in the literature; for example, in [
25], where enormous values of
kcal/mol, much larger than all the other reaction enthalpies, were obtained (see [
22,
48]). At odds with Equation (
7), the largest value of
reported in the mentioned study are smaller than BDE; none exceeds ∼150 kcal/mol (cf. Table 2 of [
25]).
Second, Theorem 2 emphasizes the particularly important role of the ionization enthalpy of the H-atom
. For this reason, in
Table 2 we present extensive data for the enthalpy of the H-atom. We used these data in
Table 3 and
Table 4 for computing
in gas and solvents using popular functionals (B3LYP, PBE0, M052x, and M062x) and frequently employed Pople basis sets.
Regarding the values of , a twofold word of caution is in order.
First, estimating
as the Kohn–Sham HOMO energy with reversed sign (Koopmans’ theorem) lamentably fails in a twofold sense; see
Table 5 and
Table 6. Because Kohn–Sham “orbitals” are mathematical objects rather than true molecular orbitals [
47,
49], these estimates are inadequate. The differences between the B3LYP- and PBE0-based Koopmans’ values and the
-DFT-based values [
50,
51,
52] are unacceptable even in the gas phase: ∼4–5 eV for B3LYP and PBE0 (cf.
Table 5) and ∼3.5 eV for M062x and M052x (cf.
Table 6). These differences grow to enormous values in polar solvents (∼12 eV, cf.
Table 5 and
Table 6). This dramatic deterioration of the description based on Koopmans’ theorem is fully in line with findings reported in previous studies on molecules in solvents [
46,
53].
Second, differences in the values of antioxidant thermodynamic descriptors in solvents as large as ∼0.5 eV (12 kcal/mol, 50 kJ/mol) reported in various publications do not necessarily reflect real physical and chemical differences. They can simply stem from the utilization of different values of the electron and/or hydrogen atom enthalpy of solvation circulated in the literature (e.g.,
kJ/mol [
54] versus
kJ/mol [
55] for a proton in water).
According to Theorem 2, the difference between and BDE should be equal to the ionization enthalpy of the H-atom in the medium (solvent) in question. It should be, but unfortunately, data in the literature exist for which this condition is not fulfilled.
In the upper part of
Table 7, we reproduce enthalpies reported at the B3LYP/6-31+G(d,p)(/IEFPCM) level of theory for the natural food colorant peonidin in gas and aqueous phases [
24].
It might be questionable whether the difference between the values = 392.87 kcal/mol and kcal/mol represents an obscure numerical artifact or a violation of Theorem 1. In fact, this should not be the case, given the fact that Theorem 1 is a mathematical identity that must be satisfied at any approximate level of theory provided that all antioxidant descriptors were correctly computed using the same method.
Anyway, letting alone this aspect, the values estimated for kcal/mol and kcal/mol differ too much from IP kcal/mol; they obviously do not satisfy Theorem 2.
Pleasantly, no objection can be raised against the enthalpies estimated in [
24] for peonidin in the gas phase. They satisfy both Theorems 1 and 2.
Let us next refer again to notorious case of [
23]. Relevant results are depicted in
Table 1. The inspection of the fifth column of
Table 1 reveals values that drastically differ from the ionization enthalpy of the H-atom in water (cf.
Table 4). Furthermore, those values also significantly depend on the molecular species, which should not be the case if they were correct.
