

Fast and Sensitive Analysis of Short- and Long-Chain Perfluoroalkyl Substances in Foods of Animal Origin


Abstract
1. Introduction
2. Results
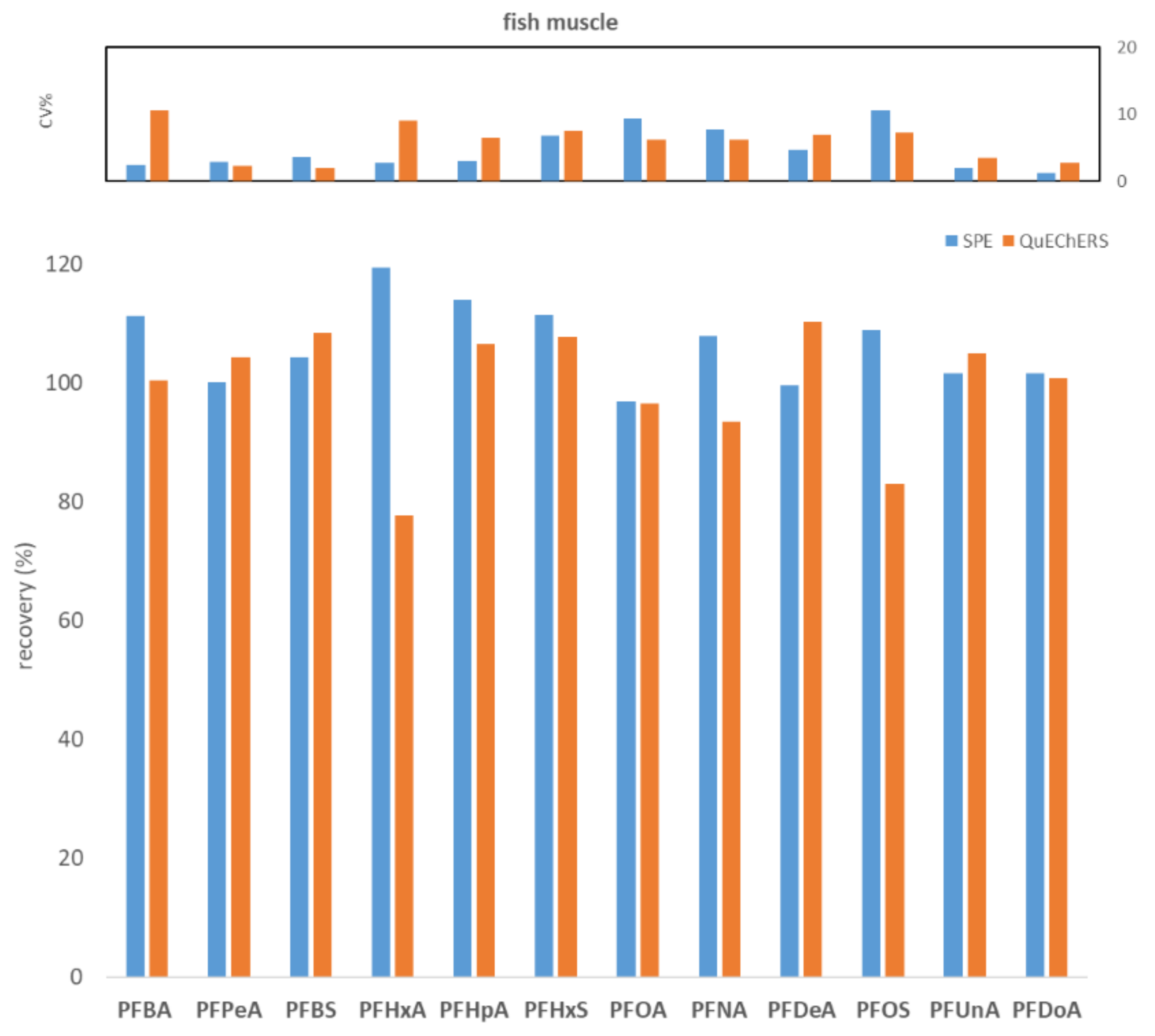
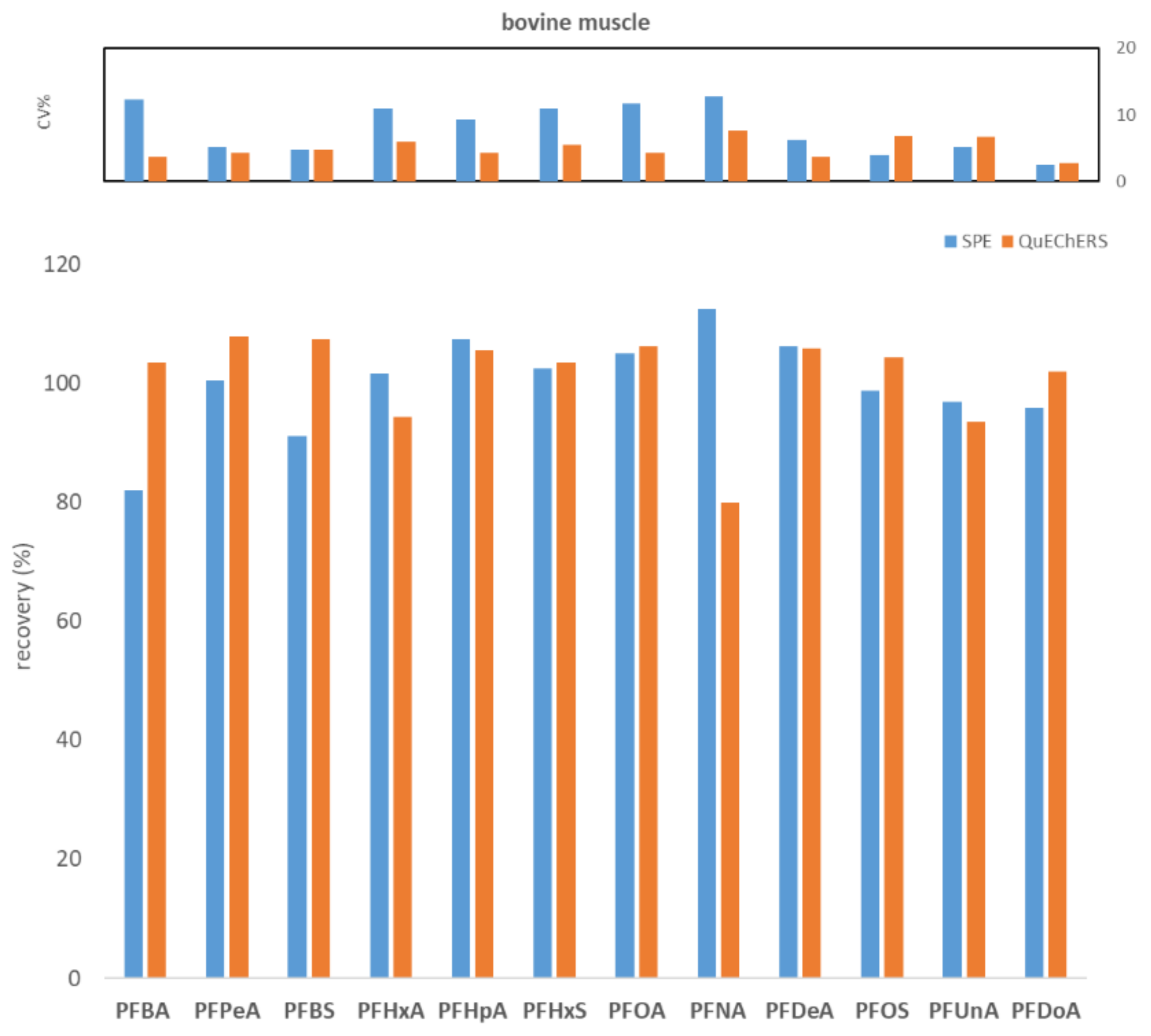
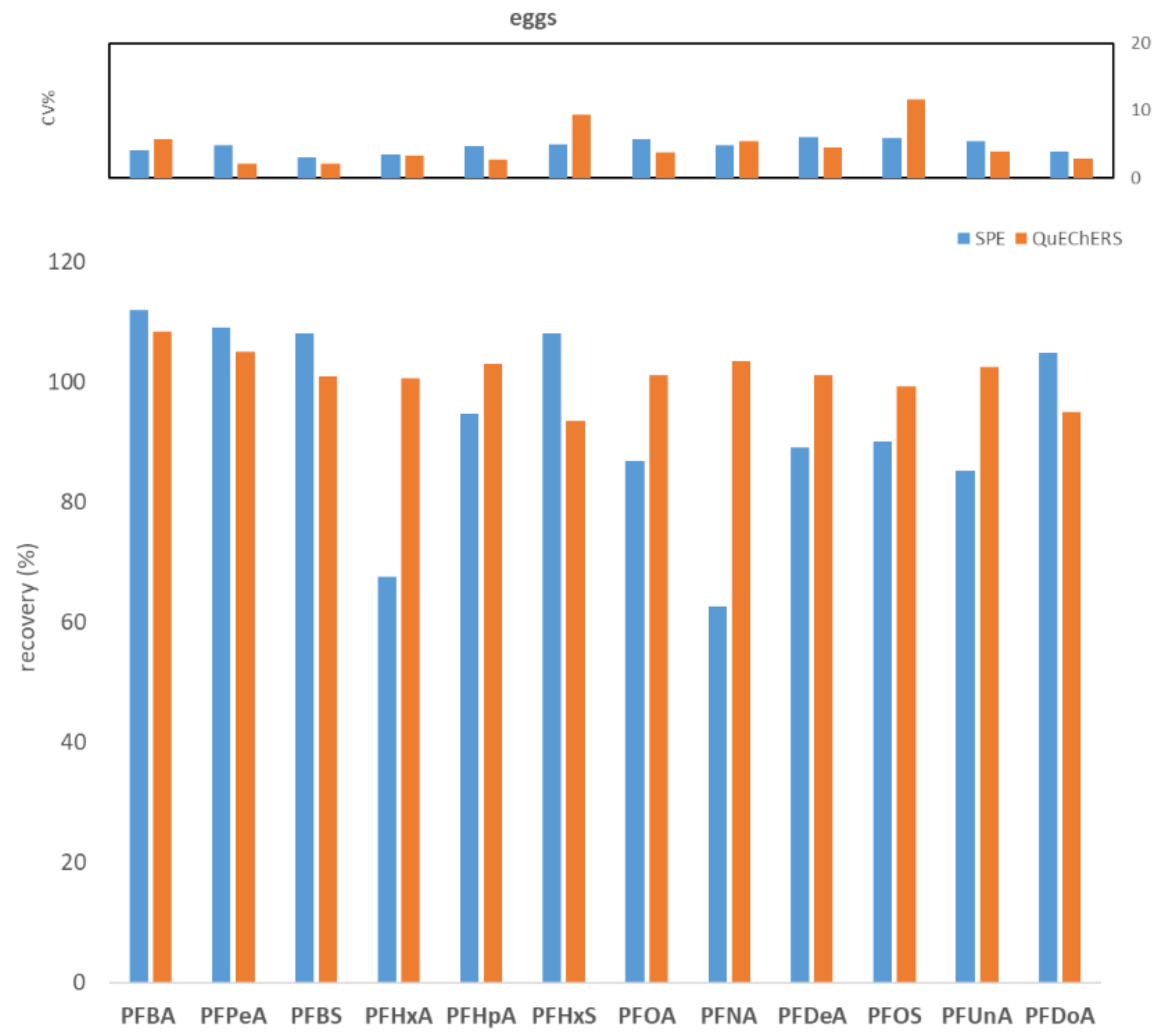
2.1. Comparison between WAX SPE and QuEChERS Methods
2.2. QuEChERS Method Validation
2.3. Analysis of Food Samples
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. QuEChERS Sample Preparation
4.3. SPE Sample Preparation
4.4. Instrumental Analysis
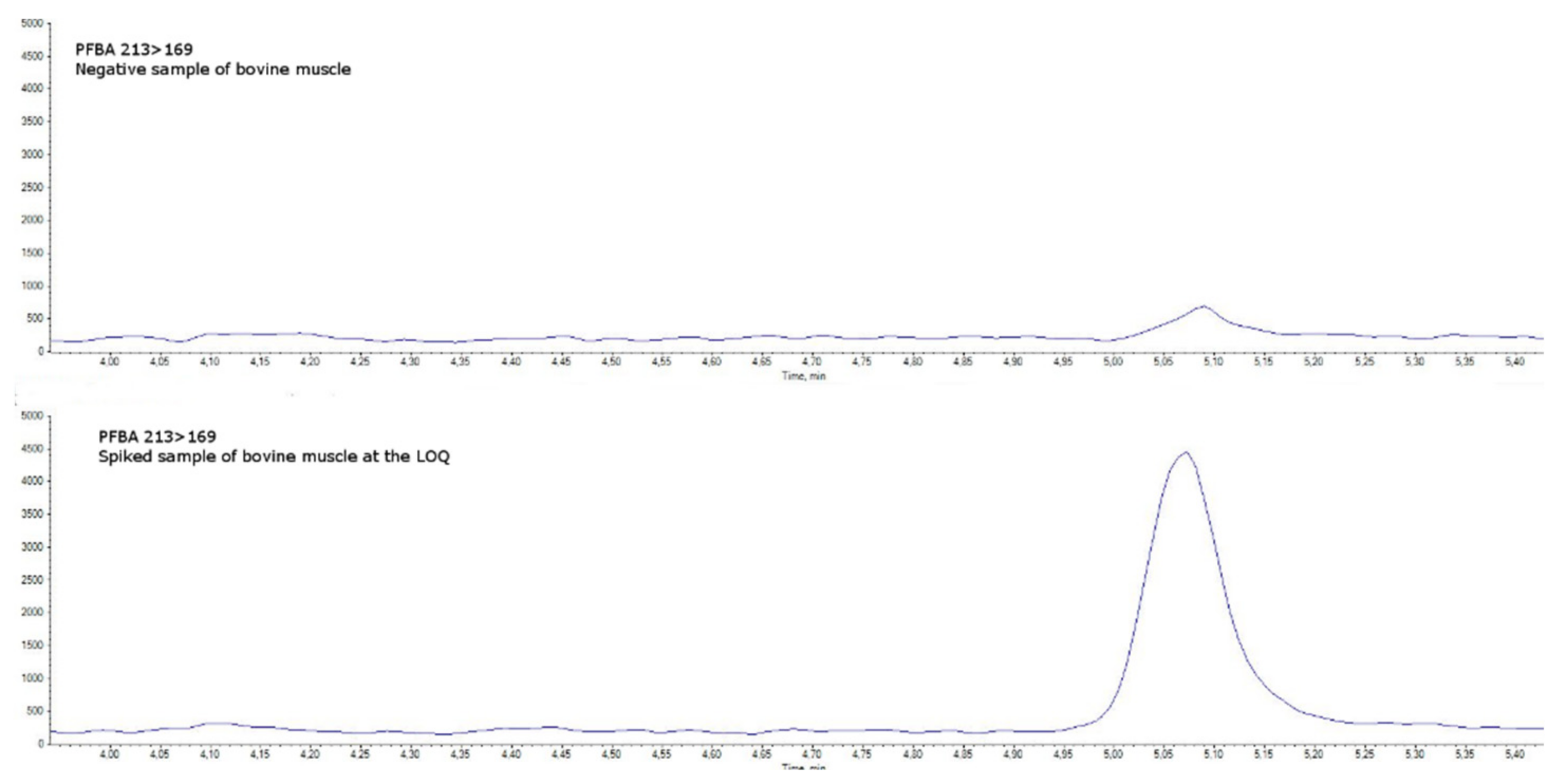
4.5. Method Validation
- p number of calibration levels;
- q number of replicate analyses per calibration level;
- tα, value from t-distribution for probability level α = 0.05 (one-sided test) and
- υ = (p × q) −2 degrees of freedom;
- tß,υ value from t-distribution for probability level ß = 0.05 (one-sided test) and
- υ = (p × q) −2 degrees of freedom;
- sy,x standard deviation of the residuals;
- b slope of the calibration curve;
- m number of replicate analyses of the test sample;
- content value corresponding to the mean calibration level;
- xi content value of the analyte at calibration level i.
- Rw within laboratory reproducibility
- u(bias) u (bias)
- RMSbias
- u(Crecovery)
- Ustd 95% confidence interval for the concentration of the standard;
- Biaspipette Volume specification for maximal bias for the pipette;
- ur,pipette Volume specification for maximal repeatability for the pipette.
4.6. Analysis of Real Samples and Quality Controls
