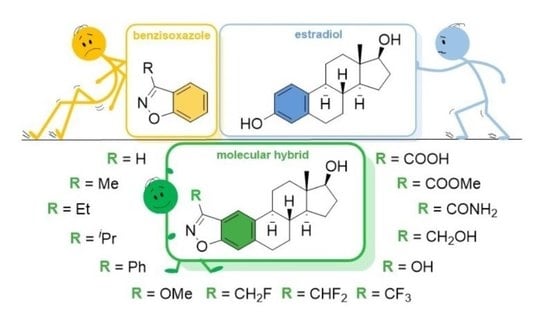
3.2. Chemistry
3.2.1. 3-Methoxyestra-1,3,5(10)-triene-17β-acetate (1)
E2Me (2.86 g, 10.0 mmol) was suspended in Ac2O (10 mL), and after adding a catalytic amount of H2SO4 (1 drop), the mixture was sonicated for 5 min. The resulting homogenous solution was poured into ice-cold water (100 mL), and the precipitate was filtered off, washed with water and dried. The crude product was purified by CC (EtOAc/hexane = 5:95). Recrystallized from MeOH/H2O. Yield (1): 3.12 g (95%, white crystals); Mp: 100–102 °C; Anal. Calcd. for C21H28O3 (328.45) C 76.79; H 8.59. Found C 76.48; H 8.77. 1H NMR (500 MHz, CDCl3): δ 0.83 (3H, s, 18-CH3), 1.23–1.64 (7H, overlapping m), 1.70–1.80 (1H, m), 1.89 (2H, m), 2.06 (3H, s, Ac-CH3), 2.16–2.27 (2H, m), 2.25–2.33 (1H, m), 2.79–2.93 (2H, m), 3.78 (3H, s), 4.69 (1H, dd, J 7.8, 9.2, 17-αH), 6.63 (1H, d, J 2.8, 4-H), 6.71 (1H, dd, J 2.8, 8.6, 2-H), 7.20 (1H, dd, J 1.1, 8.6, 1-H); 13C NMR (125 MHz, CDCl3) δ 12.2 (18-CH3), 21.3 (Ac-CH3), 23.4 (CH2), 26.4 (CH2), 27.4 (CH2), 27.7 (CH2), 29.9 (CH2), 37.1 (CH2), 38.7 (8-CH), 43.1 (13-C), 44.0 (9-CH), 50.0 (14-CH), 55.3 (3-MeO), 82.9 (17-CH), 111.6 (2-CH), 114.0 (4-CH), 126.5 (1-CH), 132.7 (10-C), 138.0 (5-C), 157.6 (3-C), 171.4 (Ac-C=O); ESI-MS m/z 329.2 [M + H]+, 329.2 calcd. for [C21H29O3]+.
3.2.2. General Procedure for the Synthesis of Compounds 2c–e and 2g-17Ac
Step 1. To a solution of 1 (328 mg, 1.0 mmol) in DCM (10 mL), AlCl3 (800 mg, 6 equiv.) and acyl chloride (1.5 equiv.) were added at 0 °C. After 30 min, the ice bath was removed, the reaction mixture was allowed to warm to RT and stirred for another 4 h. The mixture was poured into diluted HCl (1 M, 20 mL), stirred for 10 min and extracted with EtOAc (3 × 20 mL). The combined organic phase was washed with water (30 mL) and brine (30 mL), dried over anhydrous Na2SO4 and then concentrated in vacuo. The crude product was purified by CC to afford the 17-Ac derivatives of 2c–e or 2g.
Step 2. Subsequent deacetylation was perfomed (for 2c-17Ac, 2d-17Ac and 2d-17Ac) by the addition of NaOH (3 equiv.) in MeOH/DCM = 1:9 (0.1 M) under stirring at RT for 1 h. After evaporating off the solvents under reduced pressure, HCl (1 M, 10 mL) was added to the residue and extracted with EtOAc (3 × 10 mL). The combined organic phase was washed with water (10 mL) and brine (10 mL), dried over anhydrous Na2SO4 and concentrated in vacuo.
2-Propionylestra-1,3,5(10)-triene-3-ol-17β-acetate (
2c-17Ac) and 2-propionylestra-1,3,5(10)-triene-3,17β-diol (
2c): According to
Section 3.2.2, propionyl chloride (130 µL, 138 mg) was used for Step 1. Eluent (CC): EtOAc/hexane = 10:90. Recrystallized from MeOH. Yield (
2c-17Ac): 240 mg (65%, white needles); Mp: 169–171 °C; Anal. Calcd. for C
23H
30O
4 (370.49) C 74.56; H 8.16. Found C 74.43; H 8.37.
1H NMR (500 MHz, CDCl
3):
δ 0.84 (3H, s, 18-CH
3), 1.23 (3H, t,
J 7.3, CH
3 of propionyl), 1.26–1.48 (5H, overlapping m), 1.45–1.55 (1H, m), 1.52–1.62 (1H, m), 1.70–1.79 (1H, m), 1.85–1.95 (2H, m), 2.07 (3H, s, Ac-CH
3), 2.14–2.26 (2H, m), 2.26–2.34 (1H, m), 2.78–2.92 (2H, m), 2.96–3.05 (2H, m, CH
2 of propionyl), 4.70 (1H, t,
J 8.5, 17-αH), 6.69 (1H, s, 4-H), 7.63 (1H, s, 1-H), 12.12 (1H, s, 3-OH);
13C NMR (125 MHz, CDCl
3)
δ 8.5 (CH
3 of propionyl), 12.2 (18-CH
3), 21.3 (Ac-CH
3), 23.4 (CH
2), 26.4 (CH
2), 27.0 (CH
2), 27.7 (CH
2), 30.0 (CH
2), 31.5 (CH
2), 36.9 (CH
2), 38.5 (8-CH), 43.0 (13-C), 43.6 (9-CH), 50.0 (14-CH), 82.7 (17-CH), 117.5 (2-C), 117.8 (4-CH), 126.5 (1-CH), 131.4 (C), 146.9 (C), 160.2 (3-C), 171.3 (Ac-C=O), 206.8 (C=O of propionyl); ESI-MS:
m/
z 371.2 [M + H]
+, 371.2 calcd. for [C
23H
31O
4]
+.
In Step 2, 2c-17Ac (160 mg, 0.43 mmol) and NaOH (52 mg, 1.30 mmol) were used. The crude product was recrystallized from MeOH. Yield (2c): 124 mg (87%, white crystals); Mp: 137–139 °C; Anal. Calcd. for C21H28O3 (328.45) C 76.79; H 8.59. Found C 76.50; H 8.82. 1H NMR (500 MHz, CDCl3): δ 0.80 (3H, s, 18-CH3), 1.15–1.27 (1H, m), 1.20–1.28 (3H, m, CH3 of propionyl), 1.27–1.62 (7H, overlapping m), 1.66–1.76 (1H, m), 1.85–1.93 (1H, m), 1.95–2.02 (1H, m), 2.08–2.21 (2H, m), 2.29–2.37 (1H, m), 2.85 (2H, m), 2.93–3.05 (2H, m), 3.74 (1H, q, J 7.7, 7.7, 7.7), 6.69 (1H, s, 4-H), 7.65 (1H, s, 1-H), 12.12 (1H, s, 3-OH); 13C NMR (125 MHz, CDCl3) δ 8.6 (CH3 of propionyl), 11.2 (18-CH3), 23.3 (CH2), 26.5 (CH2), 27.0 (CH2), 30.0 (CH2), 30.8 (CH2), 31.5 (CH2), 36.7 (CH2), 38.8 (8-CH), 43.3 (13-C), 43.7 (9-CH), 50.2 (14-CH), 82.0 (17-CH), 117.4 (2-C), 117.8 (4-CH), 126.5 (1-CH), 131.6 (C), 147.0 (C), 160.2 (3-C), 206.8 (C=O); ESI-MS: m/z 329.2 [M + H]+, 329.2 calcd. for [C21H29O3]+.
2-Isobutyrylestra-1,3,5(10)-triene-3-ol-17β-acetate (
2d-17Ac) and 2-isobutyrylestra-1,3,5(10)-triene-3,17β-diol (
2d): According to
Section 3.2.2, isobutyryl chloride (157 µL, 160 mg) was used for Step 1. Eluent (CC): EtOAc/hexane = 10:90. Recrystallized from MeOH. Yield (
2d-17Ac): 258 mg (67%, yellowish white scales); Mp: 167–169 °C; Anal. Calcd. for C
24H
32O
4 (384.52) C 74.97; H 8.39. Found C 74.72; H 8.61.
1H NMR (500 MHz, CDCl
3):
δ 0.84 (3H, s, 18-CH
3), 1.24 (6H, dd,
J 6.4, 6.4, 2 × CH
3 of isobutyryl), 1.26–1.31 (1H, m), 1.28–1.52 (5H, overlapping m), 1.49–1.62 (1H, m), 1.70–1.80 (1H, m), 1.85–1.95 (2H, m), 2.07 (3H, s, Ac-CH
3), 2.15–2.29 (2H, m), 2.26–2.34 (1H, m), 2.79–2.93 (2H, m), 3.59 (1H, m, CH of isobutyryl), 4.70 (1H, t,
J 8.5, 17-αH), 6.71 (1H, s, 4-H), 7.66 (1H, s, 1-H), 12.29 (1H, s, 3-OH);
13C NMR (125 MHz, CDCl
3)
δ 12.2 (18-CH
3), 19.5 (one of the CH
3 of isobutyryl), 19.6 (the other CH
3 of isobutyryl), 21.3 (Ac-CH
3), 23.4 (CH
2), 26.4 (CH
2), 27.0 (CH
2), 27.7 (CH
2), 30.0 (CH
2), 34.9 (CH), 36.9 (CH
2), 38.5 (CH), 43.0 (13-C), 43.6 (9-CH), 50.0 (14-CH), 82.7 (17-CH), 116.4 (2-C), 118.0 (4-CH), 126.5 (1-CH), 131.4 (C), 147.0 (C), 160.9 (3-C), 171.3 (Ac-C=O), 210.5 (C=O of isobutyryl); ESI-MS:
m/
z 385.2 [M + H]
+, 385.2 calcd. for [C
24H
33O
4]
+.
In Step 2, 2d-17Ac (220 mg, 0.57 mmol) and NaOH (68 mg, 1.70 mmol) were used. Yield (2d): 180 mg (92%, white powder); Mp: 130–132 °C; Anal. Calcd. for C22H30O3 (342.48) C 77.16; H 8.83. Found C 76.93; H 8.99. 1H NMR (500 MHz, CDCl3): δ 0.80 (3H, s, 18-CH3), 1.16–1.27 (1H, m), 1.24 (6H, dd, J 6.8, 6.8, 2 × CH3 of isobutyryl), 1.28–1.60 (7H, overlapping m), 1.67–1.77 (1H, m), 1.85–1.93 (1H, m), 1.95–2.03 (1H, m), 2.08–2.23 (2H, m), 2.29–2.38 (1H, m), 2.79–2.93 (2H, m), 3.60 (1H, m, CH of isobutyryl), 3.75 (1H, t, J 8.7, 17-αH), 6.71 (1H, s, 4-H), 7.68 (1H, s, 1-H), 12.29 (1H, s, 3-OH); 13C NMR (125 MHz, CDCl3) δ 11.2 (18-CH3), 19.5 (one CH3 of isobutyryl), 19.6 (the other CH3 of isobutyryl), 23.3 (CH2), 26.5 (CH2), 27.0 (CH2), 30.0 (CH2), 30.8 (CH2), 34.9 (CH), 36.7 (CH2), 38.8 (CH), 43.4 (13-C), 43.7 (9-CH), 50.2 (14-CH), 82.0 (17-CH), 116.4 (2-C), 118.0 (4-CH), 126.5 (1-CH), 131.5 (C), 147.1 (C), 160.9 (3-C), 210.5 (C=O); ESI-MS: m/z 343.2 [M + H]+, 343.2 calcd. for [C22H31O3]+.
