


Development and Validation of a Reversed-Phase HPLC Method with UV Detection for the Determination of L-Dopa in Vicia faba L. Broad Beans


 , ,
, ,  ,
,  ,
,  , ,
, ,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results and Discussion
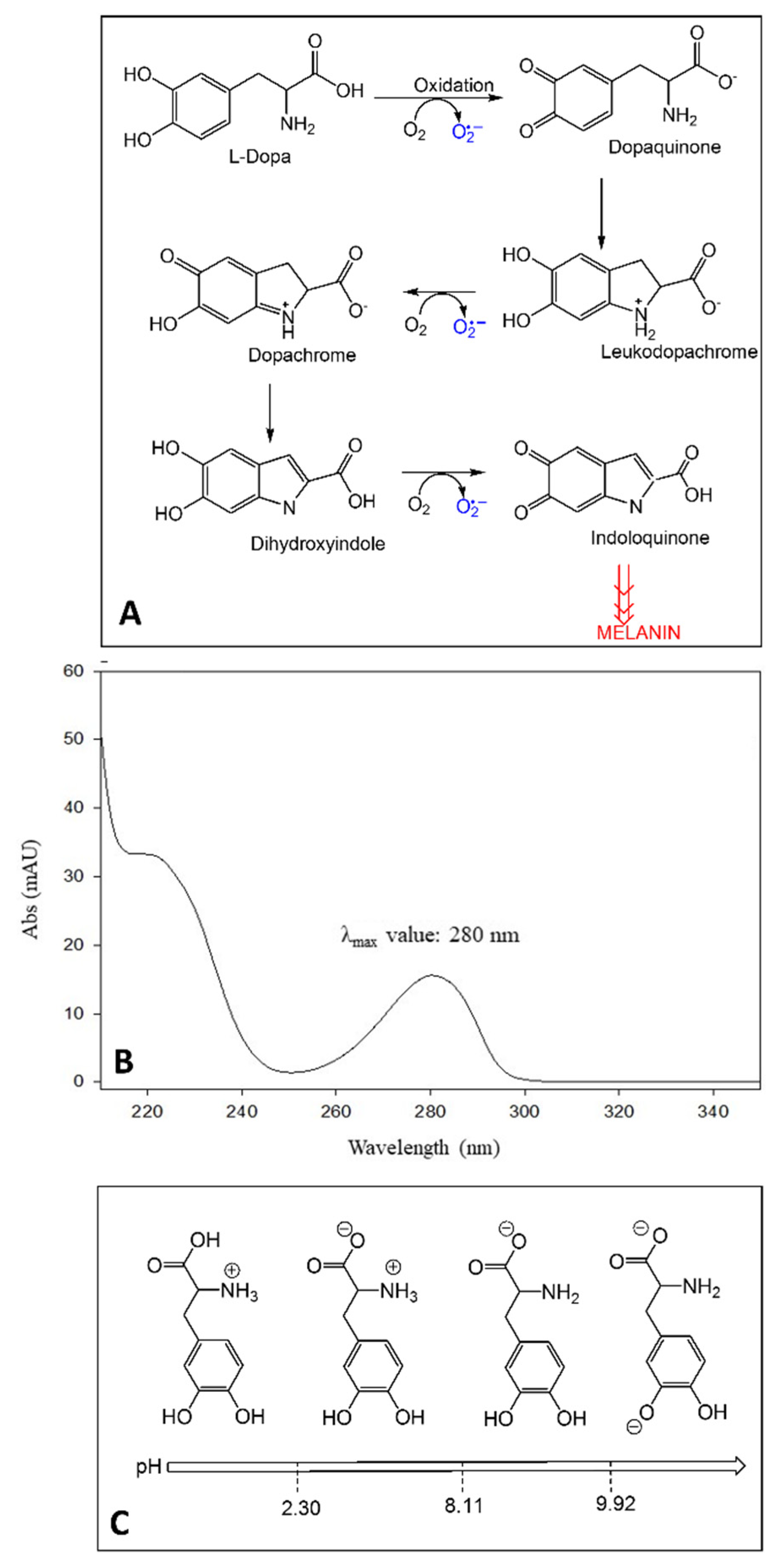
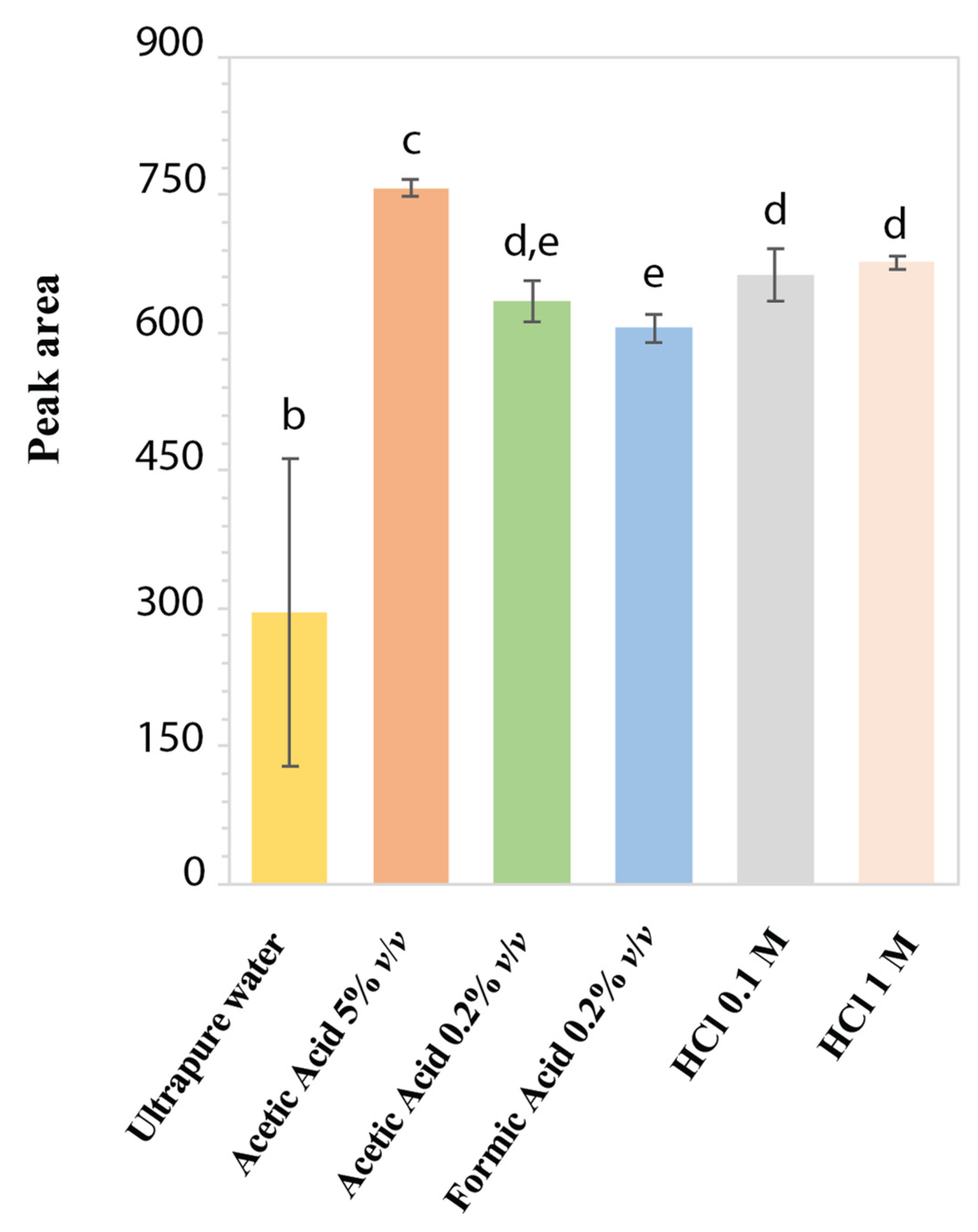
2.1. Aqueous Stability Study of LD
2.2. Chromatographic Performances
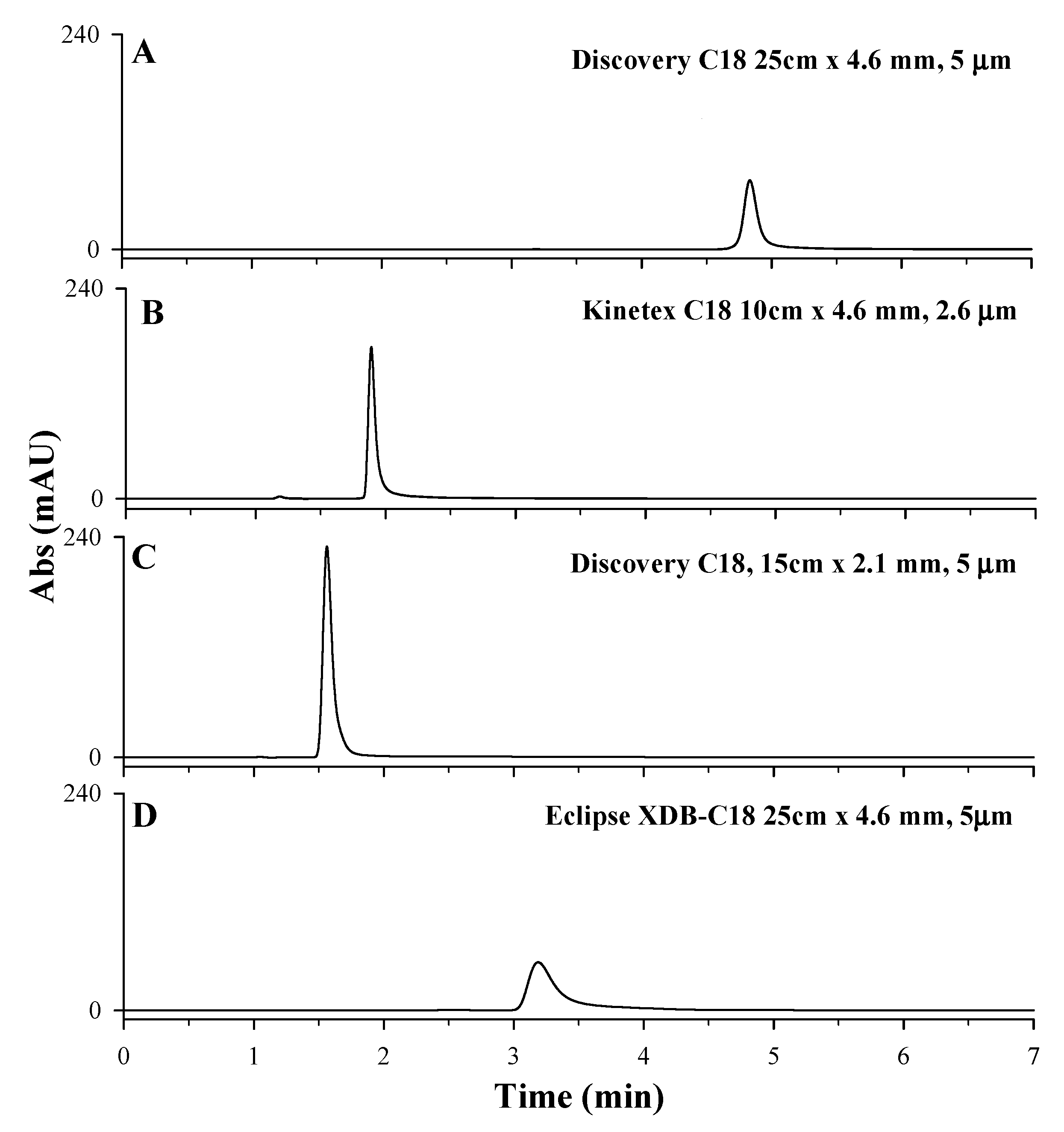
2.2.1. Choice of the Chromatographic Column
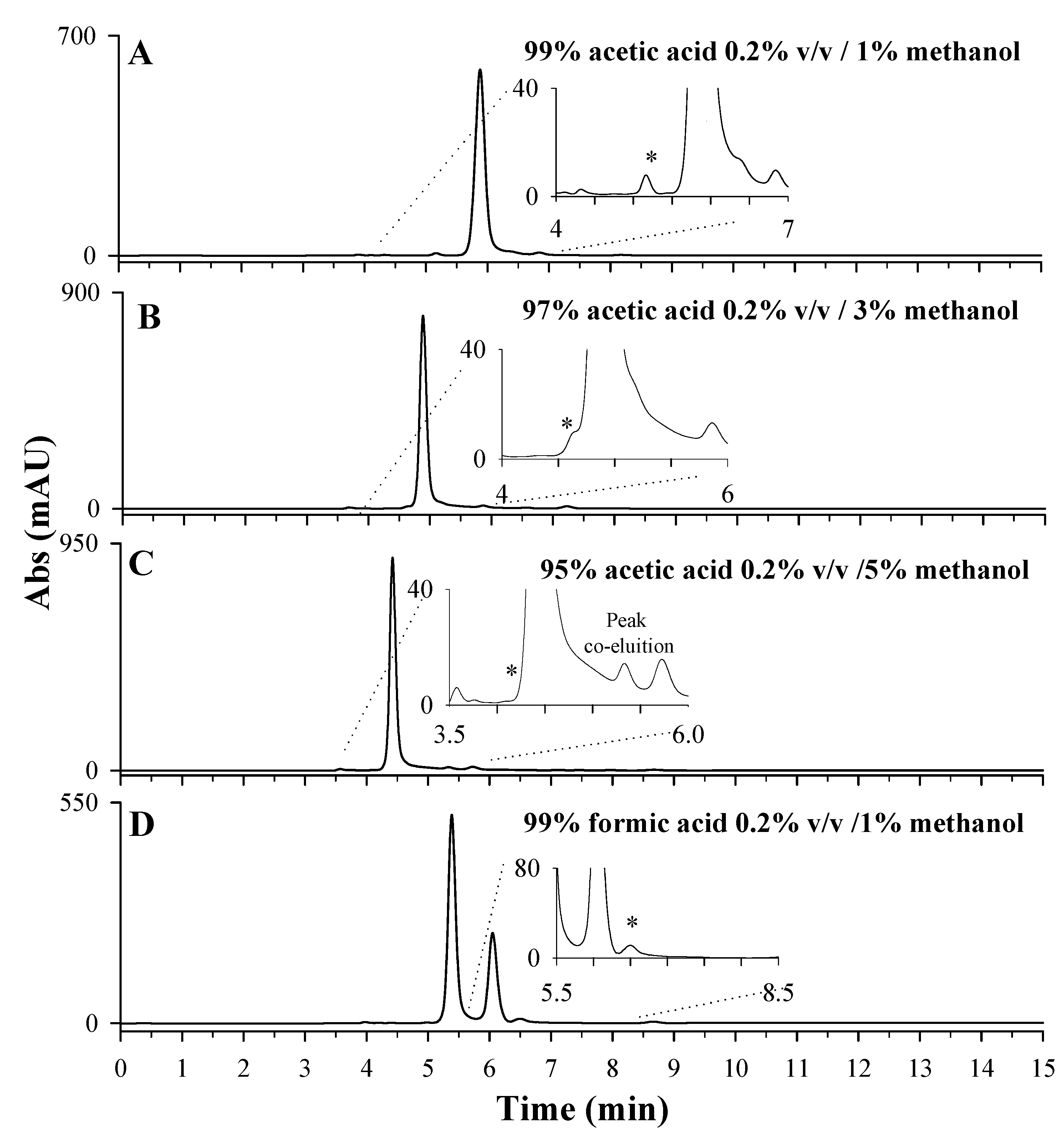
2.2.2. Optimization of the Mobile-Phase Composition
2.3. Method Validation
2.4. Quantitative Analysis of LD in Differently Stored Samples of Vicia faba L.
3. Materials and Methods
3.1. Chemicals
3.2. LD Stability Study
3.3. Analytical Method Validation
3.3.1. LC Performance
3.3.2. Validation Parameters
3.4. Vicia faba L. Broad Beans and LD Extraction
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Ribeiro, R.P.; Gasparetto, J.C.; De Oliveira Vilhena, R.; De Francisco, T.M.G.; Martins, C.A.F.; Cardoso, M.A.; Pontarolo, R. Simultaneous determination of levodopa, carbidopa, entacapone, tolcapone, 3-O-methyldopa and dopamine in human plasma by an HPLC-MS/MS method. Bioanalysis 2015, 7, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, Y.; Wu, H.; Cheng, C.; Le, W. Research advances on L-DOPA-induced dyskinesia: From animal models to human disease. Neurol. Sci. 2020, 41, 2055–2065. [Google Scholar] [CrossRef] [PubMed]
- Mortezazadeh, T.; Seyedarabi, H.; Mahmoudian, B.; Islamian, J.P. Imaging modalities in differential diagnosis of Parkinson’s disease: Opportunities and challenges. Egypt. J. Radiol. Nucl. Med. 2021, 52, 79. [Google Scholar] [CrossRef]
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.F.E.; Church, F.C. Integrative Medicine and Health Therapy for Parkinson Disease. Top. Geriatr. Rehabil. 2020, 36, 176–186. [Google Scholar] [CrossRef]