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Buck, R.C.; Franklin, J.; Berger, U.; Conder, J.M.; Cousins, I.T.; De Voogt, P.; Jensen, A.A.; Kannan, K.; Mabury, S.A.; van Leeuwen, S.P. Perfluoroalkyl and polyfluoroalkyl substances in the environment: Terminology, classification, and origins. Integr. Environ. Assess. Manag. 2011, 7, 513–541. [Google Scholar] [CrossRef] [PubMed]
- Organisation for Economic Co-operation and Development. Toward a New Comprehensive Global Database of Per-and Polyfluoroalkyl Substances (PFASs): Summary Report on Updating the OECD 2007 List of Per-and Polyfluoroalkyl Substances (PFASs); OECD: Paris, France, 2018. [Google Scholar]
- Houde, M.; Martin, J.W.; Letcher, R.J.; Solomon, K.R.; Muir, D.C. Biological monitoring of polyfluoroalkyl substances: A review. Environ. Sci. Technol. 2006, 40, 3463–3473. [Google Scholar] [CrossRef] [PubMed]
- EFSA CONTAM Panel; Schrenk, D.; Bignami, M.; Bodin, L.; Chipman, J.K.; del Mazo, J.; Grasl-Kraupp, B.; Hogstrand, C.; Hoogenboom, L.; Leblanc, J.C.; et al. Risk to human health related to the presence of perfluoroalkyl substances in food. EFSA J. 2020, 18, e06223. [Google Scholar]
- Brendel, S.; Fetter, É.; Staude, C.; Vierke, L.; Biegel-Engler, A. Short-chain perfluoroalkyl acids: Environmental concerns and a regulatory strategy under REACH. Environ. Sci. Eur. 2018, 30, 9. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Arevalo, E.; Strynar, M.; Lindstrom, A.; Richardson, M.; Kearns, B.; Pickett, A.; Smith, C.; Knappe, D.R. Legacy and emerging perfluoroalkyl substances are important drinking water contaminants in the Cape Fear River Watershed of North Carolina. Environ. Sci. Technol. Lett. 2016, 3, 415–419. [Google Scholar] [CrossRef]