2-Benzoylestra-1,3,5(10)-triene-3-ol-17β-acetate (
2e-17Ac) and 2-benzoylestra-1,3,5(10)-triene-3,17β-diol (
2e): According to
Section 3.2.2, benzoyl chloride (174 µL, 211 mg) was used for Step 1. Eluent (CC): Et
2O/hexane = 10:90. Yield (
2e-17Ac): 272 mg (65%, yellow powder); Mp: 148–150 °C; Anal. Calcd. for C
27H
30O
4 (418.53) C 77.48; H 7.23. Found C 77.40; H 7.57;
1H NMR (500 MHz, CDCl
3):
δ 0.83 (3H, s, 18-CH
3), 1.18–1.31 (1H, m), 1.28–1.36 (1H, m), 1.33–1.40 (1H, m), 1.37–1.44 (1H, m), 1.41–1.49 (1H, m), 1.50–1.61 (2H, m), 1.70–1.79 (1H, m), 1.79–1.85 (1H, m), 1.86–1.95 (1H, m), 1.98–2.06 (1H, s), 2.04 (3H, s, Ac-CH
3), 2.10–2.27 (2H, m), 2.83–2.98 (2H, m), 4.64–4.71 (1H, m, 17-αH), 6.79 (1H, s, 4-H), 7.48 (1H, s, 4-H), 7.46–7.53 (2H, overlapping m, 3′-H and 5′-H), 7.55–7.62 (1H, m, 4′-H), 7.64–7.70 (2H, m, 2′-H and 6′-H), 11.82 (1H, s, 3-OH);
13C NMR (125 MHz, CDCl
3)
δ 12.2 (18-CH
3), 21.2 (Ac-CH
3), 23.4 (CH
2), 26.2 (CH
2), 27.0 (CH
2), 27.7 (CH
2), 30.1 (CH
2), 36.9 (CH
2), 38.6 (8-CH), 43.1 (13-C), 43.6 (9-CH), 50.1 (14-CH), 82.7 (17-CH), 117.4 (2-C), 117.8 (4-CH), 128.4 (3′-CH and 5′-CH), 129.3 (2′-CH and 6′-CH), 130.6 (4′-CH), 131.3 (C), 131.9 (1-CH), 138.5 (C), 147.3 (C), 161.2 (3-C), 171.2 (Ac-C=O), 201.3 (Bz-C=O); ESI-MS:
m/
z 419.2 [M + H]
+, 419.2 calcd. for [C
27H
31O
4]
+.
In Step 2, 2e-17Ac (230 mg, 0.55 mmol) and NaOH (66 mg, 1.65 mmol) were used. Yield (2e): 186 mg (90%, white powder); Mp: 126–128 °C; Anal. Calcd. for C25H28O3 (376.50) C 79.76; H 7.50. Found C 79.56; H 7.84. 1H NMR (500 MHz, CDCl3): δ 0.77 (3H, s, 18-CH3), 1.12–1.54 (8H, overlapping m), 1.65–1.75 (1H, m), 1.84–1.94 (2H, m), 2.00–2.08 (1H, m), 2.05–2.17 (2H, m), 2.79–2.98 (2H, m), 3.66–3.75 (1H, m, 17-αH), 6.79 (1H, s, 4-H), 7.49 (1H, s, 1-H), 7.48–7.55 (2H, m, 3′-H and 5′-H), 7.55–7.62 (1H, m, 4′-H), 7.64–7.70 (2H, m, 2′-H and 6′-H), 11.85 (1H, s, 3-OH); 13C NMR (125 MHz, CDCl3) δ 11.2 (18-CH3), 23.2 (CH2), 26.2 (CH2), 27.0 (CH2), 30.2 (CH2), 30.7 (CH2), 36.6 (CH2), 38.8 (8-CH), 43.3 (13-C), 43.7 (9-CH), 50.2 (14-CH), 81.9 (17-CH), 117.3 (2-C), 117.8 (4-CH), 128.5 (2′-CH and 6′-CH), 129.2 (3′-CH and 5′-CH), 130.6 (CH), 131.4 (C), 131.9 (CH), 138.4 (C), 147.4 (C), 161.1 (3-C), 201.4 (C=O); ESI-MS: m/z 377.2 [M + H]+, 377.2 calcd. for [C25H29O3]+.
2-(Methyloxoacetyl)-estra-1,3,5(10)-triene-3-ol-17β-acetate (
2g-17Ac): The synthesis was carried out according to
Section 3.2.2, but on a larger scale using E2Me (1.64 g, 5 mmol) and methyl chlorooxoacetate (690 µL, 920 mg) for Step 1. Eluent (CC): EtOAc/hexane = 20:80. Yield (
2g-17Ac): 1.56 g (78%, yellow crystals); Mp: 134–136 °C; Anal. Calcd. for C
23H
28O
6 (400.47) C 68.98; H 7.05. Found C 68.75; H 7.22.
1H NMR (500 MHz, CDCl
3):
δ 0.83 (3H, s, 18-CH
3), 1.22–1.61 (7H, overlapping m), 1.70–1.79 (1H, m), 1.86–1.95 (2H, m), 2.06 (3H, s, Ac-CH
3), 2.12–2.28 (3H, m), 2.81–2.97 (2H, m), 3.99 (3H, s, COOC
H3), 4.65–4.72 (1H, m, 17-αH), 6.75 (1H, s, 4-H), 7.58 (1H, s, 1-H), 10.98 (1H, s, 3-OH);
13C NMR (125 MHz, CDCl
3)
δ 12.2 (18-CH
3), 21.3 (Ac-CH
3), 23.4 (CH
2), 26.1 (CH
2), 26.8 (CH
2), 27.7 (CH
2), 30.3 (CH
2), 36.7 (CH
2), 38.3 (8-CH), 43.0 (13-C), 43.4 (9-CH), 50.0 (14-CH), 53.1 (COO
CH
3), 82.7 (17-CH), 114.4 (2-C), 117.9 (4-CH), 128.9 (1-CH), 132.7 (C), 150.1 (C), 161.7 (3-C), 163.2 (
COOCH
3), 171.3 (Ac-C=O), 189.8 (C=O of ketone); ESI-MS:
m/
z 399.0 [M − H]
−, 399.2 calcd. for [C
23H
27O
6]
−.
2g-17Ac was not deacetylated in Step 2.
3.2.3. 2-Trifluoracetylestra-1,3,5(10)-triene-3,17β-diol (2f)
To a solution of E2 (272 mg, 1.0 mmol) in DCM (10 mL), AlCl3 (800 mg, 6 equiv.) and TFAA (1.5 mmol, 1.5 equiv.) were added and the mixture was stirred at 0 °C for 4 h. After completion, it was poured into HCl solution (1 M, 20 mL), stirred for 10 min and extracted with EtOAc (3 × 20 mL). The combined organic phase was washed with water (30 mL) and brine (30 mL), then dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by CC (DCM). Recrystallized from dioxane/H2O. Yield (2f): 302 mg (82%, yellow crystals); Mp: 104–106 °C; Anal. Calcd. for C20H23F3O3 (368.40) C 65.21; H 6.29. Found C 65.12; H 6.48. 1H NMR (500 MHz, CDCl3): δ 0.79 (3H, s, 18-CH3), 1.15–1.24 (1H, m), 1.24–1.33 (1H, m), 1.30–1.48 (4H, m), 1.45–1.60 (2H, m), 1.66–1.76 (1H, m), 1.87–1.95 (1H, m), 1.96–2.05 (1H, m), 2.08–2.25 (2H, m), 2.25–2.34 (1H, m), 2.82–2.98 (2H, m), 3.71–3.78 (1H, t, J 8.5, 17-αH), 6.79 (1H, s, 4-CH), 7.69 (1H, s, 1-H), 10.86 (1H, s, 3-OH); 13C NMR (125 MHz, CDCl3) δ 11.2 (18-CH3), 23.2 (CH2), 26.1 (CH2), 26.7 (CH2), 30.4 (CH2), 30.7 (CH2), 36.5 (CH2), 38.5 (8-CH), 43.3 (13-C), 43.6 (9-CH), 50.2 (14-CH), 81.9 (17-CH), 112.2 (2-C), 116.8 (1C, q, J 290.2, CF3), 118.3 (4-CH), 127.2 (1C, q, J 3.9, 1-CH), 133.2 (C), 151.1 (C), 162.4 (C), 183.9 (1C, q, J 34.8, C=O); ESI-MS: m/z 367.0 [M − H]−, 367.2 calcd. for [C20H22F3O3]−.
3.2.4. General Procedure for the Synthesis of Oximes 3a–g
To a solution of 2-substituted estradiol derivative (2a–g, 1.0 mmol) in EtOH (10 mL), hydroxylamine hydrochloride and a base were added in excess and the mixture was stirred at ambient temperature for a certain period. The solvent was removed under reduced pressure, and the residue was suspended in water (10 mL) and extracted with EtOAc (3 × 10 mL). The combined organic phase was washed with NH4Cl (1 M, 10 mL), water (10 mL) and brine (10 mL), and then dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by CC.
Estra-1,3,5(10)-triene-3,17β-diol-2-carbaldehyde oxime (
3a): According to
Section 3.2.4, 2-formyl-estradiol (
2a, 300 mg), hydroxylamine hydrochloride (104 mg, 1.5 equiv.) and sodium acetate (164 mg, 2 equiv.) were used. Conditions: RT, 1 h. CC (EtOAc/DCM = 5:95). Yield (
3a): 309 mg (98%, white powder); Mp > 180 °C (decomp.); Anal. Calcd. for C
19H
25NO
3 (315.41) C 72.35; H 7.99; N 4.44. Found C 72.30; H 8.17; N 4.33.
1H NMR (500 MHz, DMSO-
d6):
δ 0.67 (3H, s, 18-CH
3), 1.05–1.44 (7H, overlapping m), 1.53–1.63 (1H, m), 1.74–1.82 (1H, m), 1.82–1.94 (2H, m), 2.03–2.12 (1H, m), 2.20–2.28 (1H, m), 2.71–2.77 (2H, m), 3.53 (1H, td,
J 8.5, 3.9, 17-αH), 4.41 (1H, d,
J 4.7, 17-OH), 6.56 (1H, s, 4-H), 7.34 (1H, s, 1-H), 8.27 (1H, s, HC=N), 9.75 (1H, s, NOH), 11.11 (1H, s, 3-OH);
13C NMR (125 MHz, DMSO-
d6):
δ 11.1 (18-CH
3), 22.6 (CH
2), 25.9 (CH
2), 26.6 (CH
2), 28.9 (CH
2), 29.8 (CH
2), 36.4 (CH
2), 38.4 (8-CH), 42.7 (13-C), 43.2 (9-CH), 49.5 (14-CH), 79.9 (17-CH), 115.4 (4-CH), 115.5 (2-C), 125.0 (1-CH), 131.3 (10-C), 139.2 (5-C), 148.6 (HC=N), 153.7 (3-C); ESI-MS:
m/
z 314.0 [M − H]
−, 314.2 calcd. for [C
19H
24NO
3]
−.
(Estra-1,3,5(10)-triene-3,17β-diol-2-yl)ethan-1-one oxime (
3b): According to
Section 3.2.4, 2-acetyl-estradiol (
2b, 314 mg, 1.0 mmol), hydroxylamine hydrochloride (104 mg, 1.5 equiv.) and sodium acetate (164 mg, 2 equiv.) were used. Conditions: reflux, 2 h. CC (EtOAc/DCM = 5:95). Yield: 309 mg (94%, white crystals); Mp: 240–242 °C; Anal. Calcd. for C
20H
27NO
3 (C
20H
27NO
3) C 72.92; H 8.26; N 4.25. Found C 72.86; H 8.58; N 4.03.
1H NMR (500 MHz, CDCl
3):
δ 0.79 (3H, s, 18-CH
3), 1.15–1.58 (8H, overlapping m), 1.66–1.76 (1H, m), 1.84–1.93 (1H, m), 1.93–2.06 (1H, m), 2.07–2.25 (2H, m), 2.29–2.37 (1H, m), 2.32–2.36 (3H, s, CH
3 of acetoxime), 2.80–2.87 (2H, m), 3.74 (1H, t,
J 8.5, 17-αH), 6.68 (1H, s, 4-H), 7.32 (1H, s, 1-H), 7.65 (1H, s, NOH), 10.94 (1H, s, 3-OH);
13C NMR (125 MHz, CDCl
3)
δ 10.9 (CH
3), 11.2 (CH
3), 23.3 (CH
2), 26.6 (CH
2), 27.3 (CH
2), 29.5 (CH
2), 30.8 (CH
2), 36.8 (CH
2), 39.0 (8-CH), 43.4 (13-C), 44.0 (9-CH), 50.2 (14-CH), 82.1 (17-CH), 116.4 (2-C), 117.0 (4-CH), 124.5 (1-CH), 131.2 (10-C), 140.3 (5-C), 155.5 (C), 159.8 (C); ESI-MS:
m/
z 328.0 [M − H]
−, 328.2 calcd. for [C
20H
26NO
3]
−.