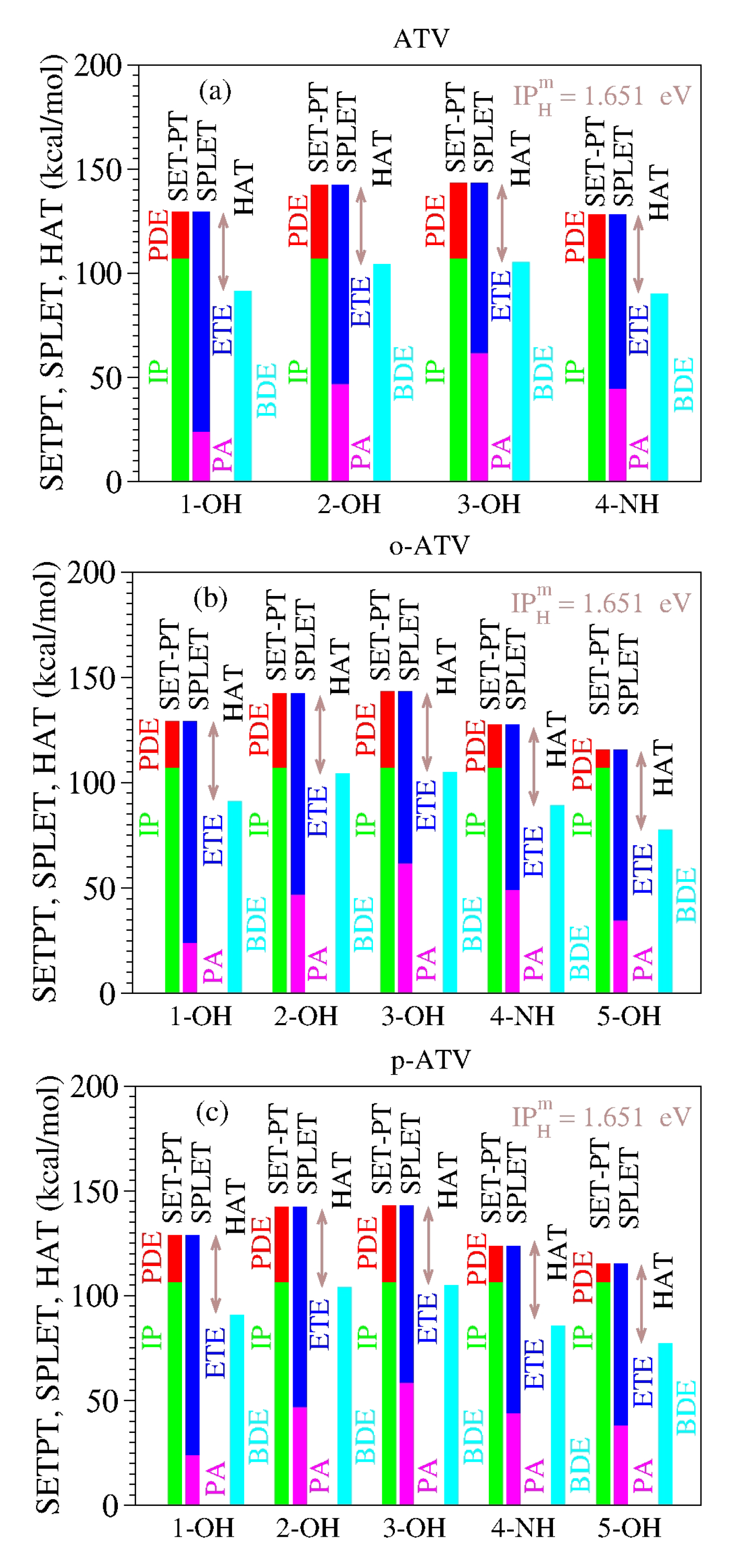
As a specific illustration of the latter aspect noted above, let us examine the results computed for atorvastatin (ATV) and its ortho- and para-hydroxy metabolites (o-ATV, p-ATV) in methanol at the B3LYP/6-31+G(d,p) level of theory [
22]. As visible in
Table 8 and
Figure 1, for each molecular species (ATV, o-ATV, and p-ATV) and any possible H-atom donation—that is, for each of the three (ATV) or four (o-ATV, p-ATV) OH-groups and the NH-group (indicated for the molecular geometries presented in [
22])—the results of the ATV-based species satisfy both theorems.