- Parsons, J.R.; Sáez, M.; Dolfing, J.; De Voogt, P. Biodegradation of perfluorinated compounds. In Reviews of Environmental Contamination and Toxicology; Springer: Berlin/Heidelberg, Germany, 2008; Volume 196, pp. 53–71. [Google Scholar]
- Wang, Z.; Cousins, I.T.; Scheringer, M.; Hungerbuehler, K. Hazard assessment of fluorinated alternatives to long-chain perfluoroalkyl acids (PFAAs) and their precursors: Status quo, ongoing challenges and possible solutions. Environ. Int. 2015, 75, 172–179. [Google Scholar] [CrossRef]
- Hopkins, Z.R.; Sun, M.; DeWitt, J.C.; Knappe, D.R. Recently detected drinking water contaminants: GenX and other per-and polyfluoroalkyl ether acids. J.-Am. Water Work. Assoc. 2018, 110, 13–28. [Google Scholar] [CrossRef]
- Strynar, M.; Dagnino, S.; McMahen, R.; Liang, S.; Lindstrom, A.; Andersen, E.; McMillan, L.; Thurman, M.; Ferrer, I.; Ball, C. Identification of novel perfluoroalkyl ether carboxylic acids (PFECAs) and sulfonic acids (PFESAs) in natural waters using accurate mass time-of-flight mass spectrometry (TOFMS). Environ. Sci. Technol. 2015, 49, 11622–11630. [Google Scholar] [CrossRef]
- Gebbink, W.A.; van Asseldonk, L.; van Leeuwen, S.P. Presence of emerging per-and polyfluoroalkyl substances (PFASs) in river and drinking water near a fluorochemical production plant in the Netherlands. Environ. Sci. Technol. 2017, 51, 11057–11065. [Google Scholar] [CrossRef]
- Pfas. Nel Po C6O4 Quasi 2000 Volte Superiori Che Sotto Miteni. I Dati Della Regione Veneto alla Procura della Repubblica. Available online: http://musei.regione.veneto.it/web/guest/comunicati-stampa/dettaglio-comunicati?_spp_detailId=3303535 (accessed on 1 April 2019).