(Estra-1,3,5(10)-triene-3,17β-diol-2-yl)propan-1-one oxime (
3c): According to
Section 3.2.4, 2-propanoyl-estradiol (
2c, 328 mg), hydroxylamine hydrochloride (104 mg, 1.5 equiv.) and sodium acetate (164 mg, 2 equiv.) were used. Conditions: reflux, overnight. CC (MeOH/DCM = 2:98). Yield (
3c): 333 mg (97%, white crystals); Mp: 240–242 °C; Anal. Calcd. for C
21H
29NO
3 (343.47) C 73.44; H 8.51; N 4.08. Found C 73.10; H 8.84; N 3.97.
1H NMR (500 MHz, DMSO-
d6):
δ 0.67 (3H, s, 18-CH
3), 1.08 (3H, t,
J 7.5, CH
3 of propionyl oxime), 1.07–1.43 (7H, overlapping m), 1.54–1.63 (1H, m), 1.75–1.82 (1H, m), 1.83–1.94 (2H, m), 2.07–2.16 (1H, m), 2.28–2.35 (1H, m), 2.71–2.86 (4H, overlapping m, 6-H
2 and CH
2 of propionyl oxime), 3.53 (1H, td,
J 8.5, 4.8, 17-αH), 4.45–4.50 (1H, m, 17-OH), 6.55 (1H, s, 4-H), 7.27 (1H, s, 1-H), 11.27 (1H, s), 11.31 (1H, s);
13C NMR (125 MHz, DMSO-
d6):
δ 11.0 (CH
3), 11.2 (CH
3), 17.6 (CH
2), 22.7 (CH
2), 26.0 (CH
2), 26.7 (CH
2), 28.8 (CH
2), 29.9 (CH
2), 36.5 (CH
2), 38.5 (8-CH), 42.8 (13-C), 43.4 (9-CH), 49.5 (14-CH), 80.0 (17-CH), 115.5 (2-C), 116.2 (4-CH), 124.1 (1-CH), 130.6 (C), 138.8 (C), 155.1 (3-C), 162.2 (C=N); ESI-MS:
m/
z 344.2 [M + H]
+, 344.2 calcd. for [C
21H
30NO
3]
+.
(Estra-1,3,5(10)-triene-3,17β-diol-2-yl)-2-methylpropan-1-one oxime isomers: According to
Section 3.2.4, 2-isobutyryl-estradiol (
2d, 342 mg), hydroxylamine hydrochloride (347 mg, 5 equiv.) and pyridine (1 mL) were used. Conditions: reflux, overnight. CC (MeOH/DCM = 2:98). Yield of the faster eluting isomer (
3d-
E): 175 mg (49%, white crystals); Mp: 211–213 °C; Anal. Calcd. for C
22H
31NO
3 (357.49) C 73.92; H 8.74; N 3.92. Found C 73.80; H 9.10; N 3.86.
1H NMR (500 MHz, DMSO-
d6):
δ 0.66 (3H, s, 18-CH
3), 1.14 (6H, dd,
J 6.9, 6.9, 2 × CH
3 of isobutyryl oxime), 1.06–1.25 (2H, m), 1.24 (1H, s), 1.24–1.35 (3H, m), 1.35–1.43 (1H, m), 1.53–1.63 (1H, m), 1.74–1.81 (1H, m), 1.81–1.94 (2H, m), 2.04–2.13 (1H, m), 2.19–2.26 (1H, m), 2.66–2.75 (2H, m), 3.38 (1H, m, CH of isobutyryl oxime), 3.52 (1H, td,
J 8.5, 4.8, 17-αH), 4.47 (1H, d,
J 4.8, 17-OH), 6.51 (1H, s, 4-H), 7.03 (1H, s, 1-H), 10.00 (1H, s), 10.85 (1H, s);
13C NMR (125 MHz, DMSO-
d6):
δ 11.2 (18-CH
3), 18.7 (2 × CH
3 of isobutyryl oxime), 22.7 (CH
2), 26.1 (CH
2), 26.8 (CH
2), 27.3 (CH of isobutyryl oxime), 28.8 (CH
2), 29.9 (CH
2), 36.5 (CH
2), 38.6 (8-CH), 42.8 (13-C), 43.3 (9-CH), 49.5 (14-CH), 80.0 (17-CH), 115.6 (4-CH), 119.2 (2-C), 126.0 (1-CH), 130.0 (C), 137.6 (C), 153.8 (C), 162.9 (C); ESI-MS:
m/
z 358.2 [M + H]
+, 358.2 calcd. for [C
22H
32NO
3]
+.
Yield of the slower eluting isomer (3d-Z): 121 mg (34%, white crystals); Mp: 214–216 °C; Anal. Calcd. for C22H31NO3 (357.49) C 73.92; H 8.74; N 3.92. Found C 73.71; H 9.03; N 3.68. 1H NMR (500 MHz, DMSO-d6): δ 0.66 (3H, s, 18-CH3), 1.02 and 1.03 (6H, overlapping d, 2 × CH3 of isobutyryl oxime), 1.06–1.43 (7H, overlapping m), 1.53–1.63 (1H, m), 1.74–1.93 (3H, m), 2.03–2.12 (1H, m), 2.15–2.22 (1H, m), 2.65–2.76 (3H, m), 3.48–3.56 (1H, m, 17-αH), 4.45–4.49 (1H, d, J 4.8, 17-OH), 6.49 (1H, s, 4-H), 6.78 (1H, s, 1-H), 8.74 (1H, bs), 10.26 (1H, bs); 13C NMR (125 MHz, DMSO-d6): δ 11.2 (18-CH3), 20.2 (one CH3 of isobutyryl oxime), 20.2 (the other CH3 of isobutyryl oxime), 22.7 (CH2), 26.1 (CH2), 26.9 (CH2), 28.9 (CH2), 29.9 (CH2), 33.4 (CH of isobutyryl oxime), 36.5 (CH2), 38.6 (8-CH), 42.8 (13-C), 43.4 (9-CH), 49.5 (14-CH), 80.0 (17-CH), 115.3 (4-CH), 120.0 (2-C), 125.5 (1-CH), 130.0 (C), 136.8 (C), 151.4 (C), 159.7 (C); ESI-MS: m/z 358.2 [M + H]+, 358.2 calcd. for [C22H32NO3]+.
(Estra-1,3,5(10)-triene-3,17β-diol-2-yl)(phenyl)methanone oxime isomers (
3e): According to
Section 3.2.4, 2-benzoyl-estradiol (
2e, 377 mg), hydroxylamine hydrochloride (347 mg, 5 equiv.) and pyridine (1 mL) were used. Conditions: reflux, 48 h. CC (EtOAc/DCM = 2:98 to 5:95). Yield of the faster eluting isomer (
3e-
E): Yield: 172 mg (44%, white powder); Mp: 280–282 °C; Anal. Calcd. for C
25H
29NO
3 (391.51) C 76.70; H 7.47; N 3.58. Found C 76.38; H 7.73; N 3.49.
1H NMR (500 MHz, DMSO-
d6):
δ 0.61 (3H, s, 18-CH
3), 0.99–1.40 (7H, overlapping m), 1.50–1.60 (1H, m), 1.61–1.71 (2H, m), 1.72–1.80 (1H, m), 1.80–1.91 (1H, m), 1.92–2.01 (1H, m), 2.71–2.77 (2H, m), 3.42–3.50 (1H, td,
J 8.5, 4.8, 17-αH), 4.41–4.46 (1H, d,
J 4.9, 17-OH), 6.62 (1H, s), 6.67 (1H, s), 7.25–7.31 (2H, m), 7.42–7.53 (3H, m), 11.00 (1H, s), 11.40 (1H, s);
13C NMR (125 MHz, DMSO-
d6):
δ 11.1 (18-CH
3), 22.7 (CH
2), 25.7 (CH
2), 26.6 (CH
2), 28.8 (CH
2), 29.9 (CH
2), 36.2 (CH
2), 38.4 (8-CH), 42.7 (13-C), 43.1 (9-CH), 49.4 (14-CH), 79.9 (17-CH), 116.1 (4-CH), 117.0 (2-C), 126.5 (1-CH), 128.2 (2′-CH and 6′-CH), 128.4 (3′-CH and 5′-CH), 128.6 (4′-CH), 130.4 (C), 132.1 (C), 139.1 (C), 155.0 (C), 159.1 (C); ESI-MS:
m/
z 390.1 [M − H]
−, 390.2 calcd. for [C
25H
28NO
3]
−.
Yield of the slower eluting isomer (3e-Z): 141 mg (36%, white powder); Mp: 277–279 °C; Anal. Calcd. for C25H29NO3 (391.51) C 76.70; H 7.47; N 3.58. Found C 76.47; H 7.69; N 3.20. 1H NMR (500 MHz, DMSO-d6): δ 0.67 (3H, s, 18-CH3), 1.02–1.43 (7H, overlapping m), 1.54–1.64 (1H, m), 1.75–1.84 (2H, m), 1.83–1.94 (1H, m), 2.06–2.18 (2H, m), 2.74–2.81 (2H, m), 3.51 (1H, td, J 8.5, 4.8, 17-αH), 4.46 (1H, d, J 4.8, 17-OH), 6.57 (1H, s), 6.83 (1H, s), 7.29–7.33 (3H, m), 7.37–7.42 (2H, m), 8.91 (1H, s), 11.04 (1H, s); 13C NMR (125 MHz, DMSO-d6): δ 11.2 (18-CH3), 22.7 (CH2), 26.1 (CH2), 26.9 (CH2), 29.0 (CH2), 29.9 (CH2), 36.5 (CH2), 38.6 (8-CH), 42.8 (13-C), 43.4 (9-CH), 49.5 (14-CH), 80.0 (17-CH), 115.4 (4-CH), 118.6 (2-CH), 126.2 (1-CH), 126.3 (2′-CH and 6′-CH), 128.1 (3′-CH and 5′-CH), 128.3 (4′-CH), 130.3 (C), 136.8 (C), 137.4 (C), 151.9 (C), 153.9 (C); ESI-MS: m/z 390.1 [M − H]−, 390.2 calcd. for [C25H28NO3]−.
(Estra-1,3,5(10)-triene-3,17β-diol-2-yl)-2,2,2-trifluoroethan-1-one oxime isomers (
3f): According to
Section 3.2.4, 2-trifluoracetyl-estradiol (
2f, 356 mg), hydroxylamine hydrochloride (695 mg, 10 equiv.) and pyridine (1.5 mL) were used. Conditions: reflux, overnight. CC (EtOAc/hexane = 20:80) yielded an inseparable mixture of
Z- and
E-isomers (1:2). Yield (
3f): 364 mg (95%, white powder); Mp: 180–182 °C; Anal. Calcd. for C
20H
24F
3NO
3 (383.41) C 62.65; H 6.32; N 3.29. Found C 62.60; H 6.57; N 3.04.