- Min, K.; Park, K.; Park, D.H.; Yoo, Y.J. Overview on the biotechnological production of l-DOPA. Appl. Microbiol. Biotechnol. 2015, 99, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Dhull, S.B.; Kidwai, M.K.; Noor, R.; Chawla, P.; Rose, P.K. A review of nutritional profile and processing of faba bean (Vicia faba L.). Legum. Sci. 2021, 4, e129. [Google Scholar] [CrossRef]
- Benfica, J.; Morais, E.S.; Miranda, J.S.; Freire, M.G.; de Cássia Superbi de Sousa, R.; Coutinho, J.A.P. Aqueous solutions of organic acids as effective solvents for levodopa extraction from Mucuna pruriens seeds. Sep. Purif. Technol. 2021, 274, 119084. [Google Scholar] [CrossRef]
- Apaydin, H.; Ertan, S.; Ozekmekçi, S. Broad bean (Vicia faba)—A natural source of L-dopa—Prolongs “on” periods in patients with Parkinson’s disease who have “on–off” fluctuations. Mov. Disord. Off. J. Mov. Disord. Soc. 2000, 15, 164–166. [Google Scholar] [CrossRef]
- Tesoro, C.; Lelario, F.; Ciriello, R.; Bianco, G.; Di Capua, A.; Acquavia, M.A. An Overview of Methods for L-Dopa Extraction and Analytical Determination in Plant Matrices. Separations 2022, 9, 224. [Google Scholar] [CrossRef]
- Polanowska, K.; Łukasik, R.M.; Kuligowski, M. Development of a Sustainable, Simple, and Robust Method for Efficient l-DOPA Extraction. Molecules 2019, 24, 2325. [Google Scholar] [CrossRef] [PubMed]
- Rathod, B.G.; Patel, N.M. Development of validated RP-HPLC method for the estimation of L-Dopa from Mucuna pruriens, its extracts and in Aphrodisiac formulation. Int. J. Pharma Sci. Res. 2014, 5, 508–513. [Google Scholar]
- Renna, M.; De Cillis, F.; Leoni, B.; Acciardi, E.; Santamaria, P. From by-product to unconventional vegetable: Preliminary evaluation of fresh fava hulls highlights richness in L-DOPa and low content of anti-nutritional factor. Foods 2020, 9, 159. [Google Scholar] [CrossRef]
- Siddhuraju, P.; Becker, K. Rapid reversed-phase high performance liquid chromatographic method for the quantification of L-Dopa (L-3,4-dihydroxyphenylalanine), non-methylated and methylated tetrahydroisoquinoline compounds from Mucuna beans. Food Chem. 2001, 72, 389–394. [Google Scholar] [CrossRef]