- Haug, L.S.; Huber, S.; Becher, G.; Thomsen, C. Characterisation of human exposure pathways to perfluorinated compounds—Comparing exposure estimates with biomarkers of exposure. Environ. Int. 2011, 37, 687–693. [Google Scholar] [CrossRef]
- Trudel, D.; Horowitz, L.; Wormuth, M.; Scheringer, M.; Cousins, I.T.; Hungerbühler, K. Estimating consumer exposure to PFOS and PFOA. Risk Anal. Int. J. 2008, 28, 251–269. [Google Scholar] [CrossRef]
- Commission Recommendation. Commission Recommendation of 17 March 2010 on the monitoring of perfluoroalkylated substances in food. 2010 (2010/161/EU). Off. J. of Eur. Union 2010, L 68/22, 22–23. Available online: https://eur-lex.europa.eu/eli/reco/2010/161/oj (accessed on 4 August 2022).
- EURL for halogenated POPs in Feed and Food: Guidance Document on Analytical Parameters for the Determination of Per-and Polyfluoroalkyl Substances (PFAS) in Food and Feed, Version 1, 1 March 2022. Available online: https://eurl-pops.eu/core-working-groups#_pfas (accessed on 4 August 2022).
- Sadia, M.; Yeung, L.W.; Fiedler, H. Trace level analyses of selected perfluoroalkyl acids in food: Method development and data generation. Environ. Pollut. 2020, 263, 113721. [Google Scholar] [CrossRef]
- Jogsten, I.E.; Perelló, G.; Llebaria, X.; Bigas, E.; Martí-Cid, R.; Kärrman, A.; Domingo, J.L. Exposure to perfluorinated compounds in Catalonia, Spain, through consumption of various raw and cooked foodstuffs, including packaged food. Food Chem. Toxicol. 2009, 47, 1577–1583. [Google Scholar] [CrossRef]
- Ballesteros-Gómez, A.; Rubio, S.; van Leeuwen, S. Tetrahydrofuran–water extraction, in-line clean-up and selective liquid chromatography/tandem mass spectrometry for the quantitation of perfluorinated compounds in food at the low picogram per gram level. J. Chromatogr. A 2010, 1217, 5913–5921. [Google Scholar] [CrossRef]
- Haug, L.S.; Salihovic, S.; Jogsten, I.E.; Thomsen, C.; van Bavel, B.; Lindström, G.; Becher, G. Levels in food and beverages and daily intake of perfluorinated compounds in Norway. Chemosphere 2010, 80, 1137–1143. [Google Scholar] [CrossRef]
- Heo, J.; Lee, J.; Kim, S.; Oh, J. Foodstuff analyses show that seafood and water are major perfluoroalkyl acids (PFAAs) sources to humans in Korea. J. Hazard. Mater. 2014, 279, 402–409. [Google Scholar] [CrossRef]
- Kim, L.; Lee, D.; Cho, H.; Choi, S. Review of the QuEChERS method for the analysis of organic pollutants: Persistent organic pollutants, polycyclic aromatic hydrocarbons, and pharmaceuticals. Trends Environ. Anal. Chem. 2019, 22, e00063. [Google Scholar] [CrossRef]
- Gallocchio, F.; Mancin, M.; Belluco, S.; Moressa, A.; Angeletti, R.; Lorenzetto, M.; Arcangeli, G.; Ferrè, N.; Ricci, A.; Russo, F. Investigation of levels of perfluoroalkyl substances in freshwater fishes collected in a contaminated area of Veneto Region, Italy. Environ. Sci. Pollut. Res. 2022, 29, 20996–21011. [Google Scholar] [CrossRef]
- ISO 13528:2015; Statistical Methods for Use in Proficiency Testing by Interlaboratory Comparison. ISO: Geneve, Switzerland, 2015.
- Su, H.; Shi, Y.; Lu, Y.; Wang, P.; Zhang, M.; Sweetman, A.; Jones, K.; Johnson, A. Home produced eggs: An important pathway of human exposure to perfluorobutanoic acid (PFBA) and perfluorooctanoic acid (PFOA) around a fluorochemical industrial park in China. Environ. Int. 2017, 101, 1–6. [Google Scholar] [CrossRef]
- Wang, F.; Zhao, C.; Gao, Y.; Fu, J.; Gao, K.; Lv, K.; Wang, K.; Yue, H.; Lan, X.; Liang, Y. Protein-specific distribution patterns of perfluoroalkyl acids in egg yolk and albumen samples around a fluorochemical facility. Sci. Total Environ. 2019, 650, 2697–2704. [Google Scholar] [CrossRef] [PubMed]
- Mikolajczyk, S.; Pajurek, M.; Warenik-Bany, M. Perfluoroalkyl substances in hen eggs from different types of husbandry. Chemosphere 2022, 303, 134950. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Commission Regulation (EC) No 333/2007 of 28 March 2007 laying down the methods of sampling and analysis for the official control of the levels of lead, cadmium, mercury, inorganic tin, 3-MCPD and benzo(a)pyrene in foodstuffs. Off. J. Eur. Union 2007, L88, 29. [Google Scholar]
- Pihlström, T.; Fernández-Alba, A.R.; Ferrer Amate, C.; Poulsen, M.E.; Hardebusch, B.; Anastassiades, M.; Lippold, R.; Cabrera, L.C.; de Kok, A.; O’Regan, F.; et al. Document Nº SANTE/11312/2021. Analytical Quality Control and Method Validation Procedures for Pesticide Residues Analysis in Food and Feed. Available online: https://www.eurl-pesticides.eu/userfiles/file/EurlALL/SANTE_11312_2021.pdf (accessed on 4 August 2022).