1H NMR (500 MHz, DMSO-
d6):
δ 0.66 (3H, s, 18-CH
3), 1.05–1.43 (7H, overlapping m), 1.53–1.63 (1H, m), 1.75–1.94 (3H, m), 2.04–2.13 (1H, td,
J 11.2, 3.9), 2.14–2.25 (1H, m), 2.68–2.81 (2H, dd,
J 10.0, 5.9), 3.48–3.58 (1H, td,
J 8.5, 4.7, 17-αH), 4.45–4.49 (1H, d,
J 4.8, 17-OH), 6.56 and 6.58 (1H, s, 4-H of
E- and
Z-isomers), 6.88 and 6.98 (1H, s, 1-H of
E- and
Z-isomers), 9.45 and 9.61 (1H, bs and s, 3-OH of
E- and
Z-isomers), 12.21 and 12.53 (1H, bs and s, NOH of
E- and
Z-isomers);
13C NMR (125 MHz, DMSO-
d6):
δ 11.2 (18-CH
3), 22.7 (CH
2), 26.0 (CH
2), 26.7 (CH
2), 29.0 (6-CH
2 of
Z-isomer), 29.0 (6-CH
2 of
E-isomer), 29.9 (CH
2), 36.5 (12-CH
2 of
E-isomer), 36.5 (12-CH
2 of
Z-isomer) 38.4 (8-CH), 42.8 (13-C), 43.2 (9-CH of
Z-isomer), 43.3 (9-CH of
E-isomer), 49.5 (14-CH), 80.0 (17-CH), 112.3 (2-C), 115.1 (1-CH of
Z-isomer), 115.4 (4-CH of
E-isomer), 116.0 (2-C), 118.2. (1C, q,
J 282.2, CF
3 of
Z-isomer), 121.2 (1C, q,
J 273.7, CF
3 of
E-isomer), 125.8 (1-CH of
E-isomer), 127.4 (1-CH of
Z-isomer), 130.6 (10-C of
E-isomer), 130.7 (10-C of
Z-isomer, 139.5 (5-C of
E-isomer), 139.8 (5-C of
Z-isomer), 144.5 (1C, q,
J 32.1, C=N of
E-isomer), 145.1 (1C, q,
J 30.3, C=N of
Z-isomer), 152.7 (3-C of
E-isomer), 153.9 (3-C of
Z-isomer); ESI-MS:
m/
z 382.0 [M − H]
−, 382.2 calcd. for [C
20H
23F
3NO
3]
−.
Methyl-2-(estra-1,3,5(10)-triene-3-hydroxy-17β-acetoxy-2-yl)-2-hydroxyimino acetate (
3g): According to
Section 3.2.4, 2-(methyloxoacetyl)estradiol-17β-acetate (
2g-17Ac, 400 mg), hydroxylamine hydrochloride (104 mg, 1.5 equiv.) and pyridine (1 mL) were used. The synthesis was repeated in duplicate. Conditions: RT, overnight. CC (EtOAc/hexane = 20:80). Yield (
3g): 378 mg (91%, white powder, as a single isomer); Mp: 188–189 °C. Anal. Calcd. for C
23H
29NO
6 (415.49) C 66.49; H 7.04; N 3.37. Found C 66.35; H 7.42; N 3.24.
1H NMR (500 MHz, DMSO-
d6):
δ 0.77 (3H, s, 17-CH
3), 1.22–1.41 (6H, overlapping m), 1.44–1.55 (1H, m), 1.63–1.72 (1H, m), 1.74–1.84 (2H, m), 2.01 (3H, s, Ac-CH
3), 2.04–2.20 (3H, m), 2.71–2.78 (2H, m), 3.77 (3H, s, COOC
H3), 4.57–4.64 (1H, t,
J 8.5, 17-αH), 6.56 (1H, s, 4-H), 7.22 (1H, s, 1-H), 9.74 (1H, s, 3-OH), 11.72 (1H, s, NOH);
13C NMR (125 MHz, DMSO-
d6):
δ 11.8 (18-CH
3), 20.8 (Ac-CH
3), 22.7 (CH
2), 25.7 (CH
2), 26.5 (CH
2), 27.1 (CH
2), 28.8 (CH
2), 36.3 (CH
2), 37.9 (8-CH), 42.4 (13-C), 42.8 (9-CH), 49.0 (14-CH), 51.8 (COO
CH
3), 81.8 (17-CH), 114.7 (2-C), 116.0 (4-CH), 124.0 (1-CH), 131.1 (10-C), 140.0 (5-C), 149.6 (C=N), 153.3 (3-C), 163.9 (
COOCH
3), 170.3 (Ac-C=O); ESI-MS:
m/
z 414.1 [M − H]
−, 414.2 calcd. for [C
23H
28NO
6]
−.
3.2.5. General Procedure for the Synthesis of Compounds 4a–g
DDQ (1.5 equiv.) was added slowly to a solution of PPh3 (1.5 equiv.) in DCM (0.5 M), and stirred for 1 min. This suspension was added to a solution of oxime (3a–g, 1.0 equiv.) and Et3N (2.0 equiv.) in DCM (0.2 M), and stirred for 10 min at RT. The solvent was removed under reduced pressure. The residue was dissolved in EtOAc/MeOH, and Celite® was added (~10× weight of the crude sample), which was then concentrated in vacuo and purified by CC.
Isoxazolo[4′,5′:2,3]estra-1,3,5(10)-triene-17β-ol (
4a): According to
Section 3.2.5,
3a (158 mg, 0.50 mmol) was used for the reaction. Eluent (CC): EtOAc/hexane = 30:70. Yield (
4a): 134 mg (90%, white powder); Mp: 176–178 °C; Anal. Calcd. for C
19H
23NO
2 (297.40) C 76.74; H 7.80; N 4.71. Found C 76.55; H 7.96; N 4.33.
1H NMR (500 MHz, CDCl
3):
δ 0.80 (3H, s, 18-CH
3), 1.18–1.57 (7H, overlapping m), 1.58–1.68 (1H, m), 1.70–1.78 (1H, m), 1.89–2.08 (2H, m), 2.09–2.20 (1H, m), 2.27–2.35 (1H, m), 2.35–2.44 (1H, m), 3.00–3.06 (2H, m), 3.76 (1H, t,
J 8.5, 17-αH), 7.32 (1H, s, 4-H), 7.61 (1H, s, 1-H), 8.60 (1H, d,
J 1.1, 3′-H);
13C NMR (125 MHz, CDCl
3)
δ 11.2 (18-CH
3), 23.4 (CH
2), 26.5 (CH
2), 27.0 (CH
2), 30.4 (CH
2), 30.7 (CH
2), 36.7 (CH
2), 38.6 (8-CH), 43.3 (13-C), 44.2 (9-CH), 50.5 (14-CH), 81.9 (17-CH), 108.8 (4-CH), 117.8 (2-CH), 119.7 (1-C), 137.6 (10-C), 141.0 (5-C), 146.2 (3′-CH), 161.1 (3-C); ESI-MS:
m/
z 296.0 [M − H]
−, 296.2 calcd. for [C
19H
22NO
2]
−.
3′-Methylisoxazolo[4′,5′:2,3]estra-1,3,5(10)-triene-17β-ol (
4b): According to
Section 3.2.5,
3b (150 mg, 0.46 mmol) was used for the reaction. Eluent (CC): EtOAc/hexane = 30:70. Yield (
4b): 132 mg (93%, white crystals); Mp: 168–170 °C; Anal. Calcd. for C
20H
25NO
2 (311.42) C 77.14; H 8.09; N 4.50. Found C 77.10; H 8.34; N 4.26.
1H NMR (500 MHz, CDCl
3):
δ 0.80 (3H, s 18-CH
3), 1.19–1.32 (1H, m), 1.31–1.56 (6H, overlapping m), 1.59–1.79 (2H, m), 1.89–1.99 (1H, m), 2.02 (1H, m), 2.09–2.20 (1H, m), 2.27–2.36 (1H, m), 2.39–2.47 (1H, m), 2.54 (3H, s, 3′-CH
3), 2.99–3.05 (2H, m), 3.72–3.80 (1H, m, 17-αH), 7.24 (1H, s, 4-H), 7.50 (1H, s, 1-H);
13C NMR (125 MHz, CDCl
3)
δ 10.2 (CH
3), 11.2 (CH
3), 23.4 (CH
2), 26.6 (CH
2), 27.1 (CH
2), 30.4 (CH
2), 30.8 (CH
2), 36.7 (CH
2), 38.7 (8-CH), 43.3 (13-C), 44.2 (9-CH), 50.5 (14-CH), 82.0 (17-CH), 109.0 (4-CH), 116.9 (1-CH), 120.6 (2-C), 136.9 (10-C), 140.5 (5-C), 154.9 (C=N), 161.6 (3-C); ESI-MS:
m/
z 312.2 [M − H]
+, 312.2 calcd. for [C
20H
26NO
2]
+.
3′-Ethylisoxazolo[4′,5′:2,3]estra-1,3,5(10)-triene-17β-ol (
4c): According to
Section 3.2.5, 3
c (50 mg, 0.15 mmol) was used for the reaction. Eluent (CC): DCM. Yield (
4c): 43 mg (93%, white crystals); Mp: 166–168 °C; Anal. Calcd. for C
21H
27NO
2 (325.45) C 77.50; H 8.36; N 4.30. Found C 77.38; H 8.67; N 4.16.
1H NMR (500 MHz, CDCl
3):
δ 0.82 (3H, s, 18-CH
3), 1.21–1.32 (1H, m), 1.31–1.56 (6H, overlapping m), 1.45 (3H, t,
J 7.4, 7.4, CH
3 of ethyl), 1.60–1.73 (1H, m), 1.70–1.80 (1H, m), 1.90–1.98 (1H, m), 1.99–2.07 (1H, m), 2.10–2.21 (1H, m), 2.28–2.37 (1H, m), 2.39–2.48 (1H, m), 2.95–3.06 (4H, m), 3.73–3.81 (1H, m, 17-αH), 7.27 (1H, s, 4-CH), 7.54 (1H, s, 1-CH);
13C NMR (125 MHz, CDCl
3)
δ 11.2 (18-CH
3), 12.4 (CH
3 of ethyl), 19.1 (CH
2), 23.4 (CH
2), 26.6 (CH
2), 27.1 (CH
2), 30.4 (CH
2), 30.8 (CH
2), 36.7 (CH
2), 38.7 (8-CH), 43.4 (13-C), 44.3 (9-CH), 50.5 (14-CH), 82.0 (17-CH), 109.1 (4-CH), 117.0 (1-CH), 119.9 (C), 136.8 (C), 140.4 (C), 159.6 (C), 161.8 (C); ESI-MS:
m/
z 326.2 [M + H]
+, 326.2 calcd. for [C
21H
28NO
2]
+.
3′-Isopropylisoxazolo[4′,5′:2,3]estra-1,3,5(10)-triene-17β-ol (
4d): According to
Section 3.2.5,
3d (80 mg, 0.22 mmol, 1:1 mixture of
E and
Z-oximes) was used for the reaction. Eluent (CC): EtOAc/hexane = 40:60. Yield (
4d): 68 mg (91%, white crystals); Mp: 168–169 °C; Anal. Calcd. for C
22H
29NO
2 (339.48) C 77.84; H 8.61; N 4.13. Found C 77.58; H 8.74; N 4.02.
1H NMR (500 MHz, CDCl
3):
δ 0.81 (3H, s, 18-CH
3), 1.21–1.30 (1H, m), 1.31–1.47 (4H, overlapping m), 1.49 (6H, d,
J 7.1, 2 × CH
3 of
iPr), 1.47–1.56 (2H, m), 1.59–1.79 (2H, m), 1.90–1.97 (1H, m), 1.99–2.06 (1H, m), 2.10–2.21 (1H, m), 2.28–2.37 (1H, m), 2.39–2.46 (1H, m), 2.99–3.05 (2H, m), 3.38 (1H, m, CH of
iPr), 3.73–3.81 (1H, m, 17-αH), 7.27 (1H, s, 4-H), 7.58 (1H, s, 1-H);
13C NMR (125 MHz, CDCl
3)
δ 11.2 (18-CH
3), 21.3 (one CH
3 of
iPr), 21.4 (the other CH
3 of
iPr), 23.4 (CH
2), 26.6 (CH
2), 27.0 (CH of
iPr), 27.1 (CH
2), 30.4 (CH
2), 30.8 (CH
2), 36.7 (CH
2), 38.7 (8-CH), 43.4 (13-C), 44.3 (9-CH), 50.5 (14-CH), 82.0 (17-CH), 109.2 (4-CH), 117.4 (1-CH), 119.2 (2-C), 136.6 (C), 140.3 (C), 162.0 (C), 163.1 (C); ESI-MS:
m/
z 340.2 [M + H]
+, 340.2 calcd. for [C
22H
30NO
2]
+.