- Singh, R.; Saini, P.; Mathur, S.; Singh, G.; Kumar, S. Application of high performance liquid chromatography to the determination and validation of levodopa in methanolic extract of Mucuna utilis. Int. J. Green Pharm. 2010, 4, 156–158. [Google Scholar] [CrossRef]
- Vora, R.N.; Joshi, A.N.; Joshi, N.C. Comparison of L-Dopa Content in Two Varieties of Broad Beans (Vicia faba) By Different Extraction Techniques. World J. Pharm. Med. Res. 2017, 3, 271–274. [Google Scholar]
- Yang, X.; Zhang, X.; Zhou, R. Determination of L-Dopa content and other significant nitrogenous compounds in the seeds of seven Mucuna and Stizolobium species in China. Pharm. Biol. 2001, 39, 312–316. [Google Scholar] [CrossRef]
- Aware, C.; Patil, R.; Gaikwad, S.; Yadav, S.; Bapat, V.; Jadhav, J. Evaluation of L-dopa, proximate composition with in vitro anti-inflammatory and antioxidant activity of Mucuna macrocarpa beans: A future drug for Parkinson treatment. Asian Pac. J. Trop. Biomed. 2017, 7, 1097–1106. [Google Scholar] [CrossRef]
- Baranowska, I.; Płonka, J. Simultaneous Determination of Biogenic Amines and Methylxanthines in Foodstuff—Sample Preparation with HPLC-DAD-FL Analysis. Food Anal. Methods 2015, 8, 963–972. [Google Scholar] [CrossRef]
- Bulduk, İ.; Topal, N. Development and Validation of a Quantification Method for L-DOPA in Plants and Pharmaceutical Materials. Hacettepe J. Biol. Chem. 2020, 49, 1–10. [Google Scholar] [CrossRef]
- Dhanani, T.; Singh, R.; Shah, S.; Kumari, P.; Kumar, S. Comparison of green extraction methods with conventional extraction method for extract yield, L-DOPA concentration and antioxidant activity of Mucuna pruriens seed. Green Chem. Lett. Rev. 2015, 8, 43–48. [Google Scholar] [CrossRef]
- Duan, S.; Kwon, S.J.; Lim, Y.J.; Gil, C.S.; Jin, C.; Eom, S.H. L-3,4-dihydroxyphenylalanine accumulation in faba bean (Vicia faba L.) tissues during different growth stages. Agronomy 2021, 11, 502. [Google Scholar] [CrossRef]
- Etemadi, F.; Hashemi, M.; Randhir, R.; Zand Vakili, O.; Ebadi, A. Accumulation of L-DOPA in various organs of faba bean and influence of drought, nitrogen stress, and processing methods on L-DOPA yield. Crop J. 2018, 6, 426–434. [Google Scholar] [CrossRef]