- Wenzl, T.; Haedrich, J.; Schaechtele, A.; Piotr, R.; Stroka, J.; Eppe, G.; Scholl, G. Guidance Document on the Estimation of LOD and LOQ for Measurements in the Field of Contaminants in Food and Feed; Publications Office of the European Union: Luxembourg, 2016. [Google Scholar]
- Nordic Commitee on Food Analysis. Estimation and Expression of Measurement Uncertainity in Chemical Analysis. NMKL Procedure N°5 (2019); NMKL: Bergen, Norway, 2019. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
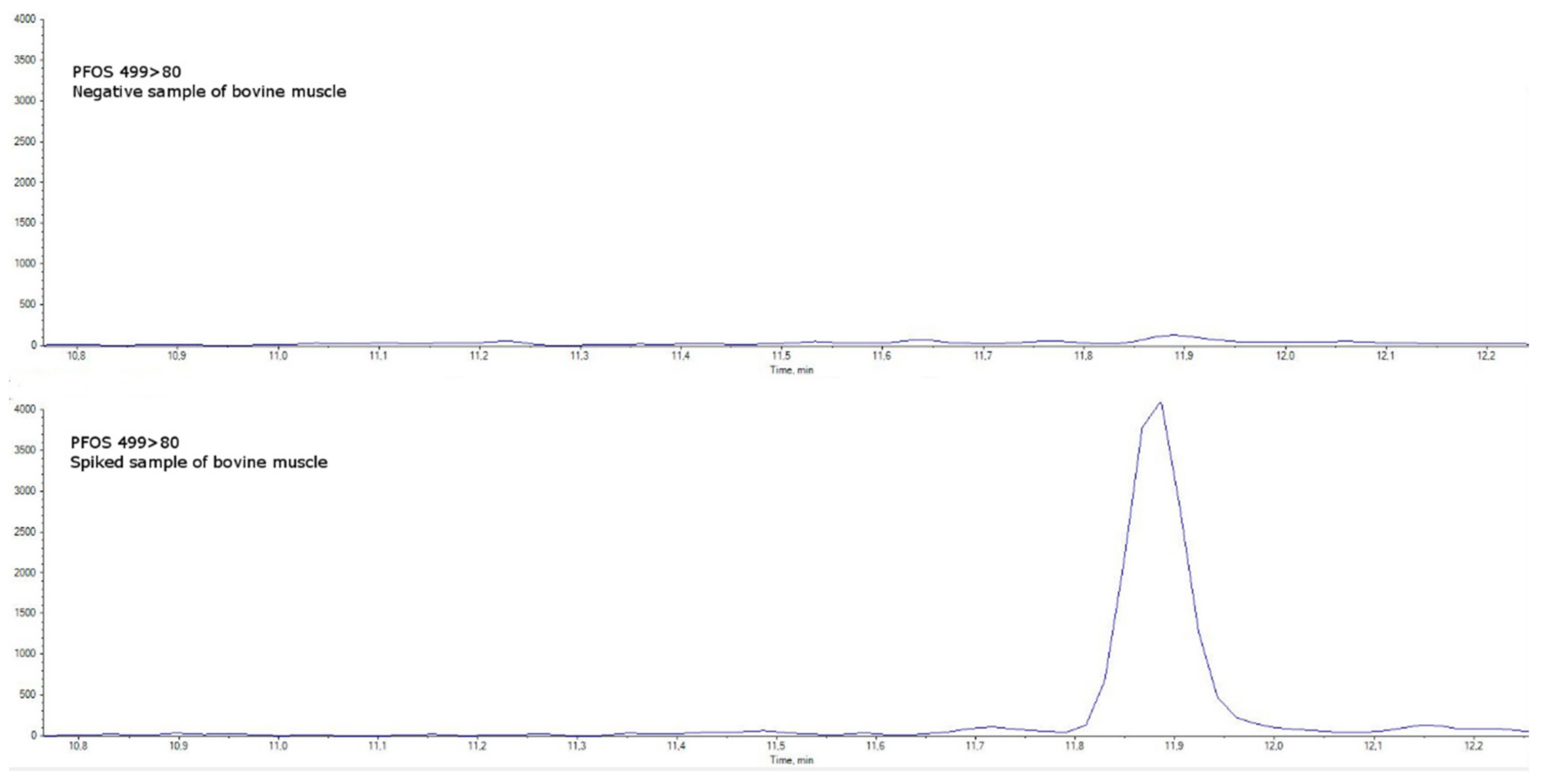{kind=link}
| Compound | Nominal Concentration (ng/kg) | Recovery (%) (n = 7) | Precision (CV %) (n = 7) | ||||
|---|---|---|---|---|---|---|---|
| Hen Eggs | Cow’s Milk | Bovine Liver | Hen Eggs | Cow’s Milk | Bovine Liver | ||
| PFBA | 50 * | 96.9 | 97.3 | 103.0 | 7.6 | 3.9 | 3.8 |
| 100 | 108.5 | 106.5 | 98.6 | 5.7 | 1.6 | 8.3 | |
| 500 | 106.5 | 102.6 | 101.5 | 2.6 | 2.5 | 2.4 | |
| 1000 | 104.6 | 103.2 | 103.3 | 0.9 | 1.7 | 3.8 | |
| PFPeA | 50 * | 105.0 | 99.8 | 99.2 | 1.2 | 4.0 | 5.4 |
| 100 | 105.1 | 102.8 | 101.0 | 2.1 | 3.4 | 6.0 | |
| 500 | 104.0 | 103.8 | 101.8 | 2.2 | 1.6 | 2.5 | |
| 1000 | 103.1 | 105.8 | 97.5 | 0.7 | 1.6 | 4.4 | |
| PFBS | 50 * | 99.2 | 103.0 | 102.3 | 1.2 | 6.1 | 4.8 |
| 100 | 101.0 | 107.5 | 105.1 | 2.1 | 5.0 | 4.6 | |
| 500 | 101.8 | 103.3 | 108.2 | 2.2 | 3.7 | 5.0 | |
| 1000 | 97.5 | 104.8 | 105.0 | 0.7 | 2.6 | 3.9 | |
| PFHxA | 50 * | 95.5 | 115.8 | 106.2 | 1.8 | 6.7 | 7.6 |
| 100 | 100.7 | 112.4 | 105.8 | 3.4 | 4.5 | 3.9 | |
| 500 | 104.7 | 105.6 | 107.7 | 2.3 | 6.0 | 4.3 | |
| 1000 | 103.6 | 105.7 | 105.1 | 1.5 | 1.2 | 3.0 | |
| PFHpA | 50 * | 106.0 | 97.4 | 106.0 | 2.2 | 5.1 | 9.8 |
| 100 | 103.0 | 104.3 | 103.0 | 2.7 | 4.7 | 5.7 | |
| 500 | 103.0 | 110.2 | 103.0 | 2.8 | 1.9 | 3.3 | |
| 1000 | 107.8 | 110.1 | 107.8 | 4.1 | 1.4 | 8.7 | |
| PFHxS | 50 * | 91.4 | 100.5 | 92.7 | 8.4 | 5.5 | 7.9 |
| 100 | 93.5 | 110.7 | 104.7 | 9.4 | 5.0 | 7.22 | |
| 500 | 99.5 | 104.6 | 96.9 | 9.9 | 5.1 | 5.6 | |
| 1000 | 101.9 | 104.4 | 97.9 | 11.5 | 6.4 | 13.7 | |
| PFOA | 50 * | 97.5 | 101.8 | 105.5 | 3.8 | 3.9 | 3.6 |
| 100 | 101.1 | 102.5 | 103.4 | 3.8 | 4.2 | 6.8 | |
| 500 | 106.0 | 101.6 | 107.2 | 1.0 | 1.8 | 9.4 | |
| 1000 | 107.7 | 105.8 | 102.4 | 2.5 | 3.1 | 4.8 | |
| PFNA | 50 * | 111.7 | 121.2 | 111.7 | 6.5 | 8.9 | 6.4 |
| 100 | 103.6 | 121.8 | 103.6 | 5.4 | 3.1 | 4.5 | |
| 500 | 106.7 | 106.3 | 106.7 | 1.6 | 2.2 | 3.6 | |
| 1000 | 105.1 | 108.3 | 105.1 | 2.7 | 3.1 | 8.3 | |
| PFDeA | 50 * | 91.8 | 111.0 | 115.1 | 5.3 | 2.3 | 10.1 |
| 100 | 101.2 | 112.0 | 110.9 | 4.6 | 1.6 | 2.6 | |
| 500 | 110.1 | 102.1 | 108.5 | 3.8 | 1.4 | 3.3 | |
| 1000 | 121.1 | 106.6 | 104.1 | 5.3 | 3.1 | 6.8 | |
| PFOS | 50 * | 86.7 | 93.9 | 103.1 | 5.6 | 3.8 | 7.7 |
| 100 | 99.3 | 107.0 | 93.2 | 11.6 | 9.0 | 15.4 | |
| 500 | 107.8 | 100.3 | 94.1 | 6.1 | 6.3 | 8.4 | |
| 1000 | 105.0 | 105.5 | 81.0 | 3.7 | 5.0 | 11.7 | |
| PFUnA | 50 * | 105.8 | 109.2 | 105.8 | 2.8 | 3.9 | 4.4 |
| 100 | 102.5 | 104.2 | 102.5 | 4.0 | 2.9 | 2.0 | |