3.5. Detailed Analysis of a Specific Case: Vitamin B3
Let us next examine in detail the case of vitamin B3 (C
6H
5NO
2, InChI=1S/C6H5NO2/ c8-6(9)5-2-1-3-7-4-5/h1-4H,(H,8,9), CAS Registry Number: 59-67-6), whose optimized geometry is depicted in
Figure 2. Vitamin B3 is one of the molecular species investigated in [
23]. The analysis that follows shows that, as it should be in general, the antioxidant properties of vitamin B3 also obey the two theorems presented above.
In our calculations for vitamin B3, we attempted to ensure consistency with our recent [
22,
48] and ongoing investigations on antioxidation, while also gaining insight into specific sources of the erroneous results reported in [
23]. For the first reason, we used the B3LYP exchange-correlation functional, for which results for vitamin B3 are also presented in [
23] (cf. Table 1 of the supplementary material of [
23]).
Ambiguities arose regarding both the basis set—6-311++G(d,p) according to
Section 3 but 6-311G**(≡6-311G(d,p)) according to Table 1 in the supplementary material of [
23]— and the—unrestricted (UB3LYP) or spin-restricted open-shell (ROB3LYP)—method employed for radicals in [
23]. (Unfortunately, our inquiry on these aspects to the authors of [
23] continues to be pending.)
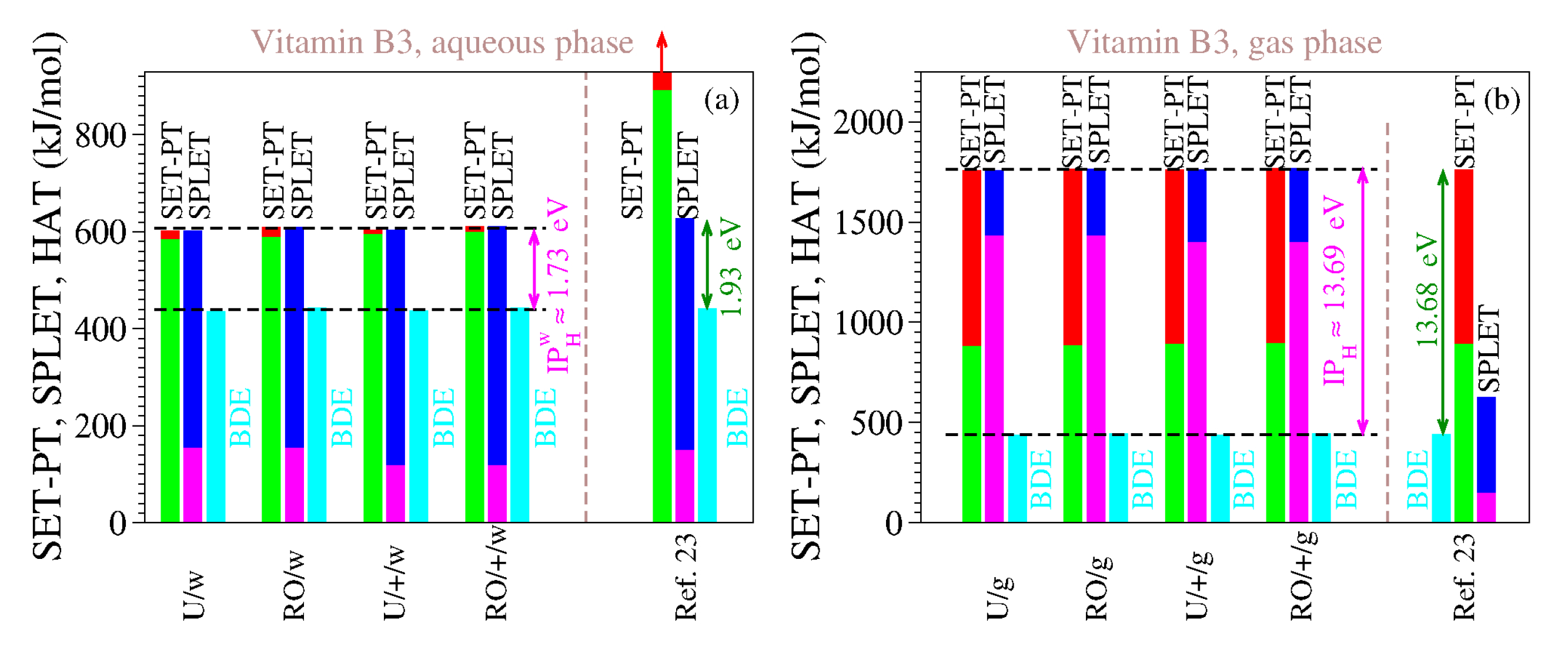
For the reasons delineated above, we computed the antioxidant descriptors of vitamin B3 using both methods (UB3LYP and ROB3LYP) and both basis sets (6-311++G(d,p) and 6-311G(d,p)). The results are presented in
Table 9 and
Table 10, and
Figure 3. The inspection of
Table 9 and
Table 10 and
Figure 3 reveals the clear differences between our estimates and those of [
23], which are also shown. As is visible, both for the gaseous phase and for the aqueous phase our values do satisfy Theorems 1 and 2.
We also performed computations for the gas phase because the enormous values of IP and PDE of [
23] (cf.
Table 9) made us suspicious that, contrary to what [
23] asserted, they were not computed for the aqueous phase but rather for the gaseous phase. It is, so far, an open question whether our assumption holds true or not, but the fact is that the IP and PDE of [
23] are completely different from our IP and PDE in water, while being considerably closer to our estimates for the gas phase. This also holds true for all the other vitamins investigated in [