3′-Phenylisoxazolo[4′,5′:2,3]estra-1,3,5(10)-triene-17β-ol (
4e): Method A (
Scheme 1, iii)
: According to
Section 3.2.5,
3e (100 mg, 0.26 mmol, 1:1 mixture of
E and
Z-oximes) was used for the reaction. Eluent (CC): EtOAc/DCM = 2:98. Yield (
4e): 24 mg (25%, yellowish white solid); Method B (
Scheme 1, vii/viii):
2e (62 mg, 0.16 mmol) was dissolved in NH
3 (6 M in MeOH, 0.5 mL) and stirred for 2 h at RT. The solvent was then removed under reduced pressure, and the residue (
5e) was redissolved in THF (1 mL). NCS (33 mg, 1.5 equiv.) and K
2CO
3 (69 mg, 3.0 equiv.) were added to the solution, and the resulting suspension was stirred at RT overnight. After evaporating off the solvent, the residue was suspended in water (10 mL) and extracted with EtOAc (3 × 10 mL). The combined organic phase was washed with HCl (1M, 10 mL), water (10 mL) and brine (10 mL), dried over anhydrous Na
2SO
4 and concentrated in vacuo. The crude product was purified by CC (DCM). Yield (
4e): 37 mg (60%, as yellowish white solid); Mp: 196–198 °C; Anal. Calcd. for C
25H
27NO
2 (373.50) C 80.40; H 7.29; N 3.75. Found C 80.02; H 7.56; N 3.55.
1H NMR (500 MHz, CDCl
3):
δ 0.81 (3H, s, 18-CH
3), 1.20–1.53 (7H, overlapping m), 1.61–1.80 (2H, m), 1.86–1.99 (1H, m), 1.99–2.09 (1H, m), 2.09–2.21 (1H, m), 2.31–2.39 (1H, m), 2.40–2.47 (1H, m), 3.03–3.09 (2H, m), 3.74–3.80 (1H, t,
J 8.5, 17-αH), 7.35 (1H, s, 4-H), 7.47–7.60 (3H, overlapping m, 3″-H, 4″-H and 5″-H), 7.78 (1H, s, 1-H), 7.90–7.97 (2H, m, 2″-H and 6″-H);
13C NMR (125 MHz, CDCl
3)
δ 11.2 (18-CH
3), 23.4 (CH
2), 26.7 (CH
2), 27.1 (CH
2), 30.4 (CH
2), 30.8 (CH
2), 36.8 (CH
2), 38.8 (8-CH), 43.4 (13-C), 44.4 (9-CH), 50.6 (14-CH), 82.0 (17-CH), 109.3 (4-CH), 117.9 (1-CH), 119.0 (2-C), 128.2 (2″-CH and 6″-CH), 129.2 (3″-CH and 5″-CH), 129.6 (1″-C), 130.2 (4″-CH), 137.7 (C), 140.7 (C), 157.3 (C), 162.8 (C); ESI-MS:
m/
z 374.2 [M + H]
+, 374.2 calcd. for [C
25H
28NO
2]
+.
3′-Trifluoromethylisoxazolo[4′,5′:2,3]estra-1,3,5(10)-triene-17β-ol (
4f): According to
Section 3.2.5,
3f (115 mg, 0.30 mmol, mixture of
E and
Z-oximes) was used for the reaction. Eluent (CC): EtOAc/hexane = 20:80. Yield (
4f): 44 mg (40%, white crystals); Mp: 106–108 °C; Anal. Calcd. for C
20H
22F
3NO
2 (365.40) C 65.74; H 6.07; N 3.83. Found C 65.60; H 6.41; N 3.75.
1H NMR (500 MHz, CDCl
3):
δ 0.81 (3H, s, 18-CH
3), 1.21–1.32 (1H, m), 1.31–1.43 (2H, m), 1.40–1.49 (3H, m), 1.48–1.57 (1H, m), 1.61–1.79 (2H, m), 1.91–1.99 (1H, m), 2.00–2.09 (1H, m), 2.09–2.20 (1H, m), 2.29–2.38 (1H, m), 2.38–2.47 (1H, m), 2.99–3.11 (2H, m), 3.73–3.80 (1H, t,
J 8.6, 17-αH), 7.39 (1H, s, 4-H), 7.66 (1H, s, 1-H);
13C NMR (125 MHz, CDCl
3)
δ 11.1 (18-CH
3), 23.4 (CH
2), 26.4 (CH
2), 26.8 (CH
2), 30.4 (CH
2), 30.7 (CH
2), 36.6 (CH
2), 38.4 (8-CH), 43.3 (13-C), 44.2 (9-CH), 50.5 (14-CH), 81.9 (17-CH), 109.4 (4-CH), 115.7 (2-C), 116.8 (1-CH), 120.5 (1C, q,
J 271.3, CF
3), 139.6 (C), 142.8 (C), 149.8 (1C, q,
J 38.2, 3′-C), 163.2 (3-C); ESI-MS:
m/
z 368.3 [M+3H]
+, 366.2 calcd. for [C
20H
23F
3NO
2]
+.
3′-Carboxymethylisoxazolo[4′,5′:2,3]estra-1,3,5(10)-triene-17β-acetate (
4g): According to
Section 3.2.5,
3g (700 mg, 1.68 mmol) was used for the reaction. Eluent (CC): EtOAc/hexane = 20:80. Yield (
4g): 601 mg (90%, yellowish white crystals); Mp: 176–178 °C; Anal. Calcd. for C
23H
27NO
5 (397.47) C 69.50; H 6.85; N 3.52. Found C 69.28; H 7.03; N 3.39.
1H NMR (500 MHz, CDCl
3):
δ 0.85 (3H, s, 18-CH
3), 1.27–1.42 (1H, m), 1.39–1.52 (4H, overlapping m), 1.50–1.71 (2H, m), 1.73–1.83 (1H, m), 1.90–2.00 (2H, m), 2.07 (3H, s, Ac-CH
3), 2.20–2.30 (1H, m), 2.30–2.39 (1H, m), 2.40–2.49 (1H, m), 2.98–3.11 (2H, m), 4.08 (3H, s, COOC
H3), 4.68–4.75 (1H, m, 17-αH), 7.36 (1H, s, 4-H), 7.99 (1H, s, 1-H);
13C NMR (125 MHz, CDCl
3)
δ 12.2 (18-CH
3), 21.3 (Ac-CH
3), 23.5 (CH
2), 26.4 (CH
2), 26.9 (CH
2), 27.7 (CH
2), 30.4 (CH
2), 36.8 (CH
2), 38.2 (8-CH), 43.0 (13-C), 44.1 (9-CH), 50.3 (14-CH), 53.0 (COO
CH
3), 82.7 (17-CH), 109.1 (4-CH), 118.2 (2-C), 118.7 (1-CH), 139.2 (C), 141.8 (C), 150.0 (3′-C), 161.0 (3-C), 163.3 (
COOCH
3), 171.3 (Ac-C=O); ESI-MS:
m/
z 398.2 [M + H]
+, 398.2 calcd. for [C
23H
28NO
5]
+.
3.2.6. 3-Hydroxy-2-trifluoracetylestra-1,3,5(10)-triene-17-one (6a)
To a suspension of E1 (1.08 g, 4.0 mmol) in DCM (30 mL), AlCl3 (3.2 g, 6 equiv.) and TFAA (670 µL, 1.2 equiv.) were added at 0 °C. After 30 min, the ice bath was removed, the reaction was gradually warmed to RT and stirred for 4 h. The reaction was poured into ice-cold HCl (1 M, 100 mL) and stirred for 10 min. The layers were separated and the aqueous phase was extracted with EtOAc (3 × 30 mL). The combined organic phase was washed with aq. NaHCO3 (10 wt. %, 40 mL), water (40 mL) and brine (40 mL), then dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by CC (EtOAc/hexane = 10:90). Yield (6a): 1.42 g (97%, yellow crystals); Mp: 179–181 °C; Anal. Calcd. for C20H21F3O3 (366.38) C 65.57; H 5.78. Found C 65.46; H 5.97. 1H NMR (500 MHz, CDCl3): δ 0.92 (3H, s, 18-CH3), 1.41–1.69 (6H, overlapping m), 1.96–2.11 (3H, m), 2.11–2.21 (1H, m), 2.21–2.33 (1H, m), 2.34–2.42 (1H, m), 2.48–2.57 (1H, m), 2.87–3.03 (2H, m), 6.82 (1H, s, 4-H), 7.69 (1H, s, 1-H), 10.87 (1H, s, 3-OH); 13C NMR (125 MHz, CDCl3) δ 13.9 (18-CH3), 21.7 (CH2), 25.7 (CH2), 26.1 (CH2), 30.2 (CH2), 31.5 (CH2), 35.9 (CH2), 38.0 (8-CH), 43.6 (13-C), 48.0 (9-CH), 50.5 (14-CH), 112.3 (2-C), 116.7 (1C, q, J 290.0, CF3), 118.4 (4-CH), 127.3 (1C, q, J 3.9, 1-CH), 132.6 (C), 150.7 (C), 162.5 (3-C), 183.9 (1C, q, J 35.0, C=O), 220.3 (17-C=O); ESI-MS: m/z 365.0 [M − H]−, 365.1 calcd. for [C20H20F3O3]−.
3.2.7. 3-Hydroxy-17-oxoestra-1,3,5(10)-triene-2-carboxylic Acid (6b)
To a solution of 6a (1.35 g, 3.68 mmol) in EtOH (20 mL), KOH (2.06 g, 10 equiv.) in water (3 mL) was added, and the mixture was kept at reflux temperature for 3 h. Then, it was acidified with HCl (6M) and the EtOH was removed under reduced pressure. To the residue HCl (1 M, 10 mL) was added and extracted with EtOAc (3 × 10 mL). The combined organic phase was washed with brine (20 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by CC (EtOAc/DCM = 10:90 with 1% AcOH additive to reduce tailing). Yield (6b): 1.09 g (94%, white powder); Mp > 190 °C (decomp.); Anal. Calcd. for C19H22O4 (314.38) C 72.59; H 7.05. Found C 72.62; H 7.24. 1H NMR (500 MHz, DMSO-d6): δ 0.82 (3H, s, 18-CH3), 1.29–1.52 (5H, overlapping m), 1.52–1.61 (1H, m), 1.73–1.80 (1H, m), 1.88–1.99 (2H, m), 2.01–2.11 (1H, m), 2.12–2.20 (1H, m), 2.26–2.34 (1H, m), 2.39–2.48 (1H, m), 2.77–2.90 (2H, m), 6.66 (1H, s, 4-H), 7.66 (1H, s, 1-H), 11.08 (1H, bs, 3-OH), 13.54 (1H, bs, COOH); 13C NMR (125 MHz, DMSO-d6): δ 13.4 (18-CH3), 21.1 (CH2), 25.4 (CH2), 25.6 (CH2), 29.0 (CH2), 31.2 (CH2), 35.3 (CH2), 37.4 (8-CH), 42.9 (13-C), 47.2 (9-CH), 49.5 (14-CH), 110.4 (2-C), 116.4 (4-CH), 126.5 (1-CH), 130.9 (C), 145.4 (C), 158.8 (3-C), 171.9 (C=O of carboxyl), 219.5 (C=O of ketone); ESI-MS: m/z 313.0 [M − H]−, 313.1 calcd. for [C19H21O4]−.
3.2.8. Three-Step Synthesis of Compound 7
Synthesis of 3,17β-dihydroxyestra-1,3,5(10)-triene-2-carboxylic acid (6c) by the reduction of 6b: To a solution of 6b (1.05 g, 3.34 mmol) in EtOH (30 mL), NaBH4 (631 mg, 5 equiv.) was added in small portions over a 10 min period and stirring was continued for 30 min at RT. The mixture was neutralized with HCl (6 M) and the EtOH was removed under reduced pressure. To the residue HCl (1 M, 10 mL) was added and extracted with EtOAc (3 × 10 mL). The combined organic phase was washed with brine (20 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by CC (EtOAc/DCM = 20:80 with 1% AcOH additive to reduce tailing). Yield (6c): 1.01 g (96%, white powder); Mp > 240 °C (decomp.); Anal. Calcd. for C19H24O4 (316.40) C 72.13; H 7.65. Found C 72.04; H 7.95. 1H NMR (500 MHz, DMSO-d6): δ 0.65 (3H, s, 18-CH3), 1.02–1.43 (7H, overlapping m), 1.52–1.62 (1H, m), 1.74–1.81 (1H, m), 1.82–1.93 (2H, m), 2.03–2.12 (1H, m), 2.18–2.26 (1H, m), 2.71–2.86 (2H, m), 3.48–3.55 (1H, t, J 8.5, 17-αH), 4.29–4.66 (1H, bs, 17-OH), 6.63 (1H, s, 4-H), 7.65 (1H, s, 1-H), 10.97 (1H, bs, 3-OH), 13.64 (1H, bs, COOH); 13C NMR (125 MHz, DMSO-d6): δ 11.1 (18-CH3), 22.7 (CH2), 25.9 (CH2), 26.4 (CH2), 29.1 (CH2), 29.8 (CH2), 36.4 (CH2), 38.2 (8-CH), 42.7 (13-C), 43.0 (9-CH), 49.5 (14-CH), 80.0 (17-CH), 110.3 (2-C), 116.3 (4-CH), 126.5 (1-CH), 131.4 (C), 145.5 (C), 158.7 (3-C), 172.0 (C=O of carboxyl); ESI-MS: m/z 315.0 [M − H]−, 315.2 calcd. for [C19H23O4]−.