| 500 | 104.8 | 101.2 | 104.8 | 2.3 | 2.5 | 3.2 | |
| 1000 | 107.1 | 104.8 | 107.1 | 3.8 | 2.5 | 4.7 | |
| PFDoA | 50 * | 87.8 | 105.0 | 104.6 | 1.8 | 2.5 | 3.0 |
| 100 | 95.0 | 107.3 | 103.8 | 2.9 | 3.0 | 3.8 | |
| 500 | 100.3 | 103.0 | 106.3 | 1.8 | 1.8 | 2.0 | |
| 1000 | 103.2 | 103.9 | 105.8 | 1.6 | 2.4 | 3.9 | |
| GenX | 100 * | 94.9 | 103.8 | 94.9 | 13.0 | 10.9 | 14.1 |
| 500 | 92.4 | 91.3 | 92.4 | 10.0 | 5.3 | 8.1 | |
| 1000 | 93.7 | 94.6 | 93.7 | 9.0 | 4.2 | 16.1 | |
| C6O4 | 100 * | 107.9 | 109.2 | 94.4 | 10.1 | 3.7 | 7.3 |
| 500 | 94.9 | 97.3 | 84.6 | 14.0 | 10.5 | 19.3 | |
| 1000 | 111.1 | 97.4 | 94.0 | 9.5 | 8.8 | 16.5 | |
| Compound | Nominal Concentration (ng/kg) | Recovery (%) (n = 7) | Precision (CV %) (n = 7) | ||||
|---|---|---|---|---|---|---|---|
| Fish Muscle | Swine Muscle | Bovine Muscle | Fish Muscle | Swine Muscle | Bovine Muscle | ||
| PFBA | 50 * | 91.9 | 83.6 | 100.0 | 13.0 | 12.2 | 4.9 |
| 100 | 100.4 | 89.4 | 103.6 | 10.5 | 11.8 | 3.7 | |
| 500 | 92.8 | 96.8 | 111.5 | 3.5 | 2.0 | 1.5 | |
| 1000 | 102.2 | 101.2 | 111.0 | 5.7 | 3.8 | 1.2 | |
| PFPeA | 50 * | 105.3 | 111.6 | 119.0 | 4.5 | 2.8 | 2.8 |
| 100 | 104.3 | 103.3 | 107.9 | 2.3 | 4.3 | 4.3 | |
| 500 | 97.2 | 101.2 | 109.4 | 3.6 | 1.3 | 1.5 | |
| 1000 | 103.1 | 105.6 | 108.7 | 5.1 | 4.5 | 2.38 | |
| PFBS | 50 * | 109.2 | 107.3 | 100.1 | 4.6 | 2.8 | 6.6 |
| 100 | 108.3 | 98.7 | 107.4 | 2.0 | 5.8 | 4.7 | |
| 500 | 99.3 | 98.6 | 111.3 | 5.2 | 2.9 | 3.6 | |
| 1000 | 103.7 | 98.5 | 108.4 | 5.5 | 4.9 | 2.1 | |
| PFHxA | 50 * | 80.0 | 90.4 | 77.8 | 9.6 | 5.5 | 14.6 |
| 100 | 77.7 | 90.2 | 94.4 | 9.11 | 5.8 | 5.9 | |
| 500 | 93.9 | 102.0 | 105.6 | 3.2 | 2.8 | 2.4 | |
| 1000 | 106.6 | 102.3 | 108.8 | 6.0 | 9.8 | 4.2 | |
| PFHpA | 50 * | 110.3 | 105.5 | 93.3 | 6.9 | 4.3 | 15.4 |
| 100 | 106.5 | 98.4 | 105.5 | 6.5 | 7.7 | 4.3 | |
| 500 | 95.1 | 100.6 | 110.2 | 7.0 | 3.9 | 2.1 | |
| 1000 | 100.9 | 105.4 | 115.2 | 8.7 | 7.2 | 5.2 | |
| PFHxS | 50 * | 107.1 | 107.2 | 106.3 | 4.8 | 5.4 | 6.8 |
| 100 | 107.7 | 103.4 | 103.6 | 7.6 | 3.7 | 5.5 | |
| 500 | 105.9 | 100.8 | 108.6 | 2.1 | 3.9 | 6.7 | |
| 1000 | 105.6 | 96.6 | 114.2 | 7.0 | 12.9 | 8.3 | |
| PFOA | 50 * | 89.6 | 96.4 | 113.7 | 6.3 | 3.0 | 6.1 |
| 100 | 96.5 | 98.2 | 106.3 | 6.1 | 2.1 | 4.3 | |
| 500 | 94.9 | 100.9 | 115.4 | 6.9 | 2.5 | 3.0 | |
| 1000 | 105.9 | 104.4 | 113.3 | 6.3 | 5.9 | 2.5 | |
| PFNA | 50 * | 67.9 | 103.2 | 82.4 | 8.7 | 5.2 | 11.1 |
| 100 | 93.4 | 104.2 | 80.0 | 6.1 | 7.3 | 7.6 | |
| 500 | 100.9 | 106.1 | 104.8 | 3.6 | 3.1 | 4.8 | |
| 1000 | 106.4 | 110.8 | 110.9 | 6.5 | 6.0 | 4.7 | |
| PFDeA | 50 * | 120.2 | 90.3 | 111.9 | 4.8 | 2.8 | 7.0 |
| 100 | 110.3 | 98.9 | 105.9 | 6.9 | 2.8 | 3.7 | |
| 500 | 95.2 | 102.7 | 116.6 | 6.4 | 1.1 | 2.0 | |
| 1000 | 102.2 | 110.9 | 120.2 | 9.4 | 3.7 | 3.4 | |
| PFOS | 50 * | 102.9 | 111.8 | 102.1 | 14.6 | 10.2 | 16.2 |
| 100 | 83.0 | 104.3 | 104.4 | 7.2 | 8.3 | 6.8 | |
| 500 | 86.3 | 103.9 | 105.1 | 3.7 | 7.6 | 5.9 | |
| 1000 | 103.6 | 105.9 | 110.4 | 12.7 | 7.8 | 9.6 | |