- Goyoaga, C.; Burbano, C.; Cuadrado, C.; Varela, A.; Guillamón, E.; Pedrosa, M.M.; Muzquiz, M. Content and distribution of vicine, convicine and l-DOPA during germination and seedling growth of two Vicia faba L. varieties. Eur. Food Res. Technol. 2008, 227, 1537–1542. [Google Scholar] [CrossRef]
- Pavón-Pérez, J.; Oviedo, C.A.; Elso-Freudenberg, M.; Henríquez-Aedo, K.; Aranda, M. LC-MS/MS Method For L-Dopa Quantification in Different Tissues of Vicia faba. J. Chil. Chem. Soc. 2019, 64, 4–6. [Google Scholar] [CrossRef]
- Shetty, P.; Atallah, M.T.; Shetty, K. Effects of UV treatment on the proline-linked pentose phosphate pathway for phenolics and L-DOPA synthesis in dark germinated Vicia faba. Process Biochem. 2002, 37, 1285–1295. [Google Scholar] [CrossRef]
- Neyra Recky, J.R.; Serrano, M.P.; Dántola, M.L.; Lorente, C. Oxidation of tyrosine: Antioxidant mechanism of L-DOPA disclosed. Free Radic. Biol. Med. 2021, 165, 360–367. [Google Scholar] [CrossRef]
- Pulikkalpura, H.; Kurup, R.; Mathew, P.J.; Baby, S. Levodopa in Mucuna pruriens and its degradation. Sci. Rep. 2015, 5, 2–10. [Google Scholar] [CrossRef]
- Omotani, H.; Yasuda, M.; Ishii, R.; Ikarashi, T.; Fukuuchi, T.; Yamaoka, N.; Mawatari, K.I.; Kaneko, K.; Nakagomi, K. Analysis of l-DOPA-derived melanin and a novel degradation product formed under alkaline conditions. J. Pharm. Biomed. Anal. 2016, 125, 22–26. [Google Scholar] [CrossRef]
- Andrade, Â.L.; Cardoso, T.D.; Thomasi, S.S.; Alvarenga, M.E.; da Silva, M.A.N.; Magalhães, E.J.; Duarte, H.A.; de Almeida, K.J. A simple and efficient method for simultaneous quantification of levodopa and carbidopa based on controlled oxidation process. Chem. Pap. 2021, 75, 3091–3102. [Google Scholar] [CrossRef]
- Gurumoorthi, P.; Janardhanan, K.; Myhrman, R.V. Effect of differential processing methods on L-Dopa and protein quality in velvet bean, an underutilized pulse. LWT 2008, 41, 588–596. [Google Scholar] [CrossRef]
- Zhou, Y.Z.; Alany, R.G.; Chuang, V.; Wen, J. Studies of the rate constant of L-DOPA oxidation and decarboxylation by HPLC. Chromatographia 2012, 75, 597–606. [Google Scholar] [CrossRef]