Synthesis of 3,17β-dihydroxyestra-1,3,5(10)-triene-2-carboxylic acid methyl ester (6d) by esterification of 6c: To a solution of 6c (950 mg, 3.00 mmol) in DMF (10 mL), Na2CO3 (382 mg, 1.2 equiv.) and MeI (280 µL, 1.5 equiv.) were added and the mixture was stirred at 50 °C for 2 h. Then, it was repeatedly concentrated under vacuum with the addition of toluene, and then water (10 mL) was added and extracted with EtOAc (3 × 10 mL). The combined organic phase was washed with aq. NaHCO3 (10 wt. %, 2 × 10 mL), water (10 mL) and brine (10 mL), and then dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by CC (EtOAc/DCM = 5:95). Yield (6d): 912 mg (92%, white crystals); Mp: 154–156 °C; Anal. Calcd. for C20H26O4 (330.42) C 72.70; H 7.93. Found C 72.46; H 8.27. 1H NMR (500 MHz, CDCl3): δ 0.78 (3H, s, 18-CH3), 1.14–1.24 (1H, m), 1.24–1.58 (7H, overlapping m), 1.64–1.75 (1H, m), 1.84–1.92 (1H, m), 1.93–2.01 (1H, m), 2.09–2.19 (2H, m), 2.31–2.39 (1H, m), 2.82–2.92 (2H, m), 3.70–3.77 (1H, t, J 8.5, 17-αH), 3.92 (3H, s, COOCH3), 6.70 (1H, s, 4-H), 7.73 (1H, s, 1-H), 10.48 (1H, s, 3-OH); 13C NMR (125 MHz, CDCl3) δ 11.2 (18-CH3), 23.3 (CH2), 26.4 (CH2), 27.0 (CH2), 30.0 (CH2), 30.7 (CH2), 36.7 (CH2), 38.8 (8-CH), 43.4 (13-C), 43.8 (9-CH), 50.2 (14-CH), 52.2 (COOCH3), 82.0 (17-CH), 110.1 (2-C), 117.1 (4-CH), 126.6 (1-CH), 132.0 (C), 146.3 (C), 159.3 (3-C), 170.8 (COOCH3); ESI-MS: m/z 331.2 [M + H]+, 331.2 calcd. for [C20H27O4]+.
Synthesis of 2-hydroxycarbamoylestra-1,3,5(10)-triene-3,17β-diol (7) by hydroxamation of 6d: To a solution of 6d (898 mg, 2.72 mmol) in MeOH/THF = 2:1 (27 mL), aq. NH2OH (50 wt. %, 2.50 mL, 15 equiv.) and KOH (1.15 g, 7.5 equiv.) were added at 0 °C and the mixture was stirred at RT for 30 min. Then, it was poured into ice-cold HCl (2 M, 50 mL), and the precipitate was filtered off and dried. The crude product was used in the next step without further purification. Yield (7): 874 mg (97%, pale tan powder); Mp > 200 °C (decomp.); Anal. Calcd. for C19H25NO4 (331.41) C 68.86; H 7.60; N 4.23. Found C 68.98; H 7.87; N 4.04. 1H NMR (500 MHz, DMSO-d6): δ 0.68 (3H, s, 18-CH3), 1.04–1.14 (1H, m), 1.12–1.44 (6H, overlapping m), 1.53–1.63 (1H, m), 1.74–1.82 (1H, m), 1.83–1.94 (2H, m), 2.03–2.11 (1H, m, 2.33–2.41 (1H, m), 2.72–2.79 (2H, m), 3.49–3.57 (1H, td, J 8.6, 4.7, 17-αH), 4.39–4.44 (1H, d, J 4.8, 17-OH), 6.57 (1H, s, 4-H), 7.57 (1H, s, 1-H), 9.14 (1H, s, NOH), 11.35 (1H, s, NH), 12.03 (1H, s, 3-OH); 13C NMR (125 MHz, DMSO-d6): δ 11.1 (18-CH3), 22.6 (CH2), 25.8 (CH2), 26.4 (CH2), 28.9 (CH2), 29.8 (CH2), 36.3 (CH2), 38.3 (8-CH), 42.7 (13-C), 43.3 (9-CH), 49.5 (14-CH), 79.9 (17-CH), 111.0 (2-C), 116.4 (4-CH), 123.3 (1-CH), 130.9 (C), 142.6 (C), 157.2 (3-C), 166.7 (C=O); ESI-MS: m/z 332.2 [M + H]+, 332.2 calcd. for [C19H26NO4]+.
3.2.9. 3′-Hydroxyisoxazolo[4′,5′:2,3]estra-1,3,5(10)-triene-17β-ol (4h)
DIAD (615 µL, 3.12 mmol) was added to a solution of PPh3 (820 mg, 3.12 mmol) in THF (25 mL), followed by the addition of 7 (828 mg, 2.5 mmol) and the mixture was stirred at RT for 2 h. Then, it was concentrated under reduced pressure and purified by CC (isopropanol/DCM = 2:98 to 5:95). Yield (4h): 697 mg (89%, white powder); Mp > 220 °C (decomp.); Anal. Calcd. for C19H23NO3 (313.40) C 72.82; H 7.40; N 4.47. Found C 72.99; H 7.72; N 4.18. 1H NMR (500 MHz, DMSO-d6): δ 0.67 (3H, s, 18-CH3), 1.05–1.45 (7H, overlapping m), 1.54–1.64 (1H, m), 1.75–1.83 (1H, m), 1.82–1.94 (2H, m), 2.12–2.28 (2H, m), 2.77–2.83 (2H, m), 3.49–3.56 (1H, t, J 8.5, 17-αH), 4.49 (1H, bs, 17-OH), 6.92 (1H, s), 6.94 (1H, s), 11.37 (1H, bs, 3′-OH); 13C NMR (125 MHz, DMSO-d6): δ 11.2 (18-CH3), 22.7 (CH2), 26.2 (CH2), 26.7 (CH2), 29.1 (CH2), 29.9 (CH2), 36.5 (CH2), 38.2 (8-CH), 42.7 (13-C), 43.8 (9-CH), 49.6 (14-CH), 80.0 (17-CH), 106.3 (4-CH), 109.1 (1-CH), 128.2 (C), 130.1 (C), 135.7 (C), 141.6 (C), 154.7 (C); ESI-MS: m/z 314.2 [M + H]+, 314.2 calcd. for [C19H24NO3]+.
3.2.10. 3′-Methoxyisoxazolo[4′,5′:2,3]estra-1,3,5(10)-triene-17β-ol (4i)
To a solution of 4h (157 mg, 0.50 mmol) in DMF (1 mL), K2CO3 (207 mg, 3 equiv.) and MeI (47 µL, 1.5 equiv.) were added, and the mixture was stirred at RT for 1 h. Then, it was repeatedly concentrated under reduced pressure with the addition of toluene, and then water (10 mL) was added to the residue and extracted with EtOAc (3 × 10 mL). The combined organic phase was washed with aq. NaHCO3 (10 wt. %, 2 × 10 mL), water (10 mL) and brine (10 mL), and then dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by CC (EtOAc/DCM = 10:90). Yield (4i): 134 mg (82%, white crystals); Mp: 220–222 °C; Anal. Calcd. for C20H25NO3 (327.42) C 73.37; H 7.70; N 4.28. Found C 73.65; H 7.89; N 4.11. 1H NMR (500 MHz, CDCl3): δ 0.80 (3H, s, 18-CH3), 1.17–1.63 (8H, overlapping m), 1.67–1.77 (1H, m), 1.86–1.94 (1H, m), 1.96–2.03 (1H, m), 2.08–2.19 (1H, m), 2.22–2.38 (2H, m), 2.82–2.96 (2H, m), 3.37 (3H, s, 3′-OMe), 3.75 (1H, t, J 8.5, 17-αH), 6.87 (1H, s, 4-H), 6.90 (1H, s, 1-H); 13C NMR (125 MHz, CDCl3) δ 11.2 (18-CH3), 23.3 (CH2), 26.9 (CH2), 27.2 (CH2), 28.2 (3′-OMe), 29.8 (CH2), 30.8 (CH2), 36.8 (CH2), 38.7 (8-CH), 43.3 (13-C), 44.5 (9-CH), 50.3 (14-CH), 82.0 (17-CH), 105.1 (4-CH), 110.1 (1-CH), 129.9 (C), 131.5 (C), 136.4 (C), 141.1 (C), 155.3 (C); ESI-MS: m/z 328.2 [M + H]+, 328.2 calcd. for [C20H26NO3]+.
3.2.11. 3′-Hydroxymethylisoxazolo[4′,5′:2,3]estra-1,3,5(10)-triene-17β-acetate (4j)
To a solution of 4g (397 mg, 1.0 mmol) in EtOH (10 mL), NaBH4 (151 mg, 4 equiv.) was added in small portions and stirred at RT for 1 h. The solution was acidified with HCl (6 M) and EtOH was evaporated. The residue was suspended in water (10 mL), extracted with EtOAc (3 × 10 mL). The combined organic phase was washed with water (10 mL), brine (10 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by CC (MeOH/DCM = 2:98). Yield (4j): 327 mg (89%, greenish white crystals); Mp: 114–116 °C; Anal. Calcd. for C22H27NO4 (369.46) C 71.52; H 7.37; N 3.79. Found C 71.43; H 7.69; N 3.46. 1H NMR (500 MHz, CDCl3): δ 0.84 (3H, s, 18-CH3), 1.22–1.68 (7H, overlapping m), 1.72–1.82 (1H, m), 1.89–1.97 (2H, m), 2.07 (3H, s, Ac-CH3), 2.11–2.18 (1H, t, J 6.3, CH2-OH), 2.18–2.29 (1H, m), 2.29–2.44 (2H, m), 2.96–3.08 (2H, m), 4.67–4.74 (1H, m, 17-αH), 5.06 (2H, d, J 6.2, CH2-OH), 7.28 (1H, s, 4-H), 7.67 (1H, d, J 1.4, 1-H); 13C NMR (125 MHz, CDCl3) δ 12.2 (18-CH3), 21.3 (Ac-CH3), 23.5 (CH2), 26.4 (CH2), 27.0 (CH2), 27.7 (CH2), 30.4 (CH2), 36.9 (CH2), 38.3 (8-CH), 43.0 (13-C), 44.1 (9-CH), 50.3 (14-CH), 57.1 (CH2-OH), 82.7 (17-H), 109.0 (4-CH), 117.5 (1-CH), 118.8 (2-C), 137.4 (10-C), 141.0 (5-C), 157.6 (3′-C), 162.3 (3-C), 171.4 (Ac-C=O); ESI-MS: m/z 370.2 [M + H]+, 370.2 calcd. for [C22H28NO4]+.