| PFUnA | 50 * | 107.9 | 112.5 | 82.3 | 4.4 | 2.2 | 4.5 |
| 100 | 105.0 | 107.1 | 93.5 | 3.4 | 5.0 | 6.7 | |
| 500 | 98.1 | 100.9 | 109.8 | 4.7 | 2.5 | 1.8 | |
| 1000 | 105.7 | 105.9 | 113.5 | 6.0 | 4.5 | 1.5 | |
| PFDoA | 50 * | 103.7 | 106.6 | 105.3 | 2.2 | 3.5 | 1.4 |
| 100 | 100.7 | 104.4 | 102.1 | 2.7 | 3.5 | 2.8 | |
| 500 | 91.1 | 99.0 | 111.1 | 4.2 | 3.5 | 2.9 | |
| 1000 | 94.7 | 102.9 | 110.5 | 5.4 | 5.6 | 2.2 | |
| GenX | 100 * | 79.9 | 80.0 | 57.2 | 11.2 | 15.9 | 5.9 |
| 500 | 88.3 | 69.2 | 66.8 | 4.8 | 4.8 | 6.6 | |
| 1000 | 97.5 | 74.4 | 81.1 | 6.2 | 8.6 | 2.2 | |
| C6O4 | 100 * | 103.8 | 82.1 | 73.5 | 10.6 | 13.1 | 7.1 |
| 500 | 99.1 | 72.9 | 85.1 | 5.6 | 4.5 | 15.6 | |
| 1000 | 96.8 | 79.8 | 85.8 | 9.3 | 13.9 | 12.2 | |
| Matrix | ||||
|---|---|---|---|---|
| Analyte | Milk | Liver | Egg | Muscle |
| LOD ng/kg | LOD ng/kg | LOD ng/kg | LOD ng/kg | |
| PFBA | 11.01 | 8.43 | 7.20 | 8.05 |
| PFPeA | 10.47 | 8.72 | 8.65 | 5.78 |
| PFBS | 12.77 | 10.78 | 14.26 | 6.87 |
| PFHxA | 8.38 | 8.65 | 7.50 | 14.76 |
| PFHpA | 4.43 | 9.09 | 8.28 | 5.81 |
| PFHxS | 4.68 | 7.84 | 10.10 | 10.39 |
| PFOA | 4.86 | 6.31 | 8.03 | 9.01 |
| PFNA | 4.94 | 9.27 | 19.17 | 8.07 |
| PFDeA | 7.79 | 6.40 | 18.45 | 9.19 |
| PFOS | 4.75 | 6.27 | 9.08 | 17.33 |
| PFUnA | 10.15 | 4.66 | 11.35 | 11.49 |
| PFDoA | 6.95 | 7.33 | 6.70 | 3.69 |
| GenX | 7.14 | 13.11 | 13.75 | 12.98 |
| C6O4 | 9.30 | 19.84 | 8.21 | 8.70 |
| Analyte | Matrix | |||
|---|---|---|---|---|
| Milk | Liver | Egg | Muscle | |
| (%) | (%) | (%) | (%) | |
| PFBA | 14.66 | 29.28 | 39.42 | 25.22 |
| PFPeA | 16.56 | 45.40 | 29.60 | 29.52 |
| PFBS | 23.36 | 41.44 | 24.18 | 18.48 |
| PFHxA | 28.80 | 40.80 | 38.10 | 46.38 |
| PFHpA | 16.86 | 32.94 | 24.44 | 33.94 |
| PFHxS | 19.26 | 32.70 | 23.38 | 19.34 |
| PFOA | 14.34 | 26.16 | 23.66 | 33.24 |
| PFNA | 31.02 | 27.12 | 45.92 | 47.60 |
| PFDeA | 23.56 | 49.44 | 17.28 | 31.56 |
| PFOS | 20.66 | 46.84 | 30.94 | 38.22 |
| PFUnA | 23.04 | 19.68 | 43.24 | 32.96 |
| PFDoA | 19.64 | 31.06 | 25.12 | 29.58 |
| GenX | 32.56 | 32.30 | 47.32 | 45.52 |
| C6O4 | 23.44 | 34.06 | 45.86 | 37.02 |
| Compound | Milk | Liver | Egg | Muscle |
|---|---|---|---|---|
| PFBA | −6% | −13% | −2% | −3% |
| PFPeA | −11% | −15% | −11% | −9% |
| PFBS | −2% | −14% | −4% | −5% |
| PFHxA | −8% | −10% | −9% | −8% |
| PFHpA | −5% | −12% | −8% | −11% |
| PFHxS | −8% | −9% | −9% | −10% |
| PFOA | −7% | −8% | −6% | −7% |
| PFNA | −5% | −10% | −6% | −8% |
| PFDeA | −4% | −13% | −2% | −5% |
| PFOS | −5% | −5% | −4% | −6% |
| PFUnA | −8% | −13% | −7% | −10% |
| PFDoA | −5% | −8% | −6% | −8% |
| GenX | −9% | −11% | −8% | −10% |
| C6O4 | −11% | −13% | −9% | −7% |
| Matrix | Species/ Category | Sample ID | PFBA ng/kg (n = 2) | PFHXS ng/kg (n = 2) | PFOA ng/kg (n = 2) | PFNA ng/kg (n = 2) | PFDA ng/kg (n = 2) | PFOS ng/kg (n = 2) | PFUNA ng/kg (n = 2) | PFDOA ng/kg (n = 2) |
|---|---|---|---|---|---|---|---|---|---|---|
| Liver | Trout | L1 | 138 | 179 | 91 | 4215 | 126 | 28 * | ||