- Kostrzewa, R.M.; Kostrzewa, J.P.; Brus, R. Neuroprotective and neurotoxic roles of levodopa (L-DOPA) in neurodegenerative disorders relating to Parkinson’s disease. Amino Acids 2002, 23, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Hörmann, P.; Delcambre, S.; Hanke, J.; Geffers, R.; Leist, M.; Hiller, K. Impairment of neuronal mitochondrial function by l-DOPA in the absence of oxygen-dependent auto-oxidation and oxidative cell damage. Cell Death Discov. 2021, 7, 151. [Google Scholar] [CrossRef] [PubMed]
- Tahvanainen, T.; Haraguchi, A. Effect of pH on phenol oxidase activity on decaying Sphagnum mosses. Eur. J. Soil Biol. 2013, 54, 41–47. [Google Scholar] [CrossRef]
- Long, W.J.; Brooks, A.E.; Biazzo, W. Analysis of Polar Compounds Using 100% Aqueous Mobile Phases with Agilent ZORBAX Eclipse Plus Phenyl-Hexyl and Other ZORBAX Phenyl Columns. Agil. Technol. Publ. 2009, 1–8. [Google Scholar]
- Cai, B.; Li, J. Evaluation of trifluoroacetic acid as an ion-pair Analysis of Polar Compounds Using 100% Aqueous Mobile Phases with Agilent ZORBAX Eclipse Plus Phenyl-Hexyl and Other ZORBAX Phenyl Columnsreagent in the separation of small ionizable molecules by reversed-phase liquid chromatography. Anal. Chim. Acta 1999, 399, 249–258. [Google Scholar] [CrossRef]
- Bidlingmeyer, B.A.; Broske, A.D. The Role of Pore Size and Stationary Phase Composition in Preventing Aqueous-Induced Retention Time Loss in Reversed-Phase HPLC. J. Chromatogr. Sci. 2004, 42, 100–106. [Google Scholar] [CrossRef]
- Buszewski, B.; Noga, S. Hydrophilic interaction liquid chromatography (HILIC)-a powerful separation technique. Anal. Bioanal. Chem. 2012, 402, 231–247. [Google Scholar] [CrossRef]
- Thiébaut, D.; Vial, J.; Michel, M.; Hennion, M.C.; Greibrokk, T. Evaluation of reversed phase columns designed for polar compounds and porous graphitic carbon in “trapping” and separating neurotransmitters. J. Chromatogr. A 2006, 1122, 97–104. [Google Scholar] [CrossRef]