3.2.12. 3′-Hydroxymethylisoxazolo[4′,5′:2,3]estra-1,3,5(10)-triene-17β-ol (4k)
To a solution of 4j (74 mg, 0.20 mmol) in MeOH/DCM = 1:9 (2 mL), NaOH (24 mg, 3 equiv.) was added and the mixture was stirred at RT for 1 h. Then, it was concentrated under reduced pressure and the crude product was purified by CC (MeOH/DCM = 2:98). Yield (4k): 62 mg (95%, white crystals); Mp: 218–220 °C; Anal. Calcd. for C20H25NO3 (327.42) C 73.37; H 7.70; N 4.28. Found C 73.20; H 8.08; N 4.09. 1H NMR (500 MHz, DMSO-d6): δ 0.68 (3H, s, 18-CH3), 1.12–1.44 (6H, overlapping m), 1.44–1.57 (1H, m), 1.58–1.66 (1H, m), 1.78–1.96 (3H, m), 2.22–2.30 (1H, m), 2.33–2.41 (1H, m), 2.90–3.03 (2H, m), 3.55 (1H, td, J 8.5, 4.5, 17-αH), 4.50 (1H, d, J 4.7, 17-OH), 4.82 (2H, d, J 4.5, CH2-OH), 5.65–5.71 (1H, m, CH2-OH), 7.38 (1H, s, 4-H), 7.82 (1H, s, 1-H); 13C NMR (125 MHz, DMSO-d6): δ 11.1 (18-CH3), 22.8 (CH2), 26.1 (CH2), 26.4 (CH2), 29.5 (CH2), 29.9 (CH2), 36.3 (CH2), 38.1 (8-CH), 42.7 (13-C), 43.6 (9-CH), 49.8 (14-CH), 54.7 (CH2-OH), 79.9 (17-CH), 108.2 (4-CH), 118.1 (1-CH), 118.9 (2-C), 136.8 (10-C), 140.5 (5-C), 158.4 (3′-C), 161.0 (3-C); ESI-MS: m/z 328.2 [M + H]+, 328.2 calcd. for [C20H26NO3]+.
3.2.13. 3′-Fluoromethylisoxazolo[4′,5′:2,3]estra-1,3,5(10)-triene-17β-acetate (4l)
To a solution of 4j (150 mg, 0.41 mmol) in DCM (2.5 mL), DAST (85 µL, 1.6 equiv.) was added under a nitrogen atmosphere and stirred at RT for 1 h. The reaction was quenched with the addition of aq. NaHCO3 (10 wt. %, 10 mL) and extracted with DCM (3 × 10 mL). The combined organic phase was washed with water (10 mL), brine (10 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by CC (EtOAc/hexane = 10:90). Yield (4l): 89 mg (59%, white crystals); Mp: 141–143 °C; Anal. Calcd. for C22H26FNO3 (371.45) C 71.14; H 7.06; N 3.77. Found C 71.01; H 7.43; N 3.68. 1H NMR (500 MHz, CDCl3): δ 0.85 (3H, s, 18-CH3), 1.24–1.69 (7H, overlapping m), 1.73–1.83 (1H, m), 1.90–2.00 (2H, m), 2.07 (3H, s, Ac-CH3), 2.18–2.30 (1H, m), 2.30–2.44 (2H, m), 2.97–3.09 (2H, m), 4.68–4.75 (1H, m, 17-αH), 5.66–5.84 (2H, d, J 47.0, CH2F), 7.32 (1H, s, 4-H), 7.68 (1H, s, 1-H); 13C NMR (125 MHz, CDCl3) δ 12.2 (18-CH3), 21.3 (Ac-CH3), 23.5 (CH2), 26.4 (CH2), 27.0 (CH2), 27.7 (CH2), 30.4 (CH2), 36.9 (CH2), 38.3 (8-CH), 43.0 (13-C), 44.1 (9-CH), 50.3 (14-CH), 76.2 (1C, d, J 166.7, CH2F), 82.7 (17-CH), 109.1 (4-CH), 117.4 (1-CH), 118.7 (2-C), 137.9 (C), 141.4 (C), 154.2 (1C, d, J 22.9, 3′-C), 162.4 (3-C), 171.4 (Ac-C=O); ESI-MS: m/z 372.2 [M + H]+, 372.2 calcd. for [C22H27FNO3]+.
3.2.14. 3′-Fluoromethylisoxazolo[4′,5′:2,3]estra-1,3,5(10)-triene-17β-ol (4m)
To a solution of 4l (62 mg, 0.17 mmol) in THF (1.7 mL), LiOH (1 M, 510 µL, 3 equiv.) was added and the mixture was stirred at RT overnight. Then, it was concentrated under reduced pressure and the crude product was purified by CC (EtOAc/DCM = 5:95). Yield (4m): 52 mg (95%, white crystals); Mp: 184–186 °C; Anal. Calcd. for C20H24FNO2 (329.42) C 72.92; H 7.34; N 4.25. Found C 72.98; H 7.46; N 4.03. 1H NMR (500 MHz, CDCl3): δ 0.80 (3H, s, 18-CH3), 1.18–1.31 (1H, m), 1.31–1.58 (6H, overlapping m), 1.61–1.79 (2H, m), 1.90–1.99 (1H, m), 1.99–2.06 (1H, m), 2.09–2.20 (1H, m), 2.28–2.37 (1H, m), 2.39–2.47 (1H, m), 2.97–3.09 (2H, m), 3.72–3.79 (1H, t, J 8.5, 17-αH), 5.69–5.84 (2H, d, J 47.0, CH2F), 7.31 (1H, s, 4-H), 7.69 (1H, s, 1-H); 13C NMR (125 MHz, CDCl3) δ 11.2 (18-CH3), 23.4 (CH2), 26.5 (CH2), 27.0 (CH2), 30.4 (CH2), 30.7 (CH2), 36.7 (CH2), 38.6 (8-CH), 43.3 (13-C), 44.2 (9-CH), 50.5 (14-CH), 76.2 (1C, d, J 166.8, CH2F) 81.9 (17-CH), 109.1 (4-CH), 117.4 (1-CH), 118.7 (2-C), 138.1 (C), 141.5 (C), 154.2 (1C, d, J 22.6, 3′-C), 162.4 (3-C). ESI-MS: m/z 330.2 [M + H]+, 330.2 calcd. for [C20H25FNO2]+.
3.2.15. 3′-Formylisoxazolo[4′,5′:2,3]estra-1,3,5(10)-triene-17β-acetate (4n)
To a solution of 4j (148 mg, 0.40 mmol) in DCM (5 mL), Dess–Martin periodinane (255 mg, 1.5 equiv.) was added under a nitrogen atmosphere and the mixture was stirred at RT for 2 h. The reaction was filtered into a separating funnel through cotton wool, and then water was added (10 mL) and extracted with DCM (3 × 10 mL). The combined organic phase was washed with aq. NaHCO3 (10 wt. %, 10 mL), water (10 mL) and brine (10 mL), and then dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by CC (hexane/DCM = 20:80). Yield (4n): 119 mg (81%, yellowish white crystals); Mp: 159–160 °C; Anal. Calcd. for C22H25NO4 (367.44) C 71.91; H 6.86; N 3.81. Found C 71.74; H 7.15; N 3.59. 1H NMR (500 MHz, CDCl3): δ 0.85 (3H, s, 18-CH3), 1.24–1.70 (7H, overlapping m), 1.72–1.82 (1H, m), 1.90–2.00 (2H, m), 2.07 (3H, s, Ac-CH3), 2.19–2.30 (1H, m), 2.30–2.40 (1H, m), 2.40–2.49 (1H, m), 2.98–3.10 (2H, m), 4.67–4.74 (1H, m, 17-αH), 7.39 (1H, s 4-H), 8.05 (1H, s, 1-H), 10.39 (1H, s, CHO); 13C NMR (125 MHz, CDCl3) δ 12.2 (18-CH3), 21.3 (Ac-CH3), 23.5 (CH2), 26.3 (CH2), 26.8 (CH2), 27.7 (CH2), 30.4 (CH2), 36.8 (CH2), 38.2 (8-CH), 43.0 (13-C), 44.1 (9-CH), 50.3 (14-CH), 82.7 (17-CH), 109.0 (4-CH), 116.3 (2-C), 118.8 (1-CH), 139.8 (C), 142.1 (C), 155.5 (C), 163.4 (C), 171.4 (Ac-C=O), 186.0 (CHO); ESI-MS: m/z 368.2 [M + H]+, 368.2 calcd. for [C22H26NO4]+.
3.2.16. 3′-Difluoromethylisoxazolo[4′,5′:2,3]estra-1,3,5(10)-triene-17β-acetate (4o)
To a solution of 4n (62 mg, 0.17 mmol) in DCM (2.0 mL), DAST (90 µL, 4.0 equiv.) was added at 0 °C under a nitrogen atmosphere and stirred at RT for 2 h. The reaction was quenched with the addition of aq. NaHCO3 (10 wt. %, 10 mL) and extracted with DCM (3 × 10 mL). The combined organic phase was washed with water (10 mL), brine (10 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by CC (EtOAc/hexane = 3:97). Yield (4o): 61 mg (92%, white crystals); Mp: 137–139 °C; Anal. Calcd. for C22H25F2NO3 (389.44) C 67.85; H 6.47; N 3.60. Found C 67.55; H 6.80; N 3.49. 1H NMR (500 MHz, CDCl3): δ 0.86 (3H, s, 18-CH3), 1.27–1.71 (7H, overlapping m), 1.72–1.83 (1 H, m), 1.91–2.00 (2H, m), 2.07 (3H, s, Ac-CH3), 2.19–2.30 (1H, m), 2.30–2.37 (1H, m), 2.37–2.45 (1H, m), 3.01–3.08 (2H, m), 4.67–4.76 (1H, t, J 8.4, 17-αH), 6.83–7.17 (1H, t, J 53.5, CHF2), 7.36 (1H, s, 4-H), 7.74 (1H, s, 1-H); 13C NMR (125 MHz, CDCl3) δ 12.2 (18-CH3), 21.3 (Ac-CH3), 23.5 (CH2), 26.3 (CH2), 26.9 (CH2), 27.8 (CH2), 30.4 (CH2), 36.9 (CH2), 38.3 (8-CH), 43.0 (13-C), 44.1 (9-CH), 50.4 (14-CH), 82.7 (17-H), 109.2 (4-CH), 110.5 (1C, t, J 236.5, CHF2), 116.1 (2-C), 117.5 (1-CH), 138.8 (C), 142.2 (C), 153.0 (1C, t, J 30.6, C=N), 162.9 (3-C), 171.3 (Ac-C=O); ESI-MS: m/z 390.2 [M + H]+, 390.2 calcd. for [C22H26F2NO3]+.
3.2.17. 3′-Difluoromethylisoxazolo[4′,5′:2,3]estra-1,3,5(10)-triene-17β-ol (4p)
To a solution of 4o (47 mg, 0.12 mmol) in THF (1.2 mL), LiOH (1 M, 360 µL, 3 equiv.) was added and the mixture was stirred at RT overnight. Then, it was concentrated under reduced pressure and the crude product was purified by CC (DCM). Yield (4p): 36 mg (86%, white crystals); Mp: 148–150 °C; Anal. Calcd. for C20H23F2NO2 (347.41) C 69.15; H 6.67; N 4.03. Found C 69.10; H 6.84; N 3.88. 1H NMR (500 MHz, CDCl3): δ 0.81 (3H, s, 18-CH3), 1.20–1.57 (7H, overlapping m), 1.62–1.79 (2H, m), 1.90–1.99 (1H, m), 1.99–2.07 (1H, m), 2.09–2.20 (1H, m), 2.28–2.37 (1H, m), 2.39–2.48 (1H, m), 2.98–3.08 (2H, m), 3.73–3.80 (1H, t, J 8.5, 17-αH), 6.82–7.14 (1H, t, J 53.5, CHF2), 7.35 (1H, s, 4-CH), 7.75 (1H, s, 1-CH); 13C NMR (125 MHz, CDCl3) δ 11.2 (18-CH3), 23.4 (CH2), 26.5 (CH2), 26.9 (CH2), 30.5 (CH2), 30.8 (CH2), 36.7 (CH2), 38.6 (8-CH), 43.4 (13-C), 44.3 (9-CH), 50.6 (14-CH), 81.9 (17-CH), 109.2 (4-CH), 110.5 (1C, t, J 236.5, CHF2), 116.1 (2-C), 117.5 (1-CH), 138.9 (C), 142.2 (C), 153.0 (1C, t, J 30.8, 3′-C), 162.9 (3-C); ESI-MS: m/z 348.2 [M + H]+, 348.2 calcd. for [C20H24F2NO2]+.