| Trout | L2 | 192 | 117 | 4083 | 128 | 27 * | ||||
| Calf | L4 | 60 | 159 | |||||||
| Calf | L5 | 41 * | 100 | |||||||
| Calf | L6 | 35 * | 50 | 90 | 25 * | 23 * | ||||
| Calf | L7 | 36 * | 48 * | 132 | 11 * | 36 * | ||||
| Calf | L8 | 30 * | 36 * | 35 * | 699 | |||||
| Calf | L9 | 68 | 64 | 301 | 30 | |||||
| Calf | L10 | 35 * | 55 | 160 | 18 | |||||
| Calf | L11 | 36 * | 56 | 149 | 19 | |||||
| Calf | L12 | 18 * | 54 | 156 | 17 | |||||
| Calf | L13 | 19 * | 29 * | 53 | 42 | |||||
| Calf | L14 | 41 * | 72 | 140 | 14 | |||||
| Bullock | L15 | 43 * | 59 | 246 | 14 | |||||
| Bullock | L16 | 36 * | 85 | 329 | 15 | |||||
| Bullock | L17 | 38 * | 62 | 186 | ||||||
| Bullock | L18 | 77 | 201 | |||||||
| Bullock | L19 | 91 | 193 | 225 | 41 * | |||||
| Bullock | L20 | 78 | 98 | 220 | 23 * | |||||
| Bullock | L21 | 35 * | 60 | 105 | 18 * | |||||
| Bullock | L22 | 38 * | 63 | 177 | 13 * | |||||
| Bullock | L23 | 38 * | 54 | 232 | 16 * | |||||
| Bullock | L24 | 23 * | ||||||||
| Bovine Adult | L25 | 123 | 284 | 409 | 1099 | 124 | 44 * | |||
| Bovine Adult | L26 | 52 | 304 | 577 | 1362 | 203 | 71 | |||
| Bovine Adult | L27 | 75 | 262 | 446 | 1220 | 144 | 53 | |||
| Bovine Adult | L28 | 50 | 255 | 411 | 1195 | 128 | 54 | |||
| Bovine Adult | L29 | 84 | 132 | 388 | 1104 | 108 | 44 * | |||
| Bovine Adult | L30 | 85 | 167 | 622 | 1609 | 179 | 58 | |||
| Bovine Adult | L31 | 83 | 137 | 314 | 1117 | 112 | 47 * | |||
| Bovine Adult | L32 | 93 | 158 | 365 | 1092 | 119 | 47 * | |||
| Swine | L33 | 39 * | 73 | 55 | 2874 | 106 | ||||
| Swine | L34 | 102 | 28 * | 63 | ||||||
| Eggs | Hens | E1 | 119 | |||||||
| Hens | E2 | 25 * | 40 * | 25 * | 60 | 150 | 910 | 71 | 170 | |
| Hens | E3 | 46 * | ||||||||
| Hens | E4 | 101 | ||||||||
| Hens | E5 | 110 | ||||||||
| Hens | E6 | 119 | ||||||||
| Muscle | Trout | M4 | 15 * | 34 * | 70 | 360 | 89 | 94 | ||
| Trout | M5 | 10 * | 36 * | 80 | 331 | 86 | 93 | |||
| Pangasius | M6 | 16 * | 34 * | 99 | 69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallocchio, F.; Moressa, A.; Zonta, G.; Angeletti, R.; Lega, F. Fast and Sensitive Analysis of Short- and Long-Chain Perfluoroalkyl Substances in Foods of Animal Origin. Molecules 2022, 27, 7899. https://doi.org/10.3390/molecules27227899
Gallocchio F, Moressa A, Zonta G, Angeletti R, Lega F. Fast and Sensitive Analysis of Short- and Long-Chain Perfluoroalkyl Substances in Foods of Animal Origin. Molecules. 2022; 27(22):7899. https://doi.org/10.3390/molecules27227899
Chicago/Turabian StyleGallocchio, Federica, Alessandra Moressa, Gloria Zonta, Roberto Angeletti, and Francesca Lega. 2022. "Fast and Sensitive Analysis of Short- and Long-Chain Perfluoroalkyl Substances in Foods of Animal Origin" Molecules 27, no. 22: 7899. https://doi.org/10.3390/molecules27227899
APA StyleGallocchio, F., Moressa, A., Zonta, G., Angeletti, R., & Lega, F. (2022). Fast and Sensitive Analysis of Short- and Long-Chain Perfluoroalkyl Substances in Foods of Animal Origin. Molecules, 27(22), 7899. https://doi.org/10.3390/molecules27227899