- Arnetoli, M.; Montegrossi, G.; Buccianti, A.; Gonnelli, C. Determination of organic acids in plants of Silene paradoxa L. by HPLC. J. Agric. Food Chem. 2008, 56, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, B.; Örnemark, U. Eurachem Guide: The Fitness for Purpose of Analytical Methods—A Laboratory Guide to Method Validation and Related Topics, 2nd ed.; Örnemark, U., Ed.; Eurachem: Gembloux, Belgium, 2014; ISBN 0-94948926-12-0. [Google Scholar]
- Pascale, R.; Bianco, G.; Coviello, D.; Cristina Lafiosca, M.; Masi, S.; Mancini, I.M.; Bufo, S.A.; Scrano, L.; Caniani, D. Validation of a liquid chromatography coupled with tandem mass spectrometry method for the determination of drugs in wastewater using a three-phase solvent system. J. Sep. Sci. 2020, 43, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Pascale, R.; Bianco, G.; Calace, S.; Masi, S.; Mancini, I.M.; Mazzone, G.; Caniani, D. Method development and optimization for the determination of benzene, toluene, ethylbenzene and xylenes in water at trace levels by static headspace extraction coupled to gas chromatography–barrier ionization discharge detection. J. Chromatogr. A 2018, 1548, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Snyder, L.R.; Kirkland, J.J.; Glajchand, J.L. Practical HPLC Method Development, 2nd ed.; John Wiley & Sons: New York, NY, USA, 1997. [Google Scholar]
- Saranya, G.; Jiby, M.V.; Jayakumar, K.S.; Pillai, P.P.; Jayabaskaran, C. L-DOPA synthesis in Mucuna pruriens (L.) DC. is regulated by polyphenol oxidase and not CYP 450/tyrosine hydroxylase: An analysis of metabolic pathway using biochemical and molecular markers. Phytochemistry 2020, 178, 112467. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, H.L.; Beyer, N.; Janssen, D.B.; Fraaije, M.W. Lyophilization conditions for the storage of monooxygenases. J. Biotechnol. 2015, 203, 41–44. [Google Scholar] [CrossRef]
- Sharma, A.; Khamar, D.; Cullen, S.; Hayden, A.; Hughes, H. Innovative Drying Technologies for Biopharmaceuticals. Int. J. Pharm. 2021, 609, 121115. [Google Scholar] [CrossRef]