3.2.18. 17β-Hydroxy-isoxazolo[4′,5′:2,3]estra-1,3,5(10)-triene-3′-carboxylic Acid (4q)
To a solution of 4g (180 mg, 0.45 mmol) in MeOH/DCM = 1:9 (4.5 mL), NaOH (108 mg, 6 equiv.) was added and the mixture was stirred at RT for 1 h. Then, it was concentrated under reduced pressure, redissolved in MeOH and poured into ice-cold HCl (2M, 20 mL). The precipitate was filtered off and dried under vacuum. Yield (4q): 144 mg (94%, white powder); Mp > 150 °C (decomp.); Anal. Calcd. for C20H23NO4 (341.41) C 70.36; H 6.79; N 4.10. Found C 70.53; H 7.01; N 3.84. 1H NMR (500 MHz, DMSO-d6): δ 0.65 (3H, s, 18-CH3), 1.03–1.42 (7H, overlapping m), 1.52–1.62 (1H, m), 1.72–1.79 (1H, m), 1.79–1.86 (1H, m), 1.83–1.93 (1H, m), 1.99–2.08 (1H, m), 2.21–2.29 (1H, m), 2.72–2.78 (2H, m), 3.47–3.54 (1H, t, J 8.5, 17-αH), 4.49 (1H, bs, 17-OH), 6.66 (1H, s, 4-H), 7.36 (1H, s, 1-H), 10.99 (1H, bs, COOH); 13C NMR (125 MHz, DMSO-d6): δ 11.2 (18-CH3), 22.7 (CH2), 25.7 (CH2), 26.4 (CH2), 29.3 (CH2), 29.8 (CH2), 36.4 (CH2), 38.1 (8-CH), 42.7 (13-C), 43.0 (9-CH), 49.4 (14-CH), 80.0 (17-CH), 96.3 (C), 115.9 (4-CH), 117.8 (C), 124.2 (C), 129.5 (1-CH), 131.4 (C), 144.1 (C), 158.4 (C); ESI-MS: m/z 342.2 [M + H]+, 342.2 calcd. for [C20H24NO4]+.
3.2.19. 17β-Hydroxy-isoxazolo[4′,5′:2,3]estra-1,3,5(10)-triene-3′-carboxylic Acid Methyl Ester (4r)
To a solution of 4q (100 mg, 0.29 mmol) in MeOH (3 mL), cc. H2SO4 (1 drop) was added and kept at reflux temperature for 36 h. Then, it was neutralized with NaHCO3 and concentrated in vacuo. The crude product was purified by CC (EtOAc/DCM = 10:90). Yield (4r): 94 mg (90%, white powder); Mp: 201–203 °C; Anal. Calcd. for C21H25NO4 (355.43) C 70.96; H 7.09; N 3.94. Found C 70.73; H 7.34; N 3.65. 1H NMR (500 MHz, CDCl3): δ 0.80 (3H, s, 18-CH3), 1.21–1.58 (7H, overlapping m), 1.62–1.79 (2H, m), 1.90–1.99 (1H, m), 1.99–2.06 (1H, m), 2.09–2.23 (1H, m), 2.29–2.38 (1H, m), 2.43–2.52 (1H, m), 2.98–3.10 (2H, m), 3.72–3.80 (1H, td, J 8.5, 3.3, 17-αH), 4.08 (3H, s, COOCH3), 7.36 (1H, s, 4-CH), 7.99 (1H, s, 1-H); 13C NMR (125 MHz, CDCl3) δ 11.2 (18-CH3), 23.4 (CH2), 26.5 (CH2), 26.9 (CH2), 30.4 (CH2), 30.7 (CH2), 36.7 (CH2), 38.5 (8-CH), 43.3 (13-C), 44.3 (9-CH), 50.5 (14-CH), 53.0 (COOCH3), 81.9 (17-CH), 109.1 (4-CH), 118.2 (2-C), 118.7 (1-CH), 139.3 (C), 141.8 (C), 150.0 (C), 161.0 (C), 163.3 (COOCH3); ESI-MS: m/z 356.2 [M + H]+, 356.2 calcd. for [C21H26NO4]+.
3.2.20. 17β-Hydroxy-isoxazolo[4′,5′:2,3]estra-1,3,5(10)-triene-3′-carboxamide (4s)
To a solution of 4r (62 mg, 0.17 mmol) in NH3 (6 M in MeOH, 2 mL), cat. KCN was added and the mixture was stirred at RT for 4 h, and then it was concentrated in vacuo. The crude product was purified by CC (EtOAc/DCM = 50:50). Yield (4s): 56 mg (94%, white crystals); Mp: 250–252 °C; Anal. Calcd. for C20H24N2O3 (340.42) C 70.57; H 7.11; N 8.23. Found C 70.37; H 7.44; N 8.06. 1H NMR (500 MHz, DMSO-d6): δ 0.67 (3H, s, 18-CH3), 1.11–1.44 (6H, overlapping m), 1.47–1.66 (2H, m), 1.78–1.95 (3H, m), 2.23–2.38 (2H, m), 2.91–3.05 (2H, m), 3.50–3.58 (1H, td, J 8.5, 4.8, 17-αH), 4.48–4.53 (1H, d, J 4.8, 17-OH), 7.51 (1H, s, 4-CH), 7.94 (1H, s, 1-H), 7.98 (1H, s, one H of NH2), 8.29 (1H, s, the other H of NH2); 13C NMR (125 MHz, DMSO-d6): δ 11.1 (18-CH3), 22.8 (CH2), 26.0 (CH2), 26.2 (CH2), 29.5 (CH2), 29.8 (CH2), 36.3 (CH2), 37.9 (8-CH), 42.7 (13-C), 43.4 (9-CH), 49.8 (14-H), 79.9 (17-CH), 108.5 (4-CH), 117.7 (2-C), 118.5 (1-CH), 138.4 (C), 141.3 (C), 151.9 (3′-C), 160.7 (3-C), 161.9 (CONH2); ESI-MS: m/z 341.2 [M + H]+, 341.2 calcd. for [C20H25N2O3]+.
3.2.21. 2-Cyanoestra-1,3,5(10)-triene-3,17β-ol (8) by Cyclization Followed by Kemp Elimination from 3a (Scheme 3, xi)
DIAD (490 µL, 2.50 mmol) was added to a solution of PPh3 (656 mg, 2.50 mmol) in ACN (10 mL), followed by the addition of 3a (315 mg, 1.00 mmol). The reaction mixture was stirred at RT for 1 h, and then it was concentrated under reduced presure. The crude product was purified by CC (EtOAc/hexane = 10:90 to 40:60). Yield (8): 258 mg (87%, white powder); Mp > 200 °C (decomp.); Anal. Calcd. for C19H23NO2 (297.40) C 76.74; H 7.80; N 4.71. Found C 76.50; H 8.14; N 4.66. 1H NMR (500 MHz, DMSO-d6): δ 0.65 (3H, s, 18-CH3), 1.02–1.45 (7H, overlapping m), 1.52–1.62 (1H, m), 1.71–1.93 (3H, m), 1.99–2.10 (1H, m), 2.22–2.30 (1H, m), 2.73–2.80 (2H, m), 3.51 (1 H, t, J 8.5, 17-αH), 4.48 (1H, s, 17-OH), 6.67 (1H, s, 4-H), 7.39 (1H, s, 1-H), 10.63 (1H, s, 3-OH); 13C NMR (125 MHz, DMSO-d6): δ 11.1 (18-CH3), 22.7 (CH2), 25.6 (CH2), 26.3 (CH2), 29.3 (CH2), 29.8 (CH2), 36.3 (CH2), 38.1 (8-CH), 42.7 (13-C), 42.9 (9-CH), 49.4 (14-CH), 79.9 (17-CH), 96.2 (2-C), 115.7 (4-CH), 117.5 (CN), 129.6 (1-CH), 132.0 (C), 144.2 (C), 157.6 (3-C); ESI-MS: m/z 298.2 [M + H]+, 298.2 calcd. for [C19H24NO2]+.
3.2.22. Estra-1,3,5(10)-triene-3,17β-diol-2-carboxamidoxime (9)
To a solution of 8 (48 mg, 0.16 mmol) in EtOH (2 mL), Na2CO3 (34 mg, 2 equiv.) and aq. NH2OH (50 wt. %, 49 µL, 5 equiv.) were added and the mixture was refluxed overnight. After being concentrated under reduced presure, water (10 mL) was added and extracted with EtOAc (3 × 10 mL). The combined organic phase was washed with water (10 mL) and brine (10 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by CC (EtOAc/DCM = 20:80). Yield (9): 48 mg (90%, white powder); Mp > 210 °C (decomp.); Anal. Calcd. for C19H26N2O3 (330.43) C 69.06; H 7.93; N 8.48. Found C 69.13; H 8.15; N 8.29. 1H NMR (500 MHz, DMSO-d6): δ 0.67 (3H, s, 18-CH3), 1.07–1.44 (8H, overlapping m), 1.54–1.62 (1H, m), 1.75–1.81 (1H, m), 1.83–1.94 (2H, m), 2.04–2.13 (1H, m), 2.70–2.76 (2H, m), 3.49–3.57 (1H, td, J 8.3, 4.7, 17-αH), 4.47 (1H, d, J 4.8, 17-OH), 6.28 (2H, s, -NH2), 6.49 (1H, s, 4-H), 7.47 (1H, s, 1-H), 9.85 (1H, s, -OH), 11.89 (1H, s, -OH); 13C NMR (125 MHz, DMSO-d6): δ 11.3 (18-CH3), 22.8 (CH2), 25.9 (CH2), 26.7 (CH2), 28.9 (CH2), 29.9 (CH2), 36.5 (CH2), 38.6 (8-CH), 42.8 (13-C), 43.7 (9-CH), 49.6 (14-CH), 80.0 (17-CH), 112.2 (2-C), 115.9 (4-CH), 122.6 (1-CH), 130.2 (C), 138.7 (C), 153.7 (C), 154.9 (C); ESI-MS: m/z 331.2 [M + H]+, 331.2 calcd. for [C19H27N2O3]+.
3.2.23. 2′-Aminooxazolo[4′,5′:2,3]estra-1,3,5(10)-triene-17β-ol (10)
DDQ (52 mg, 0.23 mmol) was added slowly to the solution of PPh3 (59 mg, 0.23 mmol) in DCM (1 mL), and the mixture was stirred for 1 min. This suspension was added to a solution of 9 (50 mg, 0.15 mmol) and Et3N (45 µL, 0.30 mmol) in DCM (1 mL), and the mixture was stirred at RT for 30 min. After 30 min, another portion of DDQ/PPh3 was added and stirred until TLC indicated the complete conversion of the starting material. The solvent was removed under vacuum; the residue was dissolved in EtOAc/MeOH, and then Celite® was added (~10 × weight of the crude sample) and the solvent was removed in vacuo. The crude product was purified by CC (EtOAc/hexane = 70:30). Yield (10): 33 mg (70%, white powder); Mp > 210 °C (decomp.); Anal. Calcd. for C19H24N2O2 (312.41) C 73.05; H 7.74; N 8.97. Found C 73.26; H 7.98; N 8.75. 1H NMR (500 MHz, DMSO-d6): δ 0.67 (3H, s, 18-CH3), 1.08–1.46 (7H, overlapping m), 1.54–1.64 (1H, m), 1.76–1.82 (1H, m), 1.83–1.94 (2H, m), 2.13–2.22 (1H, m), 2.30 (1H, m), 2.76–2.89 (2H, m, 6-H2), 3.53 (1H, td, J 8.5, 4.6, 17-αH), 4.48 (1H, d, J 4.8, 17-OH), 6.96 (1H, s, 4-H), 7.08 (1H, s, 1-H), 7.15 (2H, s, NH2); 13C NMR (125 MHz, DMSO-d6): δ 11.2 (18-CH3), 22.8 (CH2), 26.3 (CH2), 27.0 (CH2), 29.3 (CH2), 29.9 (CH2), 36.6 (CH2), 38.5 (8-CH), 42.7 (13-C), 44.1 (9-CH), 49.7 (14-CH), 80.0 (17-CH), 107.8 (4-CH), 111.8 (3-CH), 128.3 (5-C), 135.3 (10-C), 141.6 (2-C), 146.2 (3-C), 162.3 (2′-C); ESI-MS: m/z 313.2 [M + H]+, 313.2 calcd. for [C19H25N2O2]+.



 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}