23]; see
Table 1.
Parenthetically (because this is not the primary aim of the present work), we can still mention a couple more incorrect statements in [
23]. For example, the claim on page 5 of [
23], that the first step of the SET-PT mechanism (i.e., Equation (2a)) is less energetically costly than the second SET-PT step (i.e., Equation (2b)), and another claim on page 6 that the production of the alkoxide anion in the first SPLET mechanism (i.e., Equation (3a)) requires more energy than that for the electron transfer from the alkoxide anion to the free radical (i.e., Equation (3b)). In reality, the inspection of the thermochemical data for the aqueous phase (which [
23] considered) in
Table 9 illustrates that just the opposite holds true (namely,
and
).
3.6. Remark on the Dominant Antioxidant Mechanism
Theorem 1 makes it clear why the discussion on the competition between SET-PT and SPLET is incorrect in [
23]. There, the former mechanism was ruled out due to the large values of
(in fact, incorrectly computed, see
Table 9), much larger than
. In general, if this were the case, Theorem 1 would necessarily imply that
, which means that the second step of SPLET would then act as a bottleneck for the SPLET pathway. To reiterate, discussing the competition SET-PT versus SPLET merely based on the enthalpies of reaction is impossible, simply because the combined enthalpies of the two pathways (SET-PT and SPLET) are strictly equal in all cases.
The claim made in [
23] (even in the abstract) that HAT prevails is not substantiated by the values (even if they were correct) presented in that work. From the fact that
, one can/should by no means conclude that HAT prevails over SPLET. This inequality holds in
all cases (cf. Equation (
9)).
On the other hand, as emphasized recently [
22], a “small” value of
(implicitly meaning a
smaller than
) does not make SPLET a thermodynamically allowed pathway, per se. The relationship between
and
plays absolutely no role in the the first step of SPLET (proton loss). What matters in the first SPLET reaction is that the antioxidant’s PA be smaller than the PDE value of the neutralized free radical; see the discussion in [
22]. Again, whether SPLET is allowed or not cannot be settled merely in terms of the antioxidant’s properties.







{kind=link}
{kind=link}
{kind=link}