- Coviello, D.; Pascale, R.; Ciriello, R.; Salvi, A.M.; Guerrieri, A.; Contursi, M.; Scrano, L.; Bufo, S.A.; Cataldi, T.R.I.; Bianco, G. Validation of an analytical method for nitrite and nitrate determination in meat foods for infants by ion chromatography with conductivity detection. Foods 2020, 9, 1238. [Google Scholar] [CrossRef]
- Araujo, P. Key aspects of analytical method validation and linearity evaluation. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 2224–2234. [Google Scholar] [CrossRef]
- Guideline, ICH Harmonised tripartite. Validation of analytical procedures: Text and methodology Q2(R1). Guidance 2005, 1, 5. [Google Scholar]
- Ellison, S.L.R.; Williams, A. (Eds.) Eurachem/CITAC Guide: Quantifying Uncertainty in Analytical Measurement, 3rd ed.; Eurachem/CITAC: Teddington, UK, 2012. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Calibration Curve | Precision | Chromatographic Parameters | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Linear Range (t calc.) a | Linear Equation b, R2 | LoD (mg/L) LoQ (mg/L) | Level (mg/L) | Rep. RDS% (r calc.) c | Int. Precision RDS% (r calc.) d | Recovery, Rm ± u(Rm) | U (mg/L) | Rs e | T e |
| 0.5–50 mg/L (99.98) | y = (13.99 ± 0.15) × + (6.35 ± 3.52) R2 = 0.999 | 0.0414 0.0452 | 0.5 15 50 | 1.41 (0.36) 0.16 (1.31) 0.11 (2.73) | 6.87 (1.44) 2.57 (17.80) 2.20 (48.38) | 1.01 ± 0.02 1.171 ± 0.002 1.033 ± 0.004 | 0.24 0.93 2.93 | 1.64 ± 0.03 1.65 ± 0.02 1.67 ± 0.03 | 1.14 ± 0.02 1.13 ± 0.01 1.13 ± 0.01 |
| Conservation Type | LD Concentration (mg/g dw) |
|---|---|
| Vicia faba L. fresh samples | 1.21 ± 0.17 (a,b) |
| Vicia faba L. dried in the sun for 10 days | 1.26 ± 0.15 (c,b) |
| Vicia faba L. dried in the sun for 30 days | 0.81 ± 0.11 (f,g) |
| Vicia faba L. freeze-dried | 0.76 ± 0.11 (h,g) |
| Vicia faba L. frozen for 10 days | 1.03 ± 0.14 (d) |
| Vicia faba L. frozen for 30 days | 0.51 ± 0.08 (e) |
| Vicia faba L. commercial long-life frozen | <LOQ |
| Column | Column Length (cm) | Column ID (mm) | Particle Size (µm) | Pore Size (Å) | Surface Area (m2/g) | Carbon Content (%) | pH Range | Matrix Functional Group | Supplier |
|---|---|---|---|---|---|---|---|---|---|
| Discovery® C18 HPLC Column | 15 cm | 2.1 mm | 5 μm | 180 Å | 300 m2/g | 12% | 2–8 | C18 (octadecyl) phase endcapping | Supelco |
| Discovery® C18 HPLC Column | 25 cm | 4.6 mm | 5 μm | 180 Å | 300 m2/g | 12% | 2–8 | C18 (octadecyl) phase endcapping | Supelco |
| Kinetex Core-Shell Column | 10 cm | 4.6 mm | 2.6 μm | 100 Å | 200 m2/g | 12% | 1.5–10 | C18 with TMS endcapping | Phenomenex |
| Eclipse XDB-C18 Column | 25 cm | 4.6 mm | 5 μm | 80 Å | 180 m2/g | 10% | 2–9 | dimethyl-n-octadecylsilanes double endcapping | Agilent |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tesoro, C.; Ciriello, R.; Lelario, F.; Di Capua, A.; Pascale, R.; Bianco, G.; Dell’Agli, M.; Piazza, S.; Guerrieri, A.; Scrano, L.; et al. Development and Validation of a Reversed-Phase HPLC Method with UV Detection for the Determination of L-Dopa in Vicia faba L. Broad Beans. Molecules 2022, 27, 7468. https://doi.org/10.3390/molecules27217468
Tesoro C, Ciriello R, Lelario F, Di Capua A, Pascale R, Bianco G, Dell’Agli M, Piazza S, Guerrieri A, Scrano L, et al. Development and Validation of a Reversed-Phase HPLC Method with UV Detection for the Determination of L-Dopa in Vicia faba L. Broad Beans. Molecules. 2022; 27(21):7468. https://doi.org/10.3390/molecules27217468
Chicago/Turabian StyleTesoro, Carmen, Rosanna Ciriello, Filomena Lelario, Angela Di Capua, Raffaella Pascale, Giuliana Bianco, Mario Dell’Agli, Stefano Piazza, Antonio Guerrieri, Laura Scrano, and et al. 2022. "Development and Validation of a Reversed-Phase HPLC Method with UV Detection for the Determination of L-Dopa in Vicia faba L. Broad Beans" Molecules 27, no. 21: 7468. https://doi.org/10.3390/molecules27217468
APA StyleTesoro, C., Ciriello, R., Lelario, F., Di Capua, A., Pascale, R., Bianco, G., Dell’Agli, M., Piazza, S., Guerrieri, A., Scrano, L., Bufo, S. A., & Acquavia, M. A. (2022). Development and Validation of a Reversed-Phase HPLC Method with UV Detection for the Determination of L-Dopa in Vicia faba L. Broad Beans. Molecules, 27(21), 7468. https://doi.org/10.3390/molecules